

Abstract
Background
Neuroblastoma (NB) tumors expressing high levels of brain-derived neurotrophic factor (BDNF) and its receptor TrkB, or activated Akt are associated with decreased event-free or overall survival in neuroblastoma patients. The effect of perifosine, an Akt inhibitor, on chemo-sensitivity of TrkB expressing neuroblastoma cells or tumors was evaluated.
Methods
A tetracycline-regulated TrkB-expressing isogenic NB cell model system was tested and in this system NB cells were treated with etoposide and/or perifosine in vitro and in vivo. Inhibition of the target by perifosine was evaluated by Western Blotting or kinase activity assay. Cell survival and tumor growth were investigated.
Results
In vitro BDNF treatment induced Akt phosphorylation and rescued cells from etoposide-induced cell death in high TrkB-expressing cells, but not in low TrkB-expressing cells. Pretreatment of high TrkB-expressing TB3 cells with perifosine blocked BDNF/TrkB-induced Akt phosphorylation, and inhibited BDNF’s protection of TB3 cells from etoposide treatment. In vivo, tumors with high TrkB expression had elevated levels of phosphorylated Akt, and were less sensitive to etoposide treatment compared to tumors with low TrkB expression. Mice treated with a combination of perifosine and etoposide had a statistically significant decrease of tumor growth compared to mice treated with either etoposide or perifosine alone. Activation of Akt through BDNF/TrkB signaling pathway induced chemo-resistance in NB in vivo.
Conclusions
Perifosine-induced inhibition of Akt increased the sensitivity of NB to chemotherapy. Our study supports the future clinical evaluation of an Akt inhibitor combined with cytotoxic drugs for improvement of treatment efficacy.
Keywords: neuroblastoma, Akt, brain-derived neurotrophic factor, TrkB, perifosine, etoposide
Introduction
Neuroblastoma (NB) is the most common extracranial solid tumor in childhood that derives from neural crest precursor cells1. It accounts for almost 8% of pediatric malignancies, yet is responsible for about 15% of all pediatric cancer deaths2,3. Spontaneous regression, differentiation and a good response to current therapeutic regimens such as surgery and chemotherapy are common in infants or those with low risk tumors. However children, typically over 18 months, fail to have sustained responses to intensive multimodality chemotherapy and stem cell transplant4. While patients may initially response to chemotherapy, they eventually relapse with multifocal metastatic disease and resistance to chemotherapy. Despite the aggressive treatment, the prognosis of these patients is poor and their long-term survival is less than 40%4. More effective treatment strategies are urgently needed for this high-risk group of neuroblastoma patients.
A better understanding of the genetics and biologic behavior of poor prognosis NB tumors should lead to new molecular targets that can be used to develop more specific, more effective and less toxic cancer therapies. Following the reports that brain-derived neurotrophic factor (BDNF) and its tyrosine kinase receptor TrkB are often detected in NB tumors derived from patients with an unfavorable prognosis5, we identified that BDNF activation of TrkB attenuated the sensitivity of NB cells to chemotherapy6. We later identified PI3Kinase and its downstream target, Akt serine/threonine kinase as critical mediators of activated TrkB induced chemo-resistance in NB cells7,8. Besides BDNF/TrkB, specific ligand induced activation of other tyrosine kinase receptors, such as IGFR9, PDGFR10, VEGFR11, are known to activate Akt and affect NB cell survival. Studies have shown that IGF protects NB cells from chemotherapy-induced or osmolarity-induced cell death in NB cells via activation of Akt12. PDGF, a potent mesenchymal cell-derived mitogen also activates Akt in PDGFR expressing NB cells and stimulates cell proliferation, chemotaxis and neurite extension. PDGFR inhibitors block PDGR-induced phosphorylation of Akt and induce apoptosis in NB cells10. Gastrin-releasing peptide receptor (GRP-R), is a G protein coupled receptor whose up-regulation in advanced stage human NB is accompanied by increased levels of activated Akt13. Anaplastic lymphoma kinase (ALK) is a tyrosine kinase and mutation or amplification of ALK are associated with Akt activation in poor prognosis NB14. In drug resistance models, Cathespin D-induced resistance to doxorubicin is mediated by activated Akt15. Recently, a study of tumors from 116 patients with NB showed that activated Akt correlates with poor prognosis and advanced disease stage in primary NB, as well as with unfavorable biological markers in NB such as MYCN amplification and 1p36 aberrations16. It is obvious that Akt is a critical survival signaling node in many signal transduction pathways and the association of activated Akt with poor prognosis and advanced stage disease in NB makes it an important target for treatment.
Although Akt inhibitors have been developed and tested in many adult tumor model systems17, our recent study was the first to systematically evaluate an Akt inhibitor as single agent in the pediatric tumor neuroblastoma18. Since therapeutic studies implicating a role for Akt in chemo-resistance in NB have been performed for the most part using in vitro models, in this study we used a tetracycline(TET)-regulated TrkB expression system to demonstrate that increased expression of TrkB is associated with elevated levels of activated Akt in vivo and reduced sensitivity to etoposide. Moreover we show that using the Akt inhibitor perifosine, increases the sensitivity of NB tumors to etoposide and causes regression of NB tumors.
Results
1. Tetracycline-regulated TrkB expressing cell line
To study the role of BDNF/TrkB-Akt in neuroblastoma’s response to chemotherapy, we used a tetracycline (TET off)-suppressible TrkB expression cell line, TB37. The presence of tetracycline inhibited expression of TrkB, while absence of tetracycline induced expression of TrkB. In the absence of TET there was a 3.6-fold increase of TrkB mRNA levels (Fig. 1A) and 2.5-fold increase of TrkB protein levels (Fig. 1B) (densitometric analysis of the western blotting result). Consistent with previous results7, BDNF treatment induced an increase in phosphorylation of TrkB only in TrkB-expressing cells (TET-) (Fig. 1B), and this was accompanied by an increase in levels of phosphorylated Akt (Ser473 and Thr308)(3.8-fold and 2-fold, respectively) (Fig. 1C). An Akt activity assay detected an increased Akt activity indicated by phosphorylated GSK-3α/β (Ser21/9) in TrkB-expressing cells treated with BDNF (Fig. 1C). There was no difference in levels of phosphorylated-TrkB or phosphorylated-Akt (P-Akt) between TET+ and TET- conditions in the absence of BDNF. These data indicate that in our model system TET-regulated TrkB mRNA and protein levels and BDNF stimulation induced an increase in TrkB phosphorylation and induction of its downstream target Akt in high TrkB expressing NB cells.
Figure 1.

TrkB expression is regulated by tetracycline in vitro and affects the NB cell response to chemotherapy. TB3 cells were cultured for 3 days in the presence or absence of tetracycline (1μg/ml), then total RNA was extracted (A); treated with BDNF (100ng/ml) for 15min, then total protein was extracted (B, C), treated with etoposide (D) or a combination of BDNF and etoposide (E, F) for 24hrs. A. Total RNA (1μg) was reverse-transcribed into cDNA following by quantitative-PCR analysis for TrkB mRNA. B. Protein lysates (500μg/ml) were immunoprecipitated with anti-pan Trk antibody and subjected to Western Blotting for evaluation of TrkB and phosphorylated tyrosine. C. Total protein (30μg) were immunoblotted for P-Akt (Ser473), P-Akt (Thr308) and total Akt. In vitro Akt kinase activity was assessed by immunoprecipitating with P-Akt (Ser473) antibody, and then using GSK-3 as a substrate to measure Akt activity indicated by phosphorylated GSK-3α/β (Ser21/9). D, E and F: MTS assay was used to determine cell survival at the end of treatment. * P<0.05, BDNF+etoposide treatment vs etoposide treatment.
To compare the response to etoposide between high TrkB and low TrkB expressing cells, TB3 cells were treated with etoposide in the presence or absence of TET. Etoposide induced a decrease in cell survival in both high and low TrkB-expressing cells, but there was no difference in their sensitivity to etoposide (Fig. 1D). To evaluate the response to etoposide after activation of TrkB signaling pathway, we pretreated the cells with BDNF before etoposide. The addition of BDNF to the low TrkB expressing cells didn’t change the response to etoposide (Fig 1E), but the addition of BDNF to the high TrkB-expressing cells caused an almost 40% increase in cell survival after etoposide treatment (Fig. 1F).
2. Akt inhibitor perifosine blocked BDNF-induced protection of TB3 cells from etoposide
In a previous study we showed that constitutively active Akt attenuated the cytotoxic effects of etoposide on NB cells and the use of a small molecule inhibitor targeting the PH domain of Akt restored sensitivity to etoposide8. Using an Akt inhibitor perifosine which is in Phase II clinical trials in adults and FDA approved for the treatment of Neuroblastoma, we treated the high TrkB-expressing TB3 cells with perifosine and evaluated cell growth. We found that perifosine treatment induced cell death in a dose-dependent manner at 24hrs (Fig. 2A) that was accompanied by an increase in caspase 3/7 activity (Fig. 2A). We selected concentrations of perifosine (2.5μM, 5μM) that didn’t alter cell growth at 24hrs, to assess whether perifosine inhibited BDNF/TrkB-induced phosphorylation of Akt. Pre-treatment of high TrkB-expressing TB3 cells with perifosine (2.5 or 5μM) followed by BDNF treatment led to a 22% or 36% block of the increase of phosphorylated-Akt (Ser473) at 6hrs. At 24hrs 2.5μM perifosine induced a 60% and 5μM perifosine induced a 100% block in BDNF-induced increase of phosphorylated Akt (Ser473) (Fig. 2B). Similar results were found in another phosphorylated site of Akt (Thr308)(Fig. 2B). Pretreatment with perifosine blocked BDNF activation of TrkB-induced phosphorylation of S6, a downstream target of activated Akt (Fig. 2B). A 24hrs pre-treatment with perifosine completely blocked Akt activity as indicated by a decrease in phosphorylation of the Akt substrate GSK-3α/β (Ser21/9) assessed using an Akt activity assay (Fig. 2B). To determine whether perifosine altered BDNF/TrkB protection of TB3 cells from etoposide-induced cell death, we pretreated high TrkB-expressing TB3 cells with a concentration of perifosine (5μM), which inhibits activation of both Akt and its downstream target S6, followed by etoposide (1μg/ml) treatment in the absence or presence of BDNF (100ng/ml) for an additional 24hrs. The results showed that etoposide-induced cell death was blocked by pre-treatment with BDNF. However, prior treatment with perifosine blocked the BDNF/TrkB mediated rescue of TB3 cells from etoposide-induced cell death (Fig. 2C). These data indicate that perifosine blocks Akt phosphorylation and attenuates BDNF/TrkB-induced resistance to etoposide.
Figure 2.

Perifosine blocked BDNF/TrkB-induced protection of TB3 cells from etoposide. A. TB3 cells cultured in the absence of tetracycline were treated with different concentrations of perifosine for 24hrs, MTS assay was used to detect cell survival (left); or treated with perifosine for 16hrs then caspase 3/7 activity was detected (right). B. TB3 cells cultured in the absence of tetracycline were treated with perifosine (2.5μM or 5μM) for 6hrs or 24hrs followed by a 15min treatment of BDNF (100ng/ml), total proteins were extracted. The expression of P-Akt (Ser473), P-Akt (Thr308), T-Akt, P-S6 (Ser235/236) and T-S6 were analyzed by Western Blotting. In vitro Akt kinase activity was assessed as described in Fig. 1C. C. TB3 cells cultured in the absence of tetracycline were first pretreated with perifosine (5μM) for 6hrs, then treated with BDNF (100ng/ml) for 1h, followed by etoposide (1μg/ml) treatment for 24hrs. MTS assay was used to assess cell survival.
3. TET-regulated TrkB expressing Tumors in vivo
To determine whether increased TrkB expression and resistance to chemotherapy determined in in vitro models were relevant in an in vivo model system, we injected TB3 cells into nude mice that had been given access to water with TET plus sucrose (TET+) or water with sucrose alone (TET-). Our results showed that the levels of TrkB mRNA in tumors from TET- mice were almost 3-fold higher than TrkB levels in tumors from TET+ mice. The levels of TrkB protein expression and the levels of tyrosine phosphorylated TrkB in tumors from TET- mice were higher than the levels detected in tumors from TET+ mice (Fig. 3B). Hematoxyline and eosin staining of tumor tissues showed that tumors from both groups of mice had the characteristic histology of small, round, blue cell tumors of childhood typical of NB tumors (Fig. 3C). Immunohistochemical staining analyses showed that the relative levels of P-Akt were higher in tumors expressing high TrkB from TET-mice compared to those in low TrkB expressing tumors from TET+ mice (Fig. 3D). These data indicate that in the absence of TET, TrkB expression is induced in vivo and this is associated with increased expression of phosphorylated Akt.
Figure 3.

Tetracycline-regulated TrkB expressing system in vivo. Subcutaneous xenografts were established as described in “Materials and Methods”. A. mRNA was extracted from the tumor tissues. Total RNA (1μg) was reverse-transcribed into cDNA followed by quantitative-PCR analysis for TrkB mRNA levels. B. Total protein was extracted from tumor tissues, protein lysates (500μg/ml) were immunoprecipitated with anti-pan Trk antibody and subjected to Western Blotting for evaluation of TrkB and phosphorylated tyrosine (B). C and D. Tumor tissues were stained with Hematoxyline-eosin (C) or stained with P-Akt (Ser473) by immunohistochemistry staining (D). The magnifications of the pictures are 63× for upper row, and 20× for lower row.
4. High TrkB expressing tumors are more resistant to etoposide than low TrkB expressing tumors
To study the sensitivity of low and high TrkB expressing tumors to etoposide in vivo, we initiated etoposide treatment of mice (10mg/kg, 20mg/kg) when tumor volumes reached approximately 200mm3. In the mice with low TrkB expressing tumors (TET+), treatment with etoposide at either 10mg/kg or 20mg/kg caused an up to 47% decrease in tumor growth compared to placebo treatment (Fig. 4A). However in the mice with tumors expressing high TrkB (TET-), only etoposide at 20mg/kg caused an up to 39% decrease in tumor growth, while the growth of tumors treated with etoposide at 10mg/kg was not significantly different from tumor growth in the placebo treated mice (Fig. 4B). These data are consistent with a model in which the tumors with high TrkB expression are more resistant to etoposide treatment than the tumors with low TrkB expression.
Figure 4.

High TrkB expressing tumors are less sensitive to etoposide than low TrkB expressing tumors. Mice with tumors were treated with vehicle or etoposide at 10mg/kg or 20mg/kg for 21days. The size of the tumors from the etoposide-treated groups were compared to that of control group. # P<0.05 10mg/kg etoposide treated group vs control group, * P<0.05, 20mg/kg etoposide treated group vs control group.
5. Perifosine sensitized high TrkB expressing TB3 tumors to etoposide treatment
Since high TrkB expressing TB3 tumors have elevated levels of P-Akt (Fig. 3D) and are more resistant to etoposide (Fig. 4), we next tested whether Akt inhibitor perifosine would affect the sensitivity of high TrkB expressing tumors to etoposide. We first tested the response of tumors to different doses of perifosine (10mg/kg, 15mg/kg, 20mg/kg). There was an anti-tumor growth effect of perifosine at 20mg/kg, but treatment with 10 or 15mg/kg of perifosine did not cause a statistically significant difference in tumor growth compared to the placebo treatment (Fig. 5A). When we examined the tumors by immunohistochemical staining to assess expression of P-Akt (Ser 473) and P-S6 (Ser235/236) in mice treated with 15mg/kg perifosine, we found decreased expression of P-Akt and P-S6 in these tumors compared to tumors from placebo-treated mice (Fig. 5B). So we evaluated whether perifosine at a dose (15mg/kg/day) sufficient to inhibit Akt activation, would alter the sensitivity of TB3 tumors to etoposide (10mg/kg). While neither drug at these doses, as a single agent, caused a statistically significant alteration in tumor growth, the combination of perifosine (15mg/kg) and etoposide (10mg/kg) resulted in an 80% decrease in tumor growth compared to the placebo (Fig. 5C). These data indicate that perifosine treatment inhibits Akt phosphorylation in tumor tissues and increases the sensitivity of TB3 NB tumors to etoposide treatment. To determine whether perifosine could synergize with etoposide in another TrkB expressing NB cell line, we tested three cell lines (NGP, LAN5, SY5Y) and found that NGP cells express TrkB and activated Akt (Fig. 6A). The in vivo sensitivities of NGP xenografts to etoposide (Fig. 6B) and perifosine (Fig. 6C) were established. Then mice were treated with a combination of etoposide (10mg/kg) and perifosine (17mg/kg) at doses that individually didn’t have statistically significant effect on tumor growth. We found that this combination of etoposide and perifosine induced an 85% inhibition of tumor growth compared to placebo (Fig. 6D).
Figure 5.
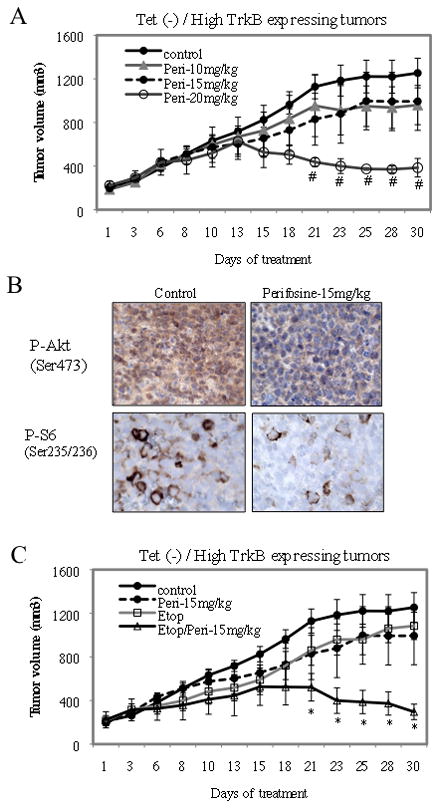
Perifosine increased the sensitivity of high TrkB expressing TB3 tumors to etoposide treatment. A. Mice were treated with vehicle or perifosine at 10mg/kg, 15mg/kg or 20mg/kg for 30 days. The tumor size in treated groups was compared to that of control group. # P<0.05, perifosine (20mg/kg) group vs control group. B. Tumor tissues were stained with P-Akt (Ser473) and P-S6 (Ser235/236) by immunohistochemistry staining. The magnifications of the pictures are 63×. C. Mice were treated with vehicle, perifosine (15mg/kg), etoposide (10mg/kg) or a combination of perifosine (15mg/kg) and etoposide (10mg/kg). The tumor size in treated groups was compared to that of control group. * P<0.05, perifosine (15mg/kg)+etoposide (10mg/kg) group vs control group.
Figure 6.
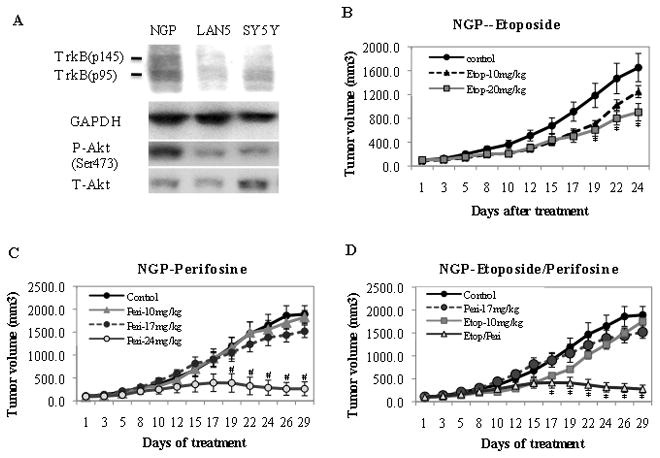
Perifosine increased the sensitivity of NGP tumors to etoposide treatment. A: Proteins from NGP, LAN5 and SY5Y cells were extracted and expressions of TrkB, GAPDH, P-Akt (Ser473) and T-Akt were analyzed by Western Blotting. B, C, D: NGP tumors were treated with etoposide (10mg/kg, 20mg/kg) (B) or perifosine (10mg/kg, 17mg/kg, 24mg/kg)(C) individually, or a combination of etopsoide (10mg/kg) and perifosine(17mg/kg) (D). The tumor size in treated groups was compared to that of control group. B: * P<0.05, Etoposide (20mg/kg) group vs control group. C: # P<0.05, Perifosine (24mg/kg) group vs control group. D: * P<0.05, perifosine (17mg/kg)+etoposide (10mg/kg) group vs control group.
Discussion
In this study we established an in vivo tumor model system using a TET repressible TrkB-expressing isogenic cell model system to study the influence of the TrkB/Akt signaling pathway on the response of NB tumors to chemotherapy. We found tumor xenografts with elevated TrkB had increased levels of activated TrkB (P-Tyr-TrkB) and Akt (P-Akt). Consistent with our in vitro studies, tumors expressing elevated activated TrkB and Akt were more resistant to etoposide. Finally the sensitivity of NB tumors expressing elevated TrkB levels to etoposide was restored if mice were treated with a concentration of perifosine sufficient to inhibit activated Akt in tumor xenografts. These findings indicate that targeting Akt in NB tumors may increase sensitivity to cytotoxic therapy.
Clinical observation has indicated that TrkB and BDNF are expressed in a subset of aggressive or unfavorable NB19. To understand how activation of the BDNF/TrkB pathway affected the biology of NB tumors, we and other investigators determined that BDNF activation of TrkB didn’t significantly alter NB cell growth, but enhanced cell survival under stress conditions such as serum-starvation20, 21. Later, we reported that BDNF activation of TrkB induced NB cell resistance to cytotoxic drug vinblastin in vitro6 and subsequent studies showed similar results using cytotoxic drugs, such as etoposide, cisplatin and paclitaxel7,8. Given the role of TrkB in the biological and clinical behavior of NB, it was reasonable to pursue inhibition of TrkB receptor as an important adjunct to therapy. Lestaurtinib (CEP-701) is a Trk-selective tyrosine kinase inhibitor that blocks activated Trk and has shown efficacy alone or in combination with conventional chemotherapy in NB cell model system utilizing constitutive TrkB expression22,23. Recently another Trk inhibitor AZ623 was found to inhibit activation of TrkB in vitro and NB cell growth in vivo24. However, neither of these studies showed the inhibition of target in vivo. Besides Trks, other receptor tyrosine kinases (RTK), such as VEGFR, EGFR, IGF1R have also been implicated in NB pathogenesis or malignant behavior 9–11. Utilization of a Trk inhibitor would only target those dependent on activated Trks, while cells not dependent on Trks could survive and even those that may initially be dependent on Trks may develop resistance or co-opt other RTKs for survival. So the advantage of targeting Akt pathway over RTK specific targeting strategies is that it would have a broader specificity against multiple RTKs that activate PI3K/Akt pathway and stimulate tumor cell survival. Therefore, it would not be necessary to know if a given tumor relied on a specific RTK, as long as its survival and/or other malignant characteristics were dependent on Akt25.
To date, several types of Akt inhibitors have been investigated, including phosphatidylinositol analog inhibitors, allosteric Akt kinase inhibitors, ATP-competitive inhibitors and alkylphospholipids26. However, use of these inhibitors is limited either by high toxicity or low bioavailability and stability in vivo26. Perifosine, an alkylphospholipid, acts at cell membrane, which is distinct from conventional chemotherapeutic drugs that mainly target DNA. In adult clinical trials perifosine has no hematological toxicity and the only dose-limiting toxicity was gastrointestinal that was readily ameliorated with medicine27. More recently, we found perifosine had anti-tumor growth effect in all NB xenograft tumor models tested and this effect was noted utilizing neuroblastoma cell lines that carried a number of genetic alterations which characterize NB tumors, such as MYCN amplification, P53 mutation, ALK mutation. The Phase I Clinical trial of perifosine in pediatric solid tumors utilized 3 doses of perifosine 25, 50 and 75mg/m2/day with serum levels reaching 17 to 32μM and no dose limiting toxicities were observed28. Our pre-clinical perifosine dosing of 24mg/kg/day would be equivalent to a Phase I dosing of 72mg/m2/day29, and in our in vitro studies doses of perifosine from 20–30μM induced at least a 70% induction of cell death in all NB cell lines evaluated. From these data one can see that doses used in our pre-clinical in vitro and in vivo models are achievable in patients. Although clinical response is not an aim of Phase I trials, 2/3 patients with stage 4 multiply – relapsed NB achieved stable disease for up to 55 weeks28.
As effective as a targeted monotherapy may be, it is usually enhanced when combined with traditional cytotoxic reagents. We found a synergistic response and an 80% decrease in tumor growth when mice bearing xenografts were given the combination of perifosine and etoposide using doses at which either agent alone had no significant effect on tumor growth. In TB3 tumors by using a dose of perifosine (15mg/kg/day, equal to 45mg/m2/day) which inhibited Akt activity but had little effect on tumor growth, we found the sensitivity of NB tumors to etoposide treatment was greatly increased. We did not observe any side effects utilizing these low doses of perifosine and etoposide (10mg/kg) in combination. An important goal of therapy is to not only increase treatment efficacy but also decrease toxicities. These results are similar to those reported using the Trk inhibitor22,23. So far no study has reported the combined usage of an Akt inhibitor and chemotherapeutic drugs in pediatric solid tumors in vivo, although perifosine has been shown to increase sensitivity of medulloblastoma to radiation in vitro30. In adult cancer studies, perifosine sensitizes endometrial cancer cells to cisplatin treatment31. In clinical trials, perifosine has synergistic anti-tumor growth effect when combined with tumor necrosis factor-related apoptosis inducing ligand (TRAIL) 32 in treatment of patients with multiple myeloma. From our pre-clinical studies and those in adults, we have reason to believe that treatment with perifosine in combination with conventional therapy will be good strategy to improve the treatment efficacy in NB.
Materials and Methods
Cell culture
Human neuroblastoma cells (TB3) that have a transfected tetracycline-regulated rat TrkB7 and NGP, LAN5, SY5Y cells were used in this study. Puromycin (0.5μg/ml) and tetracycline (1μg/ml) were added to the TB3 cells for maintenance of selection pressure and repression of TrkB gene. TB3 and NGP cells were cultured in RPMI 1640 containing 10% fetal bovine serum, 2mM glutamine and antibiotics as described previously7.
Reagents and antibodies
Recombinant human BDNF was obtained from PeproTech, Inc (Rocky Hill, NJ). Puromycin and tetracycline were purchased from Sigma, Inc. (St Louis MO). Etoposide was obtained from Bedford Laboratories (Bedford, OH). The Akt inhibitor perifosine was a gift from Keryx company. PanTrk antibody, TrkB antibody and phospho-tyrosine (P-Tyr) antibody were purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). Akt antibody, phospho-Akt (P-Akt, Ser473, Thr308) antibody, S6 antibody, phospho-S6 (P-S6, Ser235/236) antibody, GAPDH antibody and Akt kinase assay kit were obtained from Cell signaling technology (Beverly, MA).
Treatment
TB3 cells were cultured in the presence of tetracycline (1μg/ml) for 3 days in order to repress the TrkB expression. To study the activation of TrkB, TB3 cells were treated with BDNF (100ng/ml) for 15min. To study the response to etoposide or perifosine, TB3 cells were treated with different concentrations of etoposide or perifosine for 24hrs and cell survival was assessed using a MTS assay. To study the perifosine’s blockage of BDNF-induced phosphorylation of Akt, TB3 cells were pretreated with perifosine (2.5μM, 5μM) for 6hrs or 24hrs, then treated with BDNF (100ng/ml) for 15min. For the study of perifosine’s effect on BDNF-induced cell survival, TB3 cells were first treated with perifosine (5μM) for 6hrs, followed by treatment with BDNF and etoposide for 24hrs.
Real-time quantitative PCR
Total RNA was isolated using a Qiagen Extraction procedure. Equal amounts of total RNA (1μg) were reverse-transcribed using the Superscript First-Strand Synthesis System for RT-PCR (Invitrogen), and the resulting first strand cDNA was diluted and used as template in real-time quantitative-PCR analysis. All measurements were performed in duplicate. Actin served as internal control and was used to normalize variances in input cDNA. The following gene-specific primer pair was designed: TrkB-sense 5'-CTCAGCAAATCGCAGCAGG-3'; TrkB-antisense 5'-AGTAGTCGGTGCTGTATA-3'. The specificity of each primer was determined using NCBI BLAST module. Detection of TrkB expression was performed with SYBR Green (Applied Biosystems) and an ABI PRISM 7700 Sequence Detection System (Applied Biosystems).
Immunoprecipitation/Western Blotting
After treatment, TB3 cells were harvested and protein was extracted with protein lysis buffer as described previously7. Protein concentrations were determined using Bradford assay kit (Bio-Rad). For immunoprecipitation of TrkB and Phospho-tyrosine, 500μg of protein from each lysate was immnuoprecipitated with polyclonal anti-pan-Trk rabbit antibody and protein A agarose (Life Technologies, Inc. Carlsbad, CA). Immunoprecipitates were electrophoresed in 6% SDS-PAGE gels, transferred to nitrocellulose, and subsequently probed with an anti-phospho-tyrosine antibody or TrkB antibody. For Western blotting, 30μg of protein from each lysate were loaded onto 10% SDS-PAGE gels, transferred to nitrocellulose, and probed with anti-Akt antibody, anti-phospho-Akt (Ser473) antibody, anti-S6 antibody or anti-P-S6 (Ser235/236) antibody. Signals were detected using enhanced chemiluminescence reagents (Amersham Life Science, Arlington Heights, IL).
Akt kinase assay
TB3 cells were lysed in lysis buffer provided by the Akt kinase assay kit. Protein concentrations were determined using the Bradford assay kit(Bio-Rad). Akt kinase activity was assessed using Akt kinase assay kit (Cell Signaling Technology Inc., Beverly, MA) according to the manufacture’s instructions. Briefly, protein (200μg) from lysate samples were immunoprecipitated with phospho-Akt (Ser473) antibody conjugated with beads overnight at 4°C. The immunoprecipitates were washed with lysis buffer twice and kinase buffer twice. The samples were re-suspended in 50μl kinase buffer containing 1μl of 10mM ATP and kinase substrate GSK-3 fusion protein and allowed to proceed at 30°C for 30min. Reaction products were resolved by 10% SDS-PAGE gels followed by Western blotting with a phospho-GSK-3α/β antibody.
Immunohistochemistry staining
Tumor tissues were fixed in 10% formalin, embedded with paraffin, processed to slides with thickness of 5μm. The sections were stained with hematoxylin and eosin for histology observation, or slides were stained with P-Akt (Ser473, 1:50) antibody or P-S6 (Ser235/236) antibody following instruction of the company. The immune complex was visualized by DAB (3, 3’-diaminobenzidine). The sections were counterstained with hematoxyline and mounted and observed under microscope with the magnification of 20× or 63×.
Cell survival analysis
The MTS assay (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazo lium, inner salt assay) was performed according to the manufacturer's specification. The percentage of cell survival (survival rate) was calculated by dividing the absorbance value of the treated samples by the absorbance value of the untreated control within every group. All experiments were repeated two to three times.
In vivo Animal Model
TB3 and NGP cells were cultured in RPMI 1640 10% FBS media, harvested, washed with HBSS (Hank’s balanced salt solution) and re-suspended in HBSS and Matrigel (Trevigen, Gaithersburg, MD). 100μl of cell suspension containing 4×106 TB3 cells or 2×106 NGP cells were implanted into the subcutaneous tissue of the right flank of female 4–5 week old nude mice (Taconic, Germantown, NY). For TB3 xenografts, mice were given water supplemented with placebo (sucrose) or tetracycline (with sucrose) 1 week before tumor implantation and continued this way throughout the experiment. Treatment was initiated when tumors reached approximately 200mm3. Etoposide was given three times a week for about 3 weeks at doses of 10mg/kg and 20mg/kg by intraperitoneal injection. Perifosine was given by oral gavage for 30 days. For the combination of etoposide and perifosine, the mice were given etoposide (10mg/kg) three times a week, while perifosine was administered at 15mg/kg (TB3 tumors) or 17mg/kg (NGP tumors) by oral gavage daily for 30 days. Protein and RNA were extracted from tumors to detect TrkB levels in TB3 tumors. The dimensions of the resulting tumors determined at least 3 times a week using a digital caliper and the tumor volume (mm3) calculated as (L × W2)/4, where L = length (mm) and W = width (mm). All mice were euthanized by asphyxiation with regulated CO2, and their tumors were excised and immediately frozen at −80 °C or fixed in 10% formalin.
Statistic analysis
The comparison between two groups was performed using student T-Test; the comparison among three or more groups was performed using one-way ANOVA with Bonferroni correction. The results were shown as means±SE.
Acknowledgments
Funding:
Intramural Research Program, Center for Cancer Research at the National Cancer Institute,NIH.
We thank KERYX Biopharmaceuticals and Dr. Enrique Poradosu (KERYX Biopharmaceuticals) and Dr. Sherry S. Ansher (Cancer Therapy Evaluation Program) for coordinating the supply of perifosine. We thank Lauren Marks for programmatic assistance and other members of the Cellular & Molecular Biology Section, National Cancer Institute, for their thoughtful review of this study.
Footnotes
Financial disclosures: There are no financial disclosures from any authors.
References
- 1.Brodeur GM, Pritchard J, Berthold F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993;11:1466–1477. doi: 10.1200/JCO.1993.11.8.1466. [DOI] [PubMed] [Google Scholar]
- 2.Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol. 1999;17:2264–2279. doi: 10.1200/JCO.1999.17.7.2264. [DOI] [PubMed] [Google Scholar]
- 3.Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003;3:206–216. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- 4.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369(9579):2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 5.Nakagawara A, Arima-Nakagawara M, Scavarda NJ, Azar CG, Cantor AB, Brodeur GM. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N Engl J Med. 1993;328(12):847–854. doi: 10.1056/NEJM199303253281205. [DOI] [PubMed] [Google Scholar]
- 6.Scala S, Wosikowski K, Giannakakou P, Valle P, Biedler JL, Spengler BA, et al. Brain-derived neurotrophic factor protects neuroblastoma cells from vinblastine toxicity. Cancer Res. 1996;56(16):3737–3742. [PubMed] [Google Scholar]
- 7.Jaboin J, Kim CJ, Kaplan DR, Thiele CJ. Brain-derived neurotrophic factor activation of TrkB protects neuroblastoma cells from chemotherapy-induced apoptosis via phosphatidylinositol 3'-kinase pathway. Cancer Res. 2002;62 (22):6756–6763. [PubMed] [Google Scholar]
- 8.Li Z, Jaboin J, Dennis PA, Thiele CJ. Genetic and pharmacologic identification of Akt as a mediator of brain-derived neurotrophic factor/TrkB rescue of neuroblastoma cells from chemotherapy-induced cell death. Cancer Res. 2005;65(6):2070–2075. doi: 10.1158/0008-5472.CAN-04-3606. [DOI] [PubMed] [Google Scholar]
- 9.Seo JH, Ahn Y, Lee SR, Yeol Yeo C, Chung Hur K. The major target of the endogenously generated reactive oxygen species in response to insulin stimulation is phosphatase and tensin homolog and not phosphoinositide-3 kinase (PI-3 kinase) in the PI-3 kinase/Akt pathway. Mol Biol Cell. 2005;16:348–357. doi: 10.1091/mbc.E04-05-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Servidei T, Riccardi A, Sanguinetti M, Dominici C, Riccardi R. Increased sensitivity to the platelet-derived growth factor (PDGF) receptor inhibitor STI571 in chemoresistant glioma cells is associated with enhanced PDGF-BB-mediated signaling and STI571-induced Akt inactivation. J Cell Physiol. 2006;208:220–228. doi: 10.1002/jcp.20659. [DOI] [PubMed] [Google Scholar]
- 11.Beierle EA, Nagaram A, Dai W, Iyengar M, Chen MK. VEGF-mediated survivin expression in neuroblastoma cells. J Surg Res. 2005;127:21–28. doi: 10.1016/j.jss.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 12.Van Golen CM, Feldman EL. Insulin-like growth factor I is the key growth factor in serum that protects neuroblastoma cells from hyperosmotic-induced apoptosis. J Cell Physiol. 2000;182:24–32. doi: 10.1002/(SICI)1097-4652(200001)182:1<24::AID-JCP3>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 13.Kim S, Hu W, Kelly DR, Hellmich MR, Evers BM, Chung DH. Gastrin-releasing peptide is a growth factor for human neuroblastoma. Ann Surg. 2002;235 (5):621–629. doi: 10.1097/00000658-200205000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.George RE, Sanda T, Hanna M, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455(7215):975–8. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sagulenko V, Muth D, Sagulenko E, Paffhausen T, Schwab M, Westermann F. Cathepsin D protects human neuroblastoma cells from doxorubicin-induced cell death. Carcinogenesis. 2008;29:1869–1877. doi: 10.1093/carcin/bgn147. [DOI] [PubMed] [Google Scholar]
- 16.Opel D, Poremba C, Simon T, Debatin KM, Fulda S. Activation of Akt predicts poor outcome in neuroblastoma. Cancer Res. 2007;67(2):735–45. doi: 10.1158/0008-5472.CAN-06-2201. [DOI] [PubMed] [Google Scholar]
- 17.Porta C, Figlin RA. Phosphatidylinositol-3-kinase/Akt signaling pathway and kidney cancer, and the therapeutic potential of phosphatidylinositol-3-kinase/Akt inhibitors. J Urol. 2009;182:2569–2577. doi: 10.1016/j.juro.2009.08.085. [DOI] [PubMed] [Google Scholar]
- 18.Li Z, Tan F, Liewehr DJ, Steinberg SM, Thiele CJ. In vitro and in vivo inhibition of neuroblastoma tumor cell growth by AKT inhibitor perifosine. J Natl Cancer Inst. 2010;102 (11):758–770. doi: 10.1093/jnci/djq125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asgharzadeh S, Pique-Regi R, Sposto R, Wang H, Yang Y, Shimada H, et al. Prognostic significance of gene expression profiles of metastatic neuroblastomas lacking MYC gene amplification. J Natl Cancer Inst. 2006;98:1193–1203. doi: 10.1093/jnci/djj330. [DOI] [PubMed] [Google Scholar]
- 20.Nakagawara A, Azar CG, Scavarda NJ, Brodeur GM. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol Cell Biol. 1994;14:759–767. doi: 10.1128/mcb.14.1.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsumoto K, Wada RK, Yamashiro JM, Kaplan DR, Thiele CJ. Expression of brain-derived neurotrophic factor and p145TrkB affects survival, differentiation, and invasiveness of human neuroblastoma cells. Cancer Res. 1995;55(8):1798–1806. [PubMed] [Google Scholar]
- 22.Evans AE, Kisselbach KD, Liu X, Eggert A, Ikegaki N, Camoratto AM, et al. Effect of CEP-751 (KT-6587) on neuroblastoma xenografts expressing TrkB. Med Pediatr Oncol. 2001;36(1):181–184. doi: 10.1002/1096-911X(20010101)36:1<181::AID-MPO1043>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 23.Lyer R, Evans AE, Qi X, Ho R, Minturn JE, Zhao H, et al. Lestaurtinib enhances the antitumor efficacy of chemotherapy in murine xenograft models of neuroblastoma. Clin Cancer Res. 2010;16(5):1478–1485. doi: 10.1158/1078-0432.CCR-09-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zage PE, Graham TC, Zeng L, et al. The selective Trk inhibitor AZ623 inhibits brain-derived neurotrophic factor-mediated neuroblastoma cell proliferation and signaling and is synergistic with topotecan. Cancer. 2010 Oct 19; doi: 10.1002/cncr.25674. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 25.Brodeur GM. Getting into the AKT. J Natl Cancer Inst. 2010;102(11):747–749. doi: 10.1093/jnci/djq171. [DOI] [PubMed] [Google Scholar]
- 26.Barnett SF, Bilodeau MT, Lindsley CW. The Akt/PKB family of protein kinases: a review of small molecule inhibitors and progress towards target validation. Curr Top Med Chem. 2005;5(2):109–25. doi: 10.2174/1568026053507714. [DOI] [PubMed] [Google Scholar]
- 27.Knowling M, Blackstein M, Tozer R, et al. A phase II study of perifosine (D-21226) in patients with previously untreated metastatic or locally advanced soft tissue sarcoma: A National Cancer Institute of Canada Clinical Trials Group trial. Invest New Drugs. 2006;24(5):435–9. doi: 10.1007/s10637-006-6406-7. [DOI] [PubMed] [Google Scholar]
- 28.Becher OJ, Trippett TM, Kolesar J, Giheeney S, Jiang Z, Khakoo Y, et al. Phase I study of single-agent perifosine for recurrent pediatric solid tumors. American Society Clinical Oncology (ASCO); 2010. abstract #9540. [Google Scholar]
- 29.Freireich EJ, Gehan EA, Rall DP, Schmidt LH, Skipper HE. Quantitative comparison of toxicity of anticancer agents in mouse, rat, hamster, dog, monkey and man. Cancer Chemother Rep. 1966;50(4):219–244. [PubMed] [Google Scholar]
- 30.Kumar A, Fillmore HL, Kadian R, Broaddus WC, Tye GW, Van Meter TE. The alkylphospholipid perifosine induces aoptosis and p21-mediated cell cycle arrest in medulloblastoma. Mol Cancer Res. 2009;7(11):1813–1821. doi: 10.1158/1541-7786.MCR-09-0069. [DOI] [PubMed] [Google Scholar]
- 31.Engel JB, Honig A, Schönhals T, et al. Perifosine inhibits growth of human experimental endometrial cancers by blockade of AKT phosphorylation. Eur J Obstet Gynecol Reprod Biol. 2008;141 (1):64–9. doi: 10.1016/j.ejogrb.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 32.Tazzari P, Papa V, Ricci F, et al. Synergistic Proapoptotic Activity of Recombinant TRAIL Plus the Akt Inhibitor Perifosine in Acute Myelogenous Leukemia Cells-a Novel Therapeutic Approach for Leukemia Displaying Elevated Akt Signaling. Blood (ASH Annual Meeting Abstracts) 2008;112:957. [Google Scholar]