

The flagellar motor drives the rotation of flagellar filaments at hundreds of revolutions per second1,2, efficiently propelling bacteria through viscous media3. The motor uses the potential energy from an electrochemical gradient of cations4,5 across the cytoplasmic membrane to generate torque. A rapid switch from anticlockwise to clockwise rotation determines whether a bacterium runs smoothly forward or tumbles to change its trajectory6,7. A protein called FliG forms a ring in the rotor of the flagellar motor that is involved in the generation of torque8–13 through an interaction with the cation channel forming stator subunit MotA12. FliG has been suggested to adopt distinct conformations that induce switching but these structural changes and the molecular mechanism of switching are unknown. Here we report the molecular structure of the full-length FliG protein, identify conformational changes that are involved in rotational switching and uncover the structural basis for the formation of the FliG torque ring. This allows us to propose a model of the complete ring and switching mechanism in which conformational changes in FliG reverse the electrostatic charges involved in torque generation.
The structure of the full-length (FL) FliG protein (FliGFL) from Aquifex aeolicus is entirely α-helical and consists of distinct amino-terminal (N), middle (M) and carboxy-terminal (C) globular domains, which are connected by two long helices (helixNM and helixMC) (Fig. 1a). The structure of the middle to C-terminal domain of FliG from Thermotoga maritima (FliGMC) has been previously reported14 (Supplementary Fig. 1). Like FliGMC, the middle domain of FliGFL contains a single armadillo repeat motif (ARMM) whereas the C-terminal domain can be further divided into a second ARM (ARMC) and a six-helix bundle (helicesC1–6). An overview of the full-length FliG structure, assembly of the FliG ring and switching mechanism is illustrated in Supplementary Movie 1. In the C-terminal domain, charged residues that are clustered around helixC5 result in a functionally important electrostatic charge distribution (Fig. 1b) that is involved in torque generation9–12,14. Surprisingly, the fold around these charges, comprising helicesC3–6, is repeated in the N-terminal domain (helicesN1–4) (Fig. 1c). These helices are moderately conserved and contain four groups of conserved amino acid triplets (Fig. 1d). Charged residues on helixN3 seem to invert the charges on the structurally equivalent torque helixC5 (Fig. 1c, d). However, in the N-terminal domain, this does not result in an obvious polar electrostatic charge distribution, indicating that the repeated fold is not echoed with a repeated torque-generating function (Supplementary Fig. 2).
Figure 1. Structural overview of the full-length FliG monomer.

a–c, Residues are coloured from N to C terminus as a spectrum of colours from blue to red. Torque helixC5 and helixN3 are labelled with a red and blue asterisk respectively. HelicesC3–6 are shown with charged residues and the electrostatic potential on helixC5 in b adjacent to helicesN1–4 in c to highlight the conserved fold. d, Sequence alignment of the residues shown in b and c. Conserved or similar residues are highlighted, and conserved amino acid triads are underlined. HelixC5 and helixN3 are encircled and charged residues on these helices are in red (negative) and blue (positive).
FliG is required for assembly of an intact flagellar motor8 where it binds to a structural subunit called FliF, thereby coupling torque to the rest of the flagellar filament15,16 (Supplementary Fig. 3). Escherichia coli cells containing a FliF–FliG fusion mutant can form fully assembled flagella, and the deletion of groups of 10 residues within the N terminal 36 and 46 residues of FliG from Salmonella typhimurium disrupt flagellar assembly and binding to FliF, respectively17. In A. aeolicus, the equivalent residues map to the N-terminal three helices (helicesN1–3) of the protein (Fig. 1c), indicating that these promote binding to FliF.
FliGFL and FliGMC have been captured in different conformations. The most obvious conformational difference is in the middle domain. Whereas ARMM is well conserved (Cα root mean squared deviation (r.m.s.d.), 1.0 Å), helixMC is packed tightly against ARMM and helixNM in FliGFL (‘closed’ conformation, Fig. 2a), but dissociated from ARMM in FliGMC (‘open’ conformation, Fig. 2b). The closed conformation is stabilized by 15 mostly conserved hydrophobic residues, which form a highly complementary hydrophobic interface between helixMC and ARMM (Supplementary Fig. 4a, b) that is disrupted in the open conformation. A second conformational difference is between the C-terminal domains of FliGMC (CterMC) and FliGFL (CterFL). Here, ARMC and helicesC1–6 are very similar (Cα r.m.s.d., 1.0 Å and 0.77 Å, respectively) and are connected by a highly conserved loop, but their relative orientations differ between CterFL and CterMC (Fig. 2c, d). They are related by rotations of 77.8° around the F237 phi angle and 25.6° around the M236 phi angle (Supplementary Movie 2). This rotation alters the relative orientation of the torque helixC5, which has been proposed as a mechanism for rotational switching13.
Figure 2. Conformational differences between A. aeolicus FliGFL and T. maritima FliGMC.
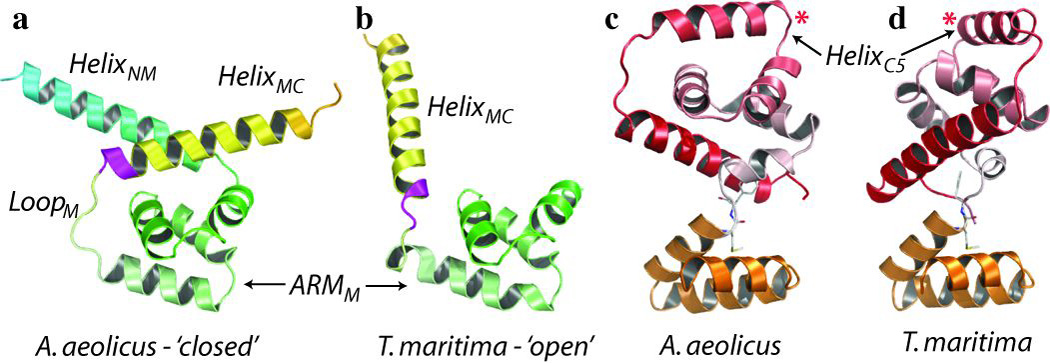
a, b, The middle domains of FliGFL (a; A. aeolicus, closed) and FLiGMC (b; T. maritima, open). The equivalent position of the extreme clockwise-biased deletion mutant is coloured in magenta. c, d, The C-terminal domains of A. aeolicus FliGFL (CterFL) (c) and T. maritima FliGMC (CterMC) (d). Torque helixC5 is labelled with a red asterisk. Hinge residues M236 and F237 are shown as sticks.
We mapped all known FliG mutations that bias the direction of rotation of the flagellar motor onto FliGFL (Supplementary Movie 3). These are clustered in three regions of the protein. Two of these clusters indicate that the conformational differences between FliGFL and FliGMC may reflect changes associated with motor switching for the following reasons. The first cluster is around helixMC, where a majority of clockwise-biased mutations occur at the interface between helixMC and the middle domain (Supplementary Fig. 4), and introduce charged or bulky residues that are likely to interfere with the closed conformation. Furthermore, deletion of a moderately conserved 169PAA171 motif (Fig. 2a, b, in magenta) in S. typhimurium results in an extreme clockwise bias18. This mutation is also likely to destabilize the closed conformation as it shortens the loop between helixMC and ARMM and alters the register of helixMC, thereby changing the orientation of the hydrophobic ridge along helixMC that binds to ARMM (Supplementary Fig. 4). In contrast to clockwise-biased mutants, anticlockwise-biased mutations do not occur at the helixMC–middle-domain interface, with the exception of the conservative I122L mutation in E. coli. Combined, these rotationally biased mutants indicate that the closed conformation represents FliG during anticlockwise rotation and that switching to clockwise rotation may involve the dissociation of helixMC from ARMM to an open conformation via a putative hinge loop between ARMM and helixMC (loopM). This may explain why mutations at almost any residue in loopM can bias the rotation direction (Supplementary Movie 3). The middle domains of FliGFL and FliGMC seem to represent anticlockwise and clockwise states respectively, indicating that CterFL and CterMC may follow the same trend. Indeed, a second cluster of mutants occurs between the two subdomains (ARMC and helicesC1–6) of the C-terminal domain. However it is less clear whether these favour either the CterFL or the CterMC conformation (Supplementary Fig. 5). A third cluster of mutations occurs at the loop between helixMC and the C-terminal domain (loopC), which is another putative hinge loop that has been implicated in switching14,19.
In the FliGFL crystal lattice (Fig. 3a), the base of ARMC is packed against the base of ARMM of an adjacent monomer (ARMM+1) (Supplementary Fig. 6a). The arrangement forms a stack of tandem ARM motifs resulting in a right-handed superhelix consisting of seven α-helices (Fig. 3b). The otherwise exposed hydrophobic patch on the base of ARMC14 is completely buried in the large interacting surface and forms part of a continuous hydrophobic core that extends over the entire superhelix. The ARMC–ARMM+1 stacking has marked parallels with eukaryotic ARM motifs20 (Supplementary Movie 4). In all structures containing ARM motifs, these form tandem repeats that interact extensively, resulting in a right-handed superhelix that creates a surface for protein–protein interactions21,22. The stacking is mediated typically by nine hydrophobic residues, usually consisting of leucine, valine or isoleucine20. Similarly, the stacking of ARMC–ARMM+1 is mediated by eleven leucine, valine or isoleucine residues, eight of which are absolutely conserved as hydrophobic residues across all known FliG sequences. This indicates that the interaction is not specific to A. aeolicus but extends to all flagellated bacteria. Indeed, in the crystal packing of the T. maritima FliGMC structure, the ARMC–ARMM+1 interaction forms an identical (Cα r.m.s.d., 1.0 Å) right-handed superhelix (Fig. 3b). Importantly, this is despite the FliGMC crystal originating from different species, constructs and crystal forms with no other conserved crystal contacts.
Figure 3. Structural basis for the formation of FliG multimers.
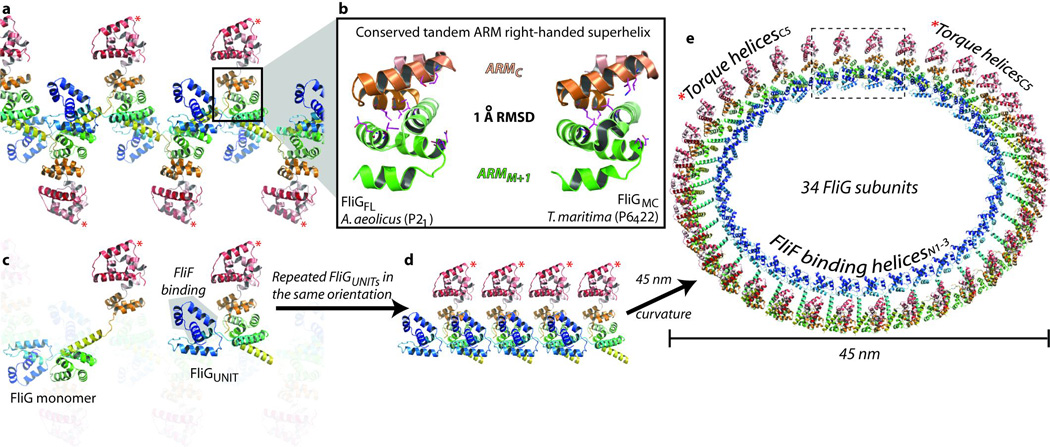
a, The FliG multimer in the FliGFL crystal lattice. The ARMC–ARMM+1 superhelix is encircled and expanded in b, which shows the ARM superhelix from FliGFL (left) adjacent to the ARM superhelix from FliGMC (right). The positions of mutations that inhibit FliM binding and flagellar assembly are shown in magenta. c, Same as a, but highlighting the orientation of a FliG monomer and a FliGUNIT in the crystal lattice. Repeated FliGUNIT protomers in the same orientation are shown linearly in d, and with the curvature of a 45-nm diameter ring in e.
Combined, these data indicate that the ARMC–ARMM+1 interaction is a real biological interaction and this has several profound implications. First, FliG forms part of the flagellar motor known as the switch complex, which contains two other proteins, FliM and FliN. All three proteins are required for flagellar assembly (Supplementary Fig. 3). Mutations at the base of ARMC and ARMM and on the face of ARMM, which contains a highly conserved EHPQR motif14, can disrupt flagellar assembly8,10,23 and FliM binding24. On the basis of the structure of a single FliG monomer, it seems evident that ARMC and ARMM are separate FliM binding sites14. However the ARMC–ARMM+1 superhelix indicates that ARMC and ARMM from adjacent monomers interact to create a surface for a single FliM binding site, which is consistent with all other known ARM superhelices that stack to form a surface for protein–protein interactions. Second, in addition to its interactions with FliF and FliM, proper assembly of the bacterial flagellum also requires a FliG–FliG interaction that is independent of other components of the flagellar motor. This is well supported by mutagenesis studies that demonstrate the requirement of at least five hydrophobic residues at the ARMC–ARMM+1 interface for flagellar assembly10,23,24 (Fig. 3b). Last, because ARMC forms a right-handed superhelix with ARMM+1, it follows that ARMC+1 has the same interaction with ARMM+2. Thus, the ARMC–ARMM+1 superhelix is the structural basis for the formation of FliG multimers (Supplementary Fig. 6a, b).
As mentioned earlier, a cluster of rotationally biased mutations highlight the importance of the putative hinge loopC in switching14,19. Similarly, such mutations also occur at almost every residue on hinge loopM, which mediates the transition from the closed to open conformation. Remarkably, in the FliG multimer, loopC and loopM+1 form an intermolecular anti-parallel β-sheet (Supplementary Fig. 6d, f) explaining how mutations in loopC may influence switching.
The FliG multimer resembles beads on a string, linked by residue A193, one of the few amino acids unrestrained by secondary structure (Supplementary Fig. 6c, e). The repeating ‘beads’ are formed by an L-shaped protomer (FliGUNIT) that contains two halves of adjacent FliG monomers (Supplementary Fig. 6a–c). Although loopC probably allows interdomain flexibility in the FliG monomer14, the FliGUNIT is a more robust arrangement of the three domains in FliG. In a FliGUNIT the intersubunit β-sheet between loopC–loopM+1, in combination with the superhelix (ARMC–ARMM+1), restrains the relative movement of the C-terminal domain (Supplementary Movie 5). The main body of the FliGUNIT contains a complete ARMC/M+1 superhelix that is tethered to the base of the C-terminal helicesC1–6 through the M236–F237 hinge loop. The torque helixC5 is perpendicular to the axis of the superhelix at the top of the FliGUNIT (Fig. 3c) and the two long helices are at the base. HelixMC connects adjacent FliGUNIT protomers and helixNM places the N-terminal domain next to the FliGUNIT, thereby forming the L-shaped protomer. In the crystal lattice, adjacent FliGUNIT protomers are in opposing orientations (Fig. 3a and Supplementary Fig. 6a–c).
To model the FliG ring, we applied a curvature to the multimer in the crystal lattice, creating a ring with a 45-nm diameter that corresponds to a region of the flagellar motor known as the C ring, where the switch complex is thought to reside (Supplementary Fig. 3). The model contains 40 FliGUNIT protomers, where adjacent protomers are in opposing orientations resulting in a 20-fold symmetry (Supplementary Fig. 7a). However, this is inconsistent with the apparent 34-fold symmetry of the C ring25. Furthermore, the opposing arrangement of alternating N- and C-terminal domains is inconsistent with the location of their respective interacting proteins, FliF and MotA. These are both on the periplasmic side of the C ring (Supplementary Fig. 3). Therefore, we generated rings by repeating the FliGUNITs in the same orientation (Fig. 3d). These rings satisfy the spatial restraints imposed by the location of the MotA stators and the FliF ring (Fig. 3e). Furthermore, the 45-nm ring contains 34 monomers, which is in agreement with the 34-fold symmetry of the C ring25 (Fig. 3e). Attempts to generate rings that match the size and symmetry of the MS ring, a 30 nm ring above the C ring that is integrated into the cytoplasmic membrane, were unsuccessful (Supplementary Fig. 7c, d).
When protomers are arranged in the same orientation, the continuous FliG polypeptide chain that spans adjacent FliGUNIT protomers is broken with a distance of around 34 Å between A193 and E192 (Fig. 4b). Remarkably, dissociation of helixMC from ARMM into an open conformation brings these residues to within a peptide-bond distance (Fig. 4a, b). Similarly, this can be achieved with FliG in the closed conformation by rotating the main body of the FliGUNIT (Fig. 4b, c). These conformational changes may explain why FliG alone does not spontaneously form an oligomer without an interaction with the FliF scaffold.
Figure 4. Molecular basis of rotational switching.
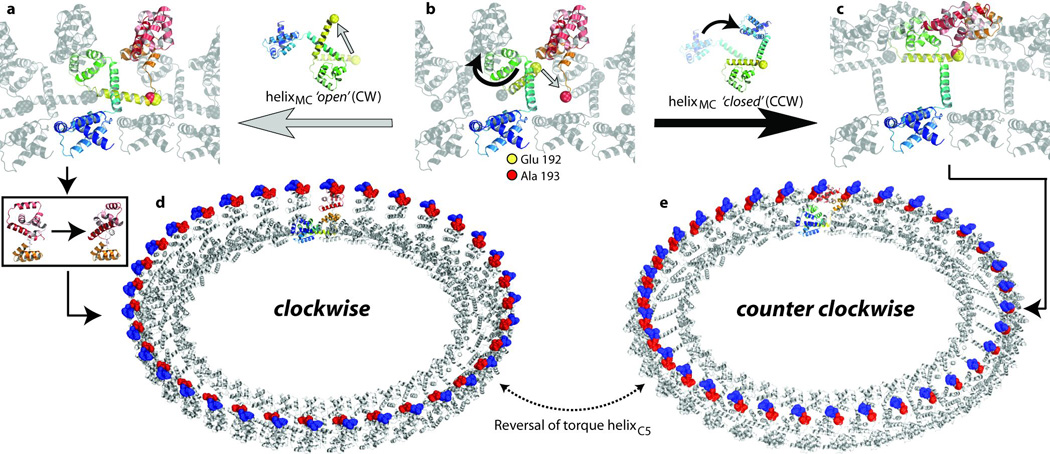
a–c, An expanded view of the encircled region in Fig. 3e is shown with one coloured FliG polypeptide chain. E192 and A193 are shown as yellow and red spheres respectively. a, b, Transition from the closed to the open conformation of helixMC. b, c, An alternative conformational change when helixMC remains in the closed conformation. d, e, The FliG ring with the monomers in the FliGMC and the FliGFL crystal structures, respectively. Charged residues on torque helixC5 are shown in blue (positive) and red (negative).
We generated two FliG rings, one with each monomer in the FliGFL (closed, anticlockwise) conformation and the other with each in the FliGMC (open, clockwise) conformation (Fig. 4d and e, respectively). In the context of the complete FliG ring, the conformational differences between FliGFL and FliGMC result in a reversal of the electrostatic charges on torque helixC5 of FliG, providing a model for rotational switching of the flagellar motor. Furthermore, to visualize the transition from FliGFL to FliGMC, we generated 100 intermediate structures between each state by interpolating the different phi and psi torsion angles. This illustrates that the entire conformational change occurs without any significant clashes in the ring (Supplementary Movie 6).
As the FliG monomer spans two adjacent FliGUNIT protomers, neighbouring protomers are restrained to the same open or closed conformation. This indicates that switching entails a bistable global conformational switch where adjacent FliG monomers are restrained to the same rotational state. However, there is sufficient flexibility in the model to allow for opposing conformations in the ring that are bridged by protomers in intermediate states (Supplementary Fig. 8). This is compatible with the conformational spread model of cooperativity26, the hallmarks of which were recently observed in the flagellar motor27.
The FliG ring is consistent with three-dimensional electron microscopy reconstructions. The highest resolution three-dimensional electron microscopy reconstruction of the flagellar motor so far has been generated from single-particle averaging of a clockwise-locked 69PAA171-deletion mutant from S. typhimurium25. We docked the FliG ring into the C ring, which has 34 distinct lobes on the periplasmic side at the outer periphery. The shape and size of these lobes complement that of the ARM superhelix and the C-terminal domain in the clockwise FliG ring (Supplementary Fig. 9a). Notably, the anticlockwise FliG ring is not consistent with the clockwise-locked electron microscopy reconstruction (Supplementary Fig. 10).
The vertical cross-section of the C ring from various electron micrographs has two lobes on the periplasmic side of the S. typhimurium C ring. The inner lobe is missing in the same micrograph of a flagellar motor containing a FliG–FliF fusion deletion mutant that does not contain the first 94 residues of FliG (N-terminal domain and part of helixNM) and the position of the C ring is shifted up towards the MS ring compared to the native or full-length FliG–FliF mutants28. We overlayed the cross-section of the FliG ring onto these micrographs and the C-terminal domain fits well on the outer lobe, the N-terminal domain on the inner lobe and the ARM superhelix in between (Supplementary Fig. 9b). Furthermore, we deleted the equivalent of the first 94 residues from FliG and overlayed this truncated structure onto electron micrographs of the vertical cross-section of the FliG–FliF deletion fusion mutant. The micrographs precisely shadow the structure of the truncated FliG ring, and the shift in the position of the C ring relative to the MS ring reflects the shortened link between these rings as a result of deleting the N-terminal domain (Supplementary Fig. 9c).
The orientation and location of the FliG rings on the periplasmic side of the C ring satisfies spatial restraints, which have been determined from structural, mutagenesis and electron microscopy data. First, in situ cryo-electron tomograms indicate that the MotA/B stator interacts with the outer circumference of the periplasmic side of the C ring29,30 (Supplementary Fig. 3). Supplementary figure 9b, 4d and e illustrate how helixC5 forms the outer circumference of the periplasmic side of the C ring and how the electrostatic charges that are involved in torque generation point towards the periplasm. Second, all FliF-binding helicesN1–3 are located in the inner circumference of the ring in an optimal position to interact with the MS ring (Fig. 3e and Supplementary Figs 3, 9b). Third, in the FliG ring, mutations that affect the interaction with FliM segregate to a single location on the cytoplasmic side of the ring on the surface of the ARM superhelix (Supplementary Fig. 9b), pointing towards the expected location of FliM (Supplementary Fig. 3). Finally, by docking structures of FliM and FliN subunits into the remaining density of the C ring we demonstrate that this density is sufficiently large to accommodate rings of both subunits in an arrangement that is consistent with biochemical studies (Supplementary Fig. 11).
METHODS SUMMARY
The full-length FliG protein from A. aeolicus was expressed in E. coli and crystallized as detailed in Supplementary Information. Crystals belong to space group P21 with one FliG monomer in the asymmetric unit. Native X-ray diffraction data were collected at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France, and single or multiple wavelength anomalous dispersion (SAD or MAD) data were collected at the Advanced Photon Source (APS) in Chicago, USA. The methods used for structure determination are detailed in Supplementary Information. X-ray data and refinement statistics are given in Supplementary Tables 1 and 2, respectively. Still images and movie frames of protein structures were generated with PyMOL (http://www.pymol.org). Morphing and modelling software were written in C++.
Supplementary Material
References
- 1.Lowe G, Meister M, Berg HC. Rapid rotation of flagellar bundles in swimming bacteria. Nature. 1987;325:637–640. [Google Scholar]
- 2.Magariyama Y, et al. Very fast flagellar rotation. Nature. 1994;371:752. doi: 10.1038/371752b0. [DOI] [PubMed] [Google Scholar]
- 3.Berg HC, Anderson RA. Bacteria swim by rotating their flagellar filaments. Nature. 1973;245:380–382. doi: 10.1038/245380a0. [DOI] [PubMed] [Google Scholar]
- 4.Manson MD, Tedesco P, Berg HC, Harold FM, Van der Drift C. A protonmotive force drives bacterial flagella. Proc Natl Acad Sci U S A. 1977;74:3060–3064. doi: 10.1073/pnas.74.7.3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirota N, Imae Y. Na+-driven flagellar motors of an alkalophilic Bacillus strain YN-1. J Biol Chem. 1983;258:10577–10581. [PubMed] [Google Scholar]
- 6.Berg HC, Brown DA. Chemotaxis in Escherichia coli analysed by three-dimensional tracking. Nature. 1972;239:500–504. doi: 10.1038/239500a0. [DOI] [PubMed] [Google Scholar]
- 7.Turner L, Ryu WS, Berg HC. Real-time imaging of fluorescent flagellar filaments. J Bacteriol. 2000;182:2793–2801. doi: 10.1128/jb.182.10.2793-2801.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irikura VM, Kihara M, Yamaguchi S, Sockett H, Macnab RM. Salmonella typhimurium fliG and fliN mutations causing defects in assembly, rotation, and switching of the flagellar motor. J Bacteriol. 1993;175:802–810. doi: 10.1128/jb.175.3.802-810.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lloyd SA, Blair DF. Charged residues of the rotor protein FliG essential for torque generation in the flagellar motor of Escherichia coli. J Mol Biol. 1997;266:733–744. doi: 10.1006/jmbi.1996.0836. [DOI] [PubMed] [Google Scholar]
- 10.Lloyd SA, Tang H, Wang X, Billings S, Blair DF. Torque generation in the flagellar motor of Escherichia coli: evidence of a direct role for FliG but not for FliM or FliN. J Bacteriol. 1996;178:223–231. doi: 10.1128/jb.178.1.223-231.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yorimitsu T, Mimaki A, Yakushi T, Homma M. The conserved charged residues of the C-terminal region of FliG, a rotor component of the Na+driven flagellar motor. J Mol Biol. 2003;334:567–583. doi: 10.1016/j.jmb.2003.09.052. [DOI] [PubMed] [Google Scholar]
- 12.Zhou J, Lloyd SA, Blair DF. Electrostatic interactions between rotor and stator in the bacterial flagellar motor. Proc Natl Acad Sci U S A. 1998;95:6436–6441. doi: 10.1073/pnas.95.11.6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lloyd SA, Whitby FG, Blair DF, Hill CP. Structure of the C-terminal domain of FliG, a component of the rotor in the bacterial flagellar motor. Nature. 1999;400:472–475. doi: 10.1038/22794. [DOI] [PubMed] [Google Scholar]
- 14.Brown PN, Hill CP, Blair DF. Crystal structure of the middle and C-terminal domains of the flagellar rotor protein FliG. EMBO J. 2002;21:3225–3234. doi: 10.1093/emboj/cdf332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Francis NR, Irikura VM, Yamaguchi S, DeRosier DJ, Macnab RM. Localization of the Salmonella typhimurium flagellar switch protein FliG to the cytoplasmic M-ring face of the basal body. Proc Natl Acad Sci U S A. 1992;89:6304–6308. doi: 10.1073/pnas.89.14.6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ueno T, Oosawa K, Aizawa S. M ring, S ring and proximal rod of the flagellar basal body of Salmonella typhimurium are composed of subunits of a single protein, FliF. J Mol Biol. 1992;227:672–677. doi: 10.1016/0022-2836(92)90216-7. [DOI] [PubMed] [Google Scholar]
- 17.Kihara M, Miller GU, Macnab RM. Deletion analysis of the flagellar switch protein FliG of Salmonella. J Bacteriol. 2000;182:3022–3028. doi: 10.1128/jb.182.11.3022-3028.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Togashi F, Yamaguchi S, Kihara M, Aizawa SI, Macnab RM. An extreme clockwise switch bias mutation in fliG of Salmonella typhimurium and its suppression by slow-motile mutations in motA and motB. J Bacteriol. 1997;179:2994–3003. doi: 10.1128/jb.179.9.2994-3003.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Way SM, Millas SG, Lee AH, Manson MD. Rusty, jammed, and well-oiled hinges: Mutations affecting the interdomain region of FliG, a rotor element of the Escherichia coli flagellar motor. J Bacteriol. 2004;186:3173–3181. doi: 10.1128/JB.186.10.3173-3181.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andrade MA, Petosa C, O'Donoghue SI, Muller CW, Bork P. Comparison of ARM and HEAT protein repeats. J Mol Biol. 2001;309:1–18. doi: 10.1006/jmbi.2001.4624. [DOI] [PubMed] [Google Scholar]
- 21.Conti E, Uy M, Leighton L, Blobel G, Kuriyan J. Crystallographic analysis of the recognition of a nuclear localization signal by the nuclear import factor karyopherin alpha. Cell. 1998;94:193–204. doi: 10.1016/s0092-8674(00)81419-1. [DOI] [PubMed] [Google Scholar]
- 22.Huber AH, Nelson WJ, Weis WI. Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell. 1997;90:871–882. doi: 10.1016/s0092-8674(00)80352-9. [DOI] [PubMed] [Google Scholar]
- 23.Brown PN, Terrazas M, Paul K, Blair DF. Mutational analysis of the flagellar protein FliG: sites of interaction with FliM and implications for organization of the switch complex. J Bacteriol. 2007;189:305–312. doi: 10.1128/JB.01281-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marykwas DL, Berg HC. A mutational analysis of the interaction between FliG and FliM, two components of the flagellar motor of Escherichia coli. J Bacteriol. 1996;178:1289–1294. doi: 10.1128/jb.178.5.1289-1294.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomas DR, Francis NR, Xu C, DeRosier DJ. The three-dimensional structure of the flagellar rotor from a clockwise-locked mutant of Salmonella enterica serovar Typhimurium. J Bacteriol. 2006;188:7039–7048. doi: 10.1128/JB.00552-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duke TA, Le Novere N, Bray D. Conformational spread in a ring of proteins: a stochastic approach to allostery. J Mol Biol. 2001;308:541–553. doi: 10.1006/jmbi.2001.4610. [DOI] [PubMed] [Google Scholar]
- 27.Bai F, et al. Conformational spread as a mechanism for cooperativity in the bacterial flagellar switch. Science. 2010;327:685–689. doi: 10.1126/science.1182105. [DOI] [PubMed] [Google Scholar]
- 28.Thomas D, Morgan DG, DeRosier DJ. Structures of bacterial flagellar motors from two FliF-FliG gene fusion mutants. J Bacteriol. 2001;183:6404–6412. doi: 10.1128/JB.183.21.6404-6412.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J, et al. Intact Flagellar Motor of Borrelia burgdorferi Revealed by Cryo-Electron Tomography: Evidence for Stator Ring Curvature and Rotor/C Ring Assembly Flexion. J Bacteriol. 2009 doi: 10.1128/JB.00340-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murphy GE, Leadbetter JR, Jensen GJ. In situ structure of the complete Treponema primitia flagellar motor. Nature. 2006;442:1062–1064. doi: 10.1038/nature05015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.