

Abstract
Evidence was acquired prior to suggest that the vesicular glutamate transporter (VGLUT) but not other glutamate transporters were inhibited by structures containing a weakly basic α-amino group. To test this hypothesis, a series of analogs using a hydantoin (pKa ~ 9.1) isostere were synthesized and analyzed as inhibitors of VGLUT and the obligate cystine-glutamate transporter (system xc−). Of the hydantoin analogs tested, a thiophene-5-carboxaldehyde analog 2l and a bis-hydantoin 4b were relatively strong inhibitors of VGLUT reducing uptake to less than 6% of control at 5 mM but few inhibited system xc− greater than 50% of control. The benzene-2,4-disulfonic acid analog 2b and p-diaminobenzene analog 2e were also good hydantoin-based inhibitors of VGLUT reducing uptake by 11% and 23% of control, respectively, but neither analog was effective as a system xc− inhibitor. In sum, a hydantoin isostere adds the requisite chemical properties needed to produce selective inhibitors of VGLUT.
Keywords: Hydantoin scaffolds, Glutamate, System xc−, Inhibitor, VGLUT
L-Glutamate (L-Glu, 1), the primary excitatory neurotransmitter in the synaptic pathways of the mammalian central nervous system (CNS), plays an important role in many integrative brain functions1–7 through its activation of a variety of ionotropic and metabotropic receptors. To maintain the proper levels of the transmitter and generate the desired receptor responses without risking the excitotoxic consequences of excessive activation, L-Glu concentrations are controlled by an array of transporters that regulate uptake and release of L-Glu in and out of cells and organelles (e.g., astrocytes, synaptic terminals, and synaptic vesicles).8, 9 For example, the excitatory amino acid transporters (EAATs) facilitate the uptake of L-Glu into neurons and astrocytes whereas the vesicular glutamate transporter (VGLUT) regulates uptake between cytosolic and luminal compartments.4, 10, 11 System xc− (Sxc−), a chloride-dependent, sodium-independent obligate exchanger, couples the export of intracellular L-Glu with the import of extracellular L-cystine that helps maintain intracellular levels of glutathione.12, 13 To decipher the individual and collective contributions that these transporters have on intra- and extracellular concentrations and CNS signaling, selective inhibitors are needed. However, since each transporter recognizes L-Glu, as well as a subset of structurally-similar inhibitors, a challenge exists to develop inhibitors that exploit individual elements of each transporter pharmacophore and thereby achieve selectivity amongst them. A number of successful approaches have been undertaken to create selective inhibitors for glutamate receptors and transporters including conformational restriction, isostere substitution, and combinations of these approaches.14, 15
The goal of this work was to identify a new class of structures that could discriminate between Sxc− and VGLUT, two transporters that share sensitivity to inhibitors possessing sulfonate groups (Fig. 1). Aryl- and heteroaryl sulfonic acid analogs, however, yielded only a few blockers with the desired selectivity.16 Further examination of Sxc− specificity indicated that substituting a heterocyclic carboxylic acid isostere in place of the γ-carboxylate (ibotenate, quisqualate) yielded potent inhibitors.17 Likewise, the inhibition of VGLUT may be accomplished with structures that incorporate a non-basic nitrogen (e.g., aniline, quinoline, etc.)18–20 in place of the α-amino group. Therefore, the substitution of a heterocyclic [carboxylic acid] isostere that contains a weakly basic α-amine group should confer the desired inhibitory effect at VGLUT, whereas placement of the same heterocycles at the γ-position would lead to improved inhibition of Sxc−.
Figure 1.

Structures of system xc− and VGLUT inhibitors and the corresponding IC50 values.
Hydantoins are readily accessible α-amino acid derivatives21–24 that are weak carboxylic acid isosteres by virtue of the imide-NH (pKa ~ 9.1). However, few if any reports utilize the hydantoin group as an isostere substituting for both the α-carboxylate and the weakly basic α-amine moiety (requisite for VGLUT inhibitors). Thus, the goal of this study was to prepare α-hydantoin-based inhibitors that would show selectivity toward VGLUT over Sxc−. Likewise, bis-hydantoins resembling cystine would be expected to show selectivity activity toward Sxc− in comparison to VGLUT.
Herein, the syntheses of C-5 substituted hydantoin and bis-hydantoin compounds were conducted including substituted aromatic and heteroaromatic rings, and arylsulfonic acids; the latter as known inhibitors of VGLUT (Schemes 1 and 2). Commercially available aldehydes (1a–q) were subjected to the Bucherer-Berg reaction25 to afford the hydantoin compounds 2a–q in 35–82% yield. The wide variation in yield (Table 1) is likely due to the difference in electron donating (lower yield) and withdrawing (higher yields) groups on the aryl aldehydes. The preparation of 4a–b was conducted similarly by reaction of commercially available dials (3a and 3b) in 59% and 68% yield, respectively.26 Each compound was fully characterized by 1H, 13C NMR, IR, and mass spectral analysis.27
Scheme 1.

Synthesis of hydantoin analogs 2a–q and 4ab (X = thiophene, phenyl). Reagents and conditions: (i) (NH4)2CO3, KCN, 1:1 MeOH, H2O, 50–60 °C, 3 h.
Scheme 2.

Synthesis of bis(5-(naphthalene-1-yl) hydantoins 5a–b.
Reagents and conditions: (i) Br(CH2)nBr, K2CO3 MeCN, reflux, 24 h.
Table 1.
Structures, yields and percent of L-glutamate uptake in system xc− and VGLUT following inhibition by compounds 2a–q, 4a–b and 5a–b
| Entry | R | Yield | Uptakea Sxc− | Uptakea VGLUT |
|---|---|---|---|---|
| 2a |
|
42 | 100±11 | 121±12 |
| 2b |
|
35 | 100±1 | 11±4 |
| 2c |
|
67 | 90±10 | 63±4 |
| 2d |
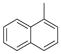
|
77 | 83±17 | 91±14 |
| 2e |
|
69 | 87±4 | 23±4 |
| 2f |
|
81 | 53±2 | 80±28 |
| 2g |
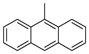
|
78 | 81±3 | 81±4 |
| 2h |
|
71 | 93±6 | 95±7 |
| 2i |
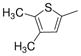
|
68 | 87±8 | 93±2 |
| 2j |
|
56 | 85±3 | 87±7 |
| 2k |
|
76 | 66±3 | 76±4 |
| 2l |
|
73 | 49±3 | 5±1 |
| 2m |
|
75 | 82±11 | 103±4 |
| 2n |
|
78 | 95±3 | 111±6 |
| 2o |
|
61 | 65±2 | 75±4 |
| 2p |
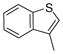
|
66 | 118±5 | 79±10 |
| 2q |
|
58 | 104±7 | 37±8 |
| 4a |
|
59 | 85±3 | 78±12 |
| 4b |
|
68 | 51±5 | 6±3 |
| 5a |
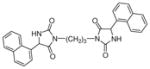
|
61 | 80±7 | 97±5 |
| 5b |
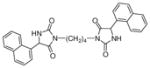
|
78 | 92±13 | 74±5 |
| L-Cystine | - | 22±3 | 90±19 | |
| Congo Red | - | 82±5 | 31±2b | |
| 0±2c |
Percent of L-glutamate uptake by system xc− and VGLUT inhibited by 2a–q, 4a–b and 5a–b. System xc− assay: 100 μM L-glutamate and 500 μM of inhibitor. VGLUT assay: 250 μM L-glutamate and 5 mM of inhibitor.
[I] = 500 μM.
[I] = 5 mM.
Target compounds 5a–b were prepared by reaction of 2d with 1,3 or 1,4 dibromoalkane in the presence of K2CO3 (Scheme 3). Compounds 5a–b lack the more acidic imide NH group thereby reducing interaction of this bioisostere with the transporters via the α-amide NH and H-bonding acceptor carbonyls. Compounds 5a–b are also highly lipophilic, which has been shown to improve inhibitory action at VGLUT.
Scheme 3.

Synthesis of 5-(2,5-dioxoimidazolidin-4-yl)thiophene-2-carboxylic acid. Reagents: (i) hexamethylenetetramine, CF3CO2H, 90 °C, 3 h, 93%; (ii) (NH4)2CO3, KCN, 1:1 EtOH, H2O, 50–60 °C, 3 h, 67%; (iii) LiOH, THF/MeOH (1:1), RT, 48 h, 53%.
The inhibition of VGLUT and Sxc− by the hydantoins were assessed by quantifying the ability of the compounds to block the specific uptake of 3H-L-glutamate by either transporter (Note: Due to well to well variability in the assay, the rate may be higher than 100% in wells where the inhibitor has no activity). Sxc− mediated uptake of L-glutamate (100 μM) was quantified in SNB19 glioma cells under Na-free conditions, corrected for non-specific uptake, and normalized to protein content.28 VGLUT mediated uptake of L-glutamate (250 μM) was measured in synaptic vesicles isolated from rat brain, corrected for non-specific uptake, and normalized to protein content.29 The ability of the hydantoins 2a–q, 4a–b and 5a–b to block uptake by the two transporters as compared to known inhibitors (Table 1).
Following the hypothesis that the hydantoins serve as a bioisostere with both acidic (imide proton) and non-basic amine (amide) moieties, we found that hydantoin-containing analogues failed as Sxc− inhibitors, but a number of structures were identified that blocked VGLUT at a level comparable to that observed with the prototypical inhibitor Congo Red (Fig. 1; standard). This observation is consistent with work demonstrating that VGLUT interacts more strongly with glutamate analogs that contain non-basic amines.16
Among those tested, compounds 2b, 2e, 2l, and 4b were the most effective at blocking the VGLUT-mediated accumulation of glutamate into the synaptic vesicles, allowing only 5–23% to be transported versus control (Congo Red; ~100% uptake blocked at 500 μM). The structures of these inhibitors each contain the hydantoin bioisostere but were attached to either and phenyl or thiophene ring. Inhibitor 2b contains a disulfonic acid motif that is present in many azo dye inhibitors20 of VGLUT and more recently shown to improve VGLUT inhibition, for example, sulfophenylglycine and sulfophenylalanine analogs.16
The finding that compound 2e blocked the uptake of glutamate at VGLUT by 77% was unexpected. Although less potent than 2b, the electron-donating p-dimethylamino substituent improves the inhibition and is more potent than the inactive phenylhydantoin (100% of control) and 1,4-bishydantoin (4a; 78% of control) - dimethylamino replaced with a hydantoin moiety. Since compounds 2l and 4b nearly blocked all glutamate uptake by VGLUT, more detailed inhibition assays were conducted. Compounds 2l and 4b exhibited a typical dose-curve with IC50’s of 355 μM and 511 μM, respectively (Figure 2). To further confirm the selectivity of compounds 2l and 4b toward VGLUT each was tested at EAATs 1–3,30 however, neither reduced uptake by greater than 12% at 100 μM (25 μM D-aspartate as substrate).
Figure 2.

Inhibition of VGLUT by 2l and 4b.
Compound 2l is a rare example of an aldehyde-containing VGLUT inhibitor and as such, we suspected that the aldehyde may have converted to the carboxylic acid during the assay. To test this possibility, the 5-COOH analog 7 corresponding to the oxidized form of aldehyde 2l was prepared (Scheme 3). Starting thiophene ester 6 was converted to the known 5-formylated intermediate31–33 using the Duff reaction – the first report using this substrate. The aldehyde was converted to the hydantoin and the ester hydrolyzed to obtain target compound 7 (Scheme 3).34 Carboxylic acid 7 also proved to be a good inhibitor of VGLUT, reducing uptake level to 7% of control. This suggests that all or part conversion of the aldehyde to the corresponding carboxylic acid could account for the observed activity.
The large difference in VGLUT inhibitory activity between compound 4b and inactive bis-hydantoins 4a and 5a/5b suggests that the thiophene sulfur atom and/or the angular difference imposed on the bis-hydantoin substituents by the five-membered ring may play a role in blocking uptake at VGLUT. Further, compounds 5a/5b lack the acidic imide protons that may be needed for effective binding to VGLUT. Molecular modeling using previously defined pharmacophore models20 provided no clear insight for hydantoin-containing VGLUT inhibitors although the addition of lipophilic groups has not yet been thoroughly explored.
As noted, the majority of the hydantoins tested were essentially inactive as inhibitors of Sxc−, with none exhibiting inhibitory activity comparable to cystine, although, compounds 2k, 2l, 2o and 4b blocked glutamate uptake from 34–51% of control at 500 μM. Interestingly, each of these compounds contains a thiophene-linked hydantoin. The only other structure shown to block uptake at Sxc− to an appreciable amount was benzylhydantoin 2f at 47% of control. This lack of activity was somewhat surprising, given the structural similarities between the hydantoins and numerous isoxazole-based inhibitors.15,26 It remains to be determined if this reflects an unfavorable interaction directly between the hydantoin group and the Sxc− binding site or the moiety’s influence on the R-group position (or a combination of both). Owing to poor inhibition of the hydantoins at Sxc−, compounds were not tested as individual enantiomers although further studies are underway with stereoisomers to refine and improve the potency as VGLUT inhibitors.
In sum, the hydantoin group has been shown to be an effective carboxylic acid isostere in the design of new inhibitors of the vesicular glutamate transporter (VGLUT), but one of questionable value in the further development of blockers of the obligate exchange transporter, Sxc−.
Acknowledgments
This research was supported by NS38248 (CMT), the Core Laboratory for Neuromolecular Production P30 NS055022 (CMT), NS058229 (ATERIS Technologies LLC), NS30570 (RJB), NS42077 (RJB) and the COBRE Center for Structural and Functional Neuroscience (NIH P20-RR015583).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Meldrum B. Epilepsia. 1984;25(Suppl 2):S140. doi: 10.1111/j.1528-1157.1984.tb05646.x. [DOI] [PubMed] [Google Scholar]
- 2.Meldrum BS. Neurology. 1994;44:S14. [PubMed] [Google Scholar]
- 3.Meldrum BS. J Nutr. 2000;130:1007S. doi: 10.1093/jn/130.4.1007S. [DOI] [PubMed] [Google Scholar]
- 4.Nakanishi S. Science. 1992;258:597. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- 5.Schoepp DD, Conn PJ. Trends Pharmacol Sci. 1993;14:13. doi: 10.1016/0165-6147(93)90107-u. [DOI] [PubMed] [Google Scholar]
- 6.Seeburg PH. Trends Neurosci. 1993;16:359. doi: 10.1016/0166-2236(93)90093-2. [DOI] [PubMed] [Google Scholar]
- 7.Watkins JC, Pook PC, Sunter DC, Davies J, Honore T. Adv Exp Med Biol. 1990;268:49. doi: 10.1007/978-1-4684-5769-8_6. [DOI] [PubMed] [Google Scholar]
- 8.Choi DW, Rothman SM. Annu Rev Neurosci. 1990;13:171. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- 9.Swanson GT, Sakai R. Prog Mol Subcell Biol. 2009;46:123. doi: 10.1007/978-3-540-87895-7_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moriyama Y, Omote H. Biol Pharm Bull. 2008;31:1844. doi: 10.1248/bpb.31.1844. [DOI] [PubMed] [Google Scholar]
- 11.Takamori S. Neurosci Res. 2006;55:343. doi: 10.1016/j.neures.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 12.Burdo J, Dargusch R, Schubert D. J Histochem Cytochem. 2006;54:549. doi: 10.1369/jhc.5A6840.2006. [DOI] [PubMed] [Google Scholar]
- 13.Domercq M, Sanchez-Gomez MV, Sherwin C, Etxebarria E, Fern R, Matute C. J Immunol. 2007;178:6549. doi: 10.4049/jimmunol.178.10.6549. [DOI] [PubMed] [Google Scholar]
- 14.Bridges RJ, Kavanaugh MP, Chamberlin AR. Curr Pharm Des. 1999;5:363. [PubMed] [Google Scholar]
- 15.Bridges RJ, Lovering FE, Koch H, Cotman CW, Chamberlin AR. Neurosci Lett. 1994;174:193. doi: 10.1016/0304-3940(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 16.Etoga JL, Ahmed SK, Patel S, Bridges RJ, Thompson CM. Bioorg Med Chem Lett. 2010;20:2680. doi: 10.1016/j.bmcl.2009.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel SA, Rajale T, O’Brien E, Burkhart DJ, Nelson JK, Twamley B, Blumenfeld A, Szabon-Watola MI, Gerdes JM, Bridges RJ, Natale NR. Bioorg Med Chem. 2010;18:202. doi: 10.1016/j.bmc.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrigan CN, Bartlett RD, Esslinger CS, Cybulski KA, Tongcharoensirikul P, Bridges RJ, Thompson CM. J Med Chem. 2002;45:2260. doi: 10.1021/jm010261z. [DOI] [PubMed] [Google Scholar]
- 19.Carrigan CN, Esslinger CS, Bartlett RD, Bridges RJ, Thompson CM. Bioorg Med Chem Lett. 1999;9:2607. doi: 10.1016/s0960-894x(99)00444-8. [DOI] [PubMed] [Google Scholar]
- 20.Thompson CM, Davis E, Carrigan CN, Cox HD, Bridges RJ, Gerdes JM. Curr Med Chem. 2005;12:2041. doi: 10.2174/0929867054637635. [DOI] [PubMed] [Google Scholar]
- 21.Barraclough P, Bolofo ML, Giles H, Gillam J, Harris CJ, Kelly MG, Leff P, McNeill A, Robertson AD, Stepney RJ, Whittle BJ. Bioorg Med Chem. 1996;4:81. doi: 10.1016/0968-0896(95)00173-5. [DOI] [PubMed] [Google Scholar]
- 22.Groutas WC, Stanga MA, Castrisos JC, Schatz EJ. J Enzyme Inhib. 1990;3:237. doi: 10.3109/14756369009035842. [DOI] [PubMed] [Google Scholar]
- 23.Meanwell NA, Roth HR, Smith EC, Wedding DL, Wright JJ, Fleming JS, Gillespie E. J Med Chem. 1991;34:2906. doi: 10.1021/jm00113a033. [DOI] [PubMed] [Google Scholar]
- 24.Sarges R, Oates PJ. Prog Drug Res. 1993;40:99. doi: 10.1007/978-3-0348-7147-1_5. [DOI] [PubMed] [Google Scholar]
- 25.Li JJ. Name Reactions. Springer; Berlin Heidelberg: 2009. p. 76. [Google Scholar]
- 26.Synthesis of compounds 4a–b. To approx 2.0 g of carbonyl compound (0.02 mole) dissolved in 50% methanol (50 mL) were added ammonium carbonate (9.1 g; 0.08 mole) and potassium cyanide (2.6 g; 0.04 mole). The mixture was warmed to 58–60 °C for 3 h, concentrated to 15 mL, and chilled to 0 °C to produce white-off yellow crystals.
- 27.Spectral data for selected compounds. Compound 2b: Yield 35%; mp > 300 °C; 1H NMR (400 MHz, DMSO-d6 ): δ (s, 1H), 7.73 (d, J = 8.25 Hz, 1H), 7.50 (d, J = 8.25 Hz, 1H), 5.77 (s, 1H); 13C: δ 169.5, 158.7, 143.4, 140.5, 136.8, 134.4, 129.7, 128.6, 54.2; ESI MS m/z = 336 (M+1); Anal. Calcd For C9H8N2O8S2: C, 32.14; H, 2.40; N, 8.33. Found: C, 32.33; H, 2.22; N, 8.62. Compound 2l : 1H NMR (400 MHz, acetone-d6). δ. 10.9 (bs, 1H), 9.81 (s, 1H), 7.81 (d, J = 3.9 Hz 1H), 7.71 (d, J = 3.9 Hz, 1H), 5.91 (s, 1H); 13C: δ 182.1, 164.6, 164.1, 148.2, 141.4, 138.4, 129.5, 64.2; ESI MS m/z = 211 (M+1); Anal. Calcd For C8H6N2O3S: C, 45.71; H, 2.88; N, 13.33. Found: C, 45.33; H, 2.91; N, 13.67. Compound 2m : Yield 75%; mp 258–261 °C; 1H NMR (400 MHz, DMSO- d6): 12.02 (bs, 1H), 10.58 (bs, 1H), 8.06 (bs, 1H), 7.59 (s, 1H), 7.09 (s, 1H), 4.99 (s, 1H); ESI MS m/z = 167 (M+1); (n max/cm−1): 3414, 3241, 2700, 1729, 1456. Anal. Calcd For C6H6N4O4: C, 43.38; H, 3.64; N, 33.72. Found: C, 43.33; H, 3.59; N, 33.62. Compound 4a: Yield 59%; mp >300 °C; 1H NMR (400 MHz, DMSO- d6): 10.78 (bs, 1H), 8.39 (bs, 1H), 7.33 (s, 4H), 5.15 (s, 2H); ESI MS m/z = 275 (M+1); (n max/cm−1): 3237, 2925, 2721, 1701, 1458, 1377, 722. Anal. Calcd For C12H10N4O4: C, 52.56; H, 3.68; N, 20.43. Found: C, 52.33; H, 3.44; N, 20.62.
- 28.Patel SA, Warren BA, Rhoderick JF, Bridges RJ. Neuropharmacology. 2004;46:273. doi: 10.1016/j.neuropharm.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 29.Kish PE, Ueda T. Methods Enzymol. 1989;174:9. doi: 10.1016/0076-6879(89)74005-2. [DOI] [PubMed] [Google Scholar]
- 30.Esslinger CS, Agarwal S, Gerdes J, Wilson PA, Davis ES, Awes AN, O’Brien E, Mavencamp T, Koch HP, Poulsen DJ, Rhoderick JF, Chamberlin AR, Kavanaugh MP, Bridges RJ. Neuropharmacology. 2005;49:850. doi: 10.1016/j.neuropharm.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 31.Chappell MDPD, Conner SE, Tripp AE, Zhu G. WO/2006/086488. Substituted Thiophene Derivatives as Glucagon Receptor Antagonists, Preparation and Therapeutic Uses. 2006
- 32.Choong IC, Lew W, Lee D, Pham P, Burdett MT, Lam JW, Wiesmann C, Luong TN, Fahr B, DeLano WL, McDowell RS, Allen DA, Erlanson DA, Gordon EM, O’Brien T. J Med Chem. 2002;45:5005. doi: 10.1021/jm020230j. [DOI] [PubMed] [Google Scholar]
- 33.Shilai M, Kondo Y, Sakamoto T. J Chem Soc Perkin Tran. 2001;1:442. [Google Scholar]
- 34.Compound 7. Ethyl thiophene-2-carboxylate (0.43 mL, 3.2 mmol) was dissolved in TFA (5 mL) and to this solution was added hexamethylenetetramine (1.34 g, 9.6 mmol) at 90 °C for 3 h. After cooling to rt, the solution was concentrated, 3 mL of water added, and the solution brought to pH 8 with Na2CO3. The solution was extracted extracted with CHCl3 (2×250 mL), dried over Na2SO4, concentrated and purified by column chromatography (EtOAc:hex, 1:4) to affford ethyl 5-formylthiophene-2-carboxylate as an off-white solid (548.4 mg, 93%). The aldehyde was converted to the hydantoin per Scheme 1. Hydantoin (0.100 g; 0.39 mmol) was placed in 1:1 MeOH/H2O (4 mL) and LiOH (19.71 mg, 0.82 mmol) for 48 h, the reaction mixture concentrated, exrtacted with EtOAc (100 mL), and the organic phase concentrated and purified by chromatography (EtOAc:hexane, 4:1) to afford 5-(2,5-dioxoimidazolidine-4-yl)thiophene-2-carboxylic acid 7 (47.1 mg, 53%). 1H NMR (400 MHz, acetone-d6). δ. 13.21(bs, 1H), 8.22 (bs, 1H), 7.55(d, J = 4.2 Hz 1H), 7.19 (d, J = 4.2 Hz, 1H), 5.88 (s, 1H); 13C: δ 163.5, 163.1, 162.2, 147.1, 139.4, 134.2, 129.5, 68.2; ESI MS m/z = 227 (M+1); Anal. Calcd For C8H6N2O4S: C, 42.48; H, 2.27; N, 12.38. Found: C, 42.11; H, 2.32; N, 12.51.