

Abstract
We have previously noted that the antinociceptive efficacy of morphine was significantly decreased in rat pups chronically infused with morphine from implanted osmotic minipumps. In this study, morphine was fully efficacious (i.e., 100% maximum possible effect, %MPE) in the 52 ºC tail-immersion test after a 72-h infusion from implanted saline-filled osmotic minipumps. However, administration of up to 1000 mg/kg s.c. morphine failed to elicit greater than a 27% MPE in rats infused with morphine at 2 mg/kg/h. Morphine was more efficacious when the water bath temperature was decreased to 49 ºC. Experiments were conducted to determine the mechanisms whereby chronic morphine administration leads to a decrease in antinociceptive efficacy. The kappa-opioid antagonist nor-binalorphimine completely blocked the antinociceptive effects of morphine in morphine-infused rat pups. The kappa agonist U50,488 elicited antinociception however, the requirement to use higher doses in morphine- than saline-infused rats indicates that kappa cross-tolerance was present. Thus, in tolerant rats the antinociceptive effects of high doses of morphine appear to be mediated through kappa-opioid receptors. The delta-opioid antagonist naltrindole was inactive in both treatment groups. DAMGO-stimulated [35S]GTPγS and [3H]naloxone binding reveal that the anatomical distribution of the mu-opioid receptor was consistent with that of the adult rat brain. In adult rats, the mu-opioid receptor is desensitized during morphine tolerance. However, desensitization was not evident in P17 rats based on the lack of significant decreases in [35S]GTPγS binding. Furthermore, [3H]naloxone binding indicated a lack of mu receptor downregulation in morphine-tolerant rat pups.
Keywords: morphine, tolerance, dependence, postnatal rats, mu opioid receptor, antinociception
1. Introduction
Studies have shown that repetitive administration of morphine in postnatal rats results in the development of tolerance [2,28,33,36]. However, continuous administration of morphine by osmotic minipumps results in a decrease in the antinociceptive efficacy of morphine and morphine is unable to elicit a percent of maximum possible effect (%MPE) greater than 50% [28]. This finding is interesting in that it is in contrast to morphine tolerance dose response curves in adult rats where morphine is fully efficacious in tolerant animals [7,15].
It is important to study and characterize the loss of morphine’s antinociceptive efficacy rat pups continuously infused with morphine, as the levels of opioid receptors in rat pups are developmentally different than adults. Mu (μ) opioid receptor levels differ in young postnatal rats vs. adult animals, with receptor expression highest in the striatum in the early neonatal period. Only later, during postnatal days14 (P14) through 21 (P21) do μ opioid receptor levels reach adult density in the thalamus and hypothalamus [16]. Chronic pre- and postnatal morphine treatment decreases μ receptor density in the brains of neonatal rats [31]. Kappa (κ) and delta (δ) opioid receptors are fully developed and functional by P17 and P21 respectively [16].
The overall objective of the present study is to fully characterize the change in morphine’s antinocicpetive efficacy in postnatal day 17 (P17) rats continuously infused with morphine and examine morphine’s activity at the μ receptor. The warm water tail immersion test was used to determine morphine’s antinociceptive efficacy in saline-infused P17 rats (control) and morphine-infused P17 rats. The observed loss of morphine’s antnocicpetive efficacy in the morphine-infused rats was dependent on the temperature of the water in the tail immersion test. In the morphine-infused rats, blocking the δ receptor did not affect the decline in morphine’s efficacy; however blocking the κ receptor resulted in a further decrease in morphine’s antinocicpetive efficacy. [35S]GTPγS and [3H]naloxone autoradiography were employed to evaluate the activity and levels of the μ receptor in both saline-infused and morphine-infused P17 rats. Results from both analyses indicate that μ opioid receptors are present and fully functional in all regions of the brain and spinal cord examined. Therefore, the overall results from this study indicate that the antinocicpetive efficacy of morphine is decreaed in P17 rats continuously infused with morphine and in these rats, morphine acts through the κ opioid receptors at high levels.
2. Results
Loss of morphine’s efficacy in morphine-tolerant P17 rats
P17 rats were tested for morphine-induced antinociception in the 52°C tail immersion test after a 72-h infusion of saline (1 μl/h) or morphine (2 mg/kg/h) from implanted osmotic minipumps. Morphine elicited dose-dependent antinociception in naïve and saline pump-implanted rat pups (Fig. 1). The respective ED50 values of 3.7 mg/kg (95% C.L. 3.2 to 4.3) and 4.3 mg/kg (95% C.L. 3.3 to 5.6) were similar, demonstrating no surgical effect of pump implantation. However, the antinociceptive efficacy of morphine was significantly decreased in the morphine-infused rats. Morphine doses from 50 to 1000 mg/kg failed to elicit above a 27% MPE value, and thus it was impossible to calculate an ED50 value for this group of rats.
Figure 1. Morphine antinociception in opioid-naïve and morphine tolerant P17 rats tested in the 52 °C tail-immersion assay.

Rats remained naïve (●) or were surgically implanted with 1003D osmotic minipumps infusing isotonic saline (■, 1 μl/h) or morphine (▲, 2 mg/kg/h). Seventy-two hours later baseline latencies were obtained in the 52 °C tail-immersion test before treatment with morphine s.c. for test latencies for dose-response curves 30-min later. Morphine produced the same degree of antinociception in naïve and saline-infused rats. The antinociceptive efficacy of morphine was reduced to 27% MPE at the highest dose tested in the morphine-infused rats. n=6 rats per dose per treatment group.
Influence of water bath temperature on the efficacy of morphine
Water bath temperatures were adjusted to determine the influence nociceptive stimulus intensity on the antinociceptive effects of morphine. Initially, morphine-infused rats were tested without the administration of morphine. Tail withdrawal latencies decreased as a function of increases in water bath temperatures from 46 °C to 52 °C (Fig. 2A). 46 °C was minimally noxious, at best. At this temperature, high variability occurred, with latencies ranging between 4.6- to 8.9-sec. The baseline latencies of 4 out of 6 pups was greater than 6.8-sec, which was above the mid-point of the 10-sec cut-off for these studies. Morphine-tolerant rats were then assessed for morphine-induced antinociception (100 mg/kg, s.c.) at each water bath temperature. As seen in figure 2B, the antinociceptive effects of morphine were diminished in a temperature-dependent fashion with increases in water bath temperature. Further studies were conducted at 49 °C, since morphine was able to elicit an intermediate level of antinociception (Fig. 3). Under these conditions, the ED50 value of morphine in saline-infused rats was 1.6 mg/kg (95% C.L. 1.3 to 1.8). By decreasing the temperature 3 °C from 52 °C to 49 °C the potency of morphine was significantly increased the saline pump-implanted rats (i.e. 52 °C = 4.3 mg/kg (95% C.L. 3.3 to 5.6) vs. 49 °C = 1.6 mg/kg (95% C.L. 1.3 to 1.8)). Furthermore, in morphine-infuesd rats, morphine was more efficacious at 49 °C than at 52 °C, and reached an average maximum %MPE value of 67%. The calculated ED50 value was 150 mg/kg (95% C.L. 60.1 to 375.8). The 95% C.L. of the ED50 value of the morphine-infused group was large due to high intra-dose variability in rat pup responses to morphine.
Figure 2.

(A) Influence of water bath temperature on baseline withdrawal latencies in chronic morphine infused P17 rats. Rats were infused with morphine (●, 2 mg/kg/h) for 72-h from implanted osmotic minipumps. Baseline latencies were then obtained by testing individual groups of rats at different water bath temperatures. As the temperature of the water bath was increased, the baseline tail withdrawal latencies increased. n=6 rats per temperature.(B) Antinociceptive effects of morphine at different water bath temperatures. Rats were infused with morphine (▧, 2 mg/kg/h) for 72-h from implanted osmotic minipumps. Individual groups of P17 rats were then treated with morphine (100 mg/kg, s.c.) and then tested with different water bath temperatures 30-min later. As the temperature of the water bath was increased, the tail withdrawal latencies increased. n=6 rats per temperature.
Figure 3. Morphine antinociception in opioid-naïve and morphine tolerant P17 rats tested in the 49 °C tail-immersion assay.

Rats were infused with isotonic saline (■, 1 μl/h) or morphine (▲, 2 mg/kg/h) for 72-h from implanted osmotic minipumps. Baseline latencies were then obtained in the 49 °C tail-immersion test before treatment with morphine s.c. for test latencies for dose-response curves 30-min later. The antinocicpetive effect of morphine was reduced in the morphine-infused rats to 67% MPE at the highest dose tested. n=6 rats per dose per treatment group.
Involvement of κ and δ opioid receptors in mediating the antinociceptive effects of morphine in tolerant rat pups
Higher doses of morphine have been demonstrated to bind not only μ opioid receptors, but to bind κ and δ opioid receptors as well [5]. This led to the hypothesis that the antinociceptive effects of morphine in tolerant rat pups is mediated by high doses of morphine acting on κ and/or δ opioid receptors. For these studies, opioid receptor subtype antagonists were tested for their ability to block morphine antinociception in both vehicle- and morphine-infused rat pups in the 49 °C tail-immersion test. The κ receptor antagonist nor-BNI (10 mg/kg, s.c.) was injected 3 h before administering morphine s.c. for construction of dose-response curves. Nor-BNI completely blocked the entire range of antinociceptive morphine doses tested (i.e., 50 to 1000 mg/kg) (Fig. 4A). Attempts to surmount the blockade by administering higher doses of morphine were not possible due to the toxicity of morphine above doses of 1000 mg/kg. Thus, the data clearly indicate that high doses of morphine stimulate κ opioid receptors to elicit antinociception in morphine-infused rat pups. Other rat pups were injected with the δ antagonist naltrindole (3 mg/kg, s.c.) 5 min before morphine. As seen in figure 4B, the ED50 value of morphine was not significantly altered by naltrindole (i.e., 429.6 mg/kg [95% C.L. 219 to 842.3]). Finally, μ opioid receptor antagonists like naloxone or naltrexone were not tested since rat pups experience withdrawal signs and loss of antinociception upon injection [28,33].
Figure 4.

(A) Role of κ opioid receptors in mediating the antinociceptive effects of morphine in tolerant rat pups. Rats were infused with isotonic saline (1 μl/h) or morphine (2 mg/kg/h) for 72-h from implanted osmotic minipumps. On the test day, saline-infused rats received either vehicle s.c. (●) or nor-BNI (■, 10 mg/kg, s.c.), while morphine-infused rats received vehicle (○) or nor-BNI (□), 2.5-h before injection of morphine s.c. The rats were tested 30-min after morphine s.c. in the 49 °C tail-immersion assay. nor-BNI pretreatment had no effect of saline-infused rats, whereas in morphine-infused rats, pretreatment with nor-BNI significantly attenuated morphine’s antinociceptive efficacy at high doses. n=6 rats per dose per treatment group. *p<0.05 compared to morphine-infused rats pretreated with vehicle. (B) Role of δ opioid receptors in mediating the antinociceptive effects of morphine in tolerant rat pups. Rats were infused with isotonic saline (1 μl/h) or morphine (2 mg/kg/h) for 72-h from implanted osmotic minipumps. On the test day, saline-infused rats received either vehicle s.c. (●) or naltrindole (▲, 3 mg/kg, s.c.), while morphine-infused rats received vehicle (○) or nor-BNI (△), 5-min before injection of morphine s.c. The rats were tested 30-min after morphine s.c. in the 49 °C tail-immersion assay. Pretreatment with naltrindole had no effect on morphine’s antinociceptive efficacy in both saline- and morphine-infused rats. n=6 rats per dose per treatment group.
To further explore the role of the κ receptor in mediating antinociception in morphine-infused rat pups, the ability of the κ receptor agonist, U50,488 to elicit antinociception in saline- and morphine-infused rat pups was evaluated (Fig 5). Using the 49 °C tail-immersion test, U50,488 was fully efficacious in both saline- and morphine-infused groups. However, the ED50 for U50,488 was four times higher for the morphine-infused rat pups. The ED50 for U50,488 was 5.5 mg/kg (95% C.L. 3.9 to 7.6) and 22.0 mg/kg (95% C.L. 16.2 to 29.7) for saline- and morphine-infused rats, respectively. Therefore, morphine-infused rats exhibited cross-tolerance to the κ receptor agonist, indicating the at high infusion doses, morphine is eliciting antinocicpetion through the κ receptors.
Figure 5. Demonstration of cross tolerance to the kappa receptor agonist, U50,488 in morphine-infused rats.

Rats were infused with isotonic saline (1 μl/h) or morphine (2 mg/kg/h) for 72-h from implanted osmotic minipumps. Baseline latencies were then obtained in the 49 °C tail-immersion test before treatment with U50,488 i.p. for test latencies for dose-response curves 30-min later. U50,488 was less potent but fully efficacious in the morphine-infused rats compared to saline-infused rats. n=6 rats per dose per treatment group.
Re-establishment of morphine antinociception after pump removal
Experiments were conducted to determine whether the antinociceptive effects of morphine would be reinstated after removal of the morphine pumps and the rat pups were no longer tolerant. After a 72-h infusion, saline- and morphine-pumps were surgically removed, and the rats were returned to the dams. We have previously demonstrated that spontaneous withdrawal occurs during the first 4-days, and that naloxone is inactive on day-5 [27]. On day 5, acute morphine administration resulted in complete dose-response curves in naïve, saline- and morphine-pumped P21 rats (Fig. 6). The ED50 value of morphine-infused rats of 1.2 mg/kg (95% C.L. 0.7 to 1.9) was similar to the naïve and saline-infused ED50 values of 1.2 mg/kg (95% C.L. 0.7 to 2.2) and 1.8 mg/kg (95% C.L. 1.2 to 2.7), respectively. Thus, the rats were able to obtain the full antinociceptive effect of morphine 5 days after the continuous infusion of morphine was stopped.
Figure 6. Re-establishment of the antinociceptive effects of morphine in P21 rats after chronic exposure to morphine.

Rats remained naïve, or were infused with isotonic saline (1 μl/h) or morphine (2 mg/kg/h) for 72-h from implanted osmotic minipumps. On P17 the pumps were removed and the rats were allowed to recover from the exposure to morphine for 5 days. On P21, naïve (○), saline- (■) and morphine-infused (▲) rats were treated with morphine s.c. and tested 30-min later in the 49 °C tail-immersion test. Morphine was fully efficacious in naïve, saline- and morphine-infused rats. n=6 rats per dose per treatment group.
Autoradiography results of DAMGO-stimulated [35S]GTPγS binding and [3H]naloxone binding
Coronal sections obtained from saline- and morphine-infused P17 rats were processed for both [35S]GTPγS binding to assess for μ receptor-activated G protein activity, and [3H]naloxone binding to assess for μ receptor binding. Representative sections of DAMGO-stimulated [35S]GTPγS binding and [3H]naloxone binding from control brains are shown in figure 7. The anatomical distribution of [35S]GTPγS binding is the same as [3H]naloxone binding. Fully developed and functional μ receptors appear in the caudate-putamen (Fig 7A), thalamus and amygdala (Fig 7B), parabrachial nuclei (Fig 7C), and the dorsal horn of the spinal cord (Fig 7D). This regional distribution and stimulated activity of the μ receptor in P17 rats is consistent with that previously reported for adult rats [22,23].
Figure 7.
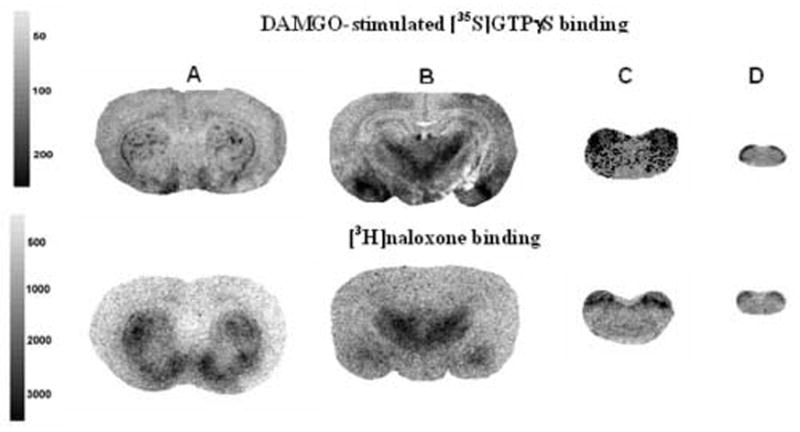
(A) Distribution of DAMGO-stimulated [35S]GTPγS binding and [3H]naloxone binding of μ receptors in opioid naïve P17 rats. Rats were infused with isotonic saline (1 μl/h) for 72-h from implanted osmotic minipumps. DAMGO-stimulated [35S]GTPγS binding or [3H]naloxone binding was conducted on coronal brain sections from a group of 10 rats to determine μ receptor activity and binding capacity within each brain region. The representative autoradiograms reveal the anatomical distribution of μ receptor activity localized in the (A) caudate-putamen, (B) thalamus/amygdala, (C) parabrachial nucleus, and (D) the dorsal horn of the spinal cord. The grayscale bars indicate amount of [35S]GTPγS incorporation and [3H]naloxone binding (nCi/g).
Table 1 shows net DAMGO-stimulated [35S]GTPγS binding and specific [3H]naloxone binding in saline- versus morphine-infused rats. [35S]GTPγS binding did not differ significantly between saline-infused and morphine-tolerant rats in any region examined. Similarly, [3H]naloxone binding did not differ significantly between saline-infused rats and morphine-tolerant rats in the same regions. These results demonstrate that the 3 day morphine infusion at 2 mg/kg/h did not affect μ receptor-stimulated G protein activation or μ receptor binding in any brain region examined.
Table 1. The effect of chronic morphine administration on DAMGO-stimulated [35S]GTPγS activity and [3H]naloxone binding in morphine tolerant P17 rat brain.
Rats were infused with isotonic saline (1 μl/h) or morphine (2 mg/kg/h) for 72-h from implanted osmotic minipumps. Brains were removed and prepared as described in Methods for coronal sections to measure DAMGO-stimulated [35S]GTPγS binding activity or [3H]naloxone binding. Exposed Kodak MR film was analyzed using the NIH IMAGE program for densitometric analysis of 10 saline and 10 morphine-treated rats.
| Net DAMGO-Stimulated | ||||
|---|---|---|---|---|
| [35S]GTPγS binding (nCi/g) | [3H]naloxone binding (nCi/g) | |||
| Region | Saline | Morphine | Saline | Morphine |
| Caudate putamen | 121 ± 14 | 92 ± 17 | 3014 ± 262 | 2853 ± 142 |
| Thalamus | 153 ± 19 | 113 ± 10 | 3451 ± 185 | 3074 ± 219 |
| Amygdala | 110 ± 22 | 125 ± 19 | 2625 ± 192 | 2657 ± 176 |
| Periaqueductal gray | 117 ± 25 | 116 ± 19 | 2279 ± 137 | 2366 ± 132 |
| Parabrachial nuclei | 289 ± 8 | 254 ± 35 | 3880 ± 494 | 3700 ± 151 |
| Spinal cord | 246 ± 19 | 212 ± 16 | 1627 ± 91 | 1698 ± 62 |
3. Discussion
Behavioral expression of morphine tolerance in P17 rats
The purpose of this study was to understand the mechanism(s) underlying the decrease in morphine’s efficacy in tolerant P17 rat pups. Other investigators have produced tolerance in infant rats by repeatedly injecting morphine under different schedules [2,36]. This study incorporated the use of osmotic minipumps to continuously infuse morphine to produce tolerance. However, instead of a producing a parallel rightward shift in the dose-response curve, the maximum antinociceptive efficacy of acutely administered morphine was limited to 27 %MPE (Fig. 1). In our previous study of P9 and P17 rats, as the minipump infusion dose was increased for each group of rats, Emax values for acutely administered morphine were decreased in a dose-dependent fashion [28]. In the previous study, the nociceptive stimulus temperature for the tail-flick test was held constant, while varying the morphine infusion dose. Thus, one goal of this study was to determine whether varying the nociceptive stimulus intensity would alter the antinociceptive effects of morphine (Fig 2A and 2B). Groups have previously tested neonatal and infant rats using immersion test temperatures from 40 °C to 55 °C [10,38,39]. Our data demonstrated that morphine was maximally active (i.e., 90% MPE or greater) in naïve and saline-infused rats tested at 52 °C, and morphine’s potency was significantly increased when water bath temperatures were decreased to 49 °C (Fig 1 vs. Fig. 3). However, morphine was minimally active (i.e., maximum 27% MPE) in tolerant rats tested at 52 °C, while testing these rats at 49 °C led to a significant increase in morphine’s efficacy to 67% MPE (Fig. 1 vs. Fig. 3). These data indicate that the higher 52 °C nociceptive stimulus intensity contributed, in part, to decreasing morphine’s efficacy in tolerant rats. However, nociceptive stimulus intensity was not the only factor altering the maximum efficacy of morphine. Many investigators have tested neonatal and infant rats using a 50 °C tail immersion temperature [1,8,11]. Yet, even when P17 rat pups were tested at 49 °C, morphine’s maximal efficacy was limited to 67 %MPE at the maximum 1000 mg/kg morphine dose (Fig. 1). These results indicated that high doses of morphine elicited antinociception in tolerant rats through a non-μ receptor mechanism.
The hypothesis was tested that high doses of morphine stimulate κ opioid receptors to elicit antinociception in tolerant rats. A dose of nor-BNI demonstrated to block κ receptor agonist-mediated antinociception [30] was shown in our study to completely block morphine antinociception in tolerant rat pups (Fig 4A). Additionally, morphine-infused rats demonstrated cross-tolerance to the κ receptor agonist, U50,488 (Fig 5). These results indicate that high doses of morphine elicit antinociception by stimulating κ opioid receptors. The κ receptor is fully developed and functional by P17 [16]. κ receptor agonists elicit antinociception, although not to the same degree as μ or δ receptor drugs [19]. If morphine was acting through κ receptors, this could explain why it was necessary to decrease the temperature of the water bath in order to demonstrate an increase in the efficacy of morphine. Interestingly, κ receptor agonists like U50,488 and dynorphin A (1–17) are often tested at a tail-immersion temperature of 49 °C [13,35].
High concentrations of morphine bind to δ and κ receptors [5]. Yet, the δ receptor antagonist naltrindole [29] was inactive in both non-tolerant and tolerant rats (Fig 4B). Accordingly, δ receptors are not fully developed in the brain and spinal cords of rats until P21 [16]. In adult rats, both μ and δ receptors are functional, and it has been unknown whether high doses of morphine can elicit antinociception through δ receptors. Recently, it was demonstrated that high doses of morphine elicit antinociception in μ knockout mice by stimulating κ, but not δ, opioid receptors [37]
Finally, a previous study demonstrated that fentanyl tolerant infant rats were desensitized to morphine as P50 juvenile and P90 adult rats [32]. In addition, in utero exposure to morphine led to a decrease in morphine’s antinociceptive potency in infant and juvenile rats [14]. This led to the possibility that the decrease in morphine’s antinociceptive efficacy induced by continuous morphine administration might result in desensitization after their recovery from the morphine treatment. Yet at P21, morphine-infused rats were no different from saline-infused rats, although desensitization could develop later in life (Fig. 5). However, in the absence of biochemical and behavioral data, conducting these studies would be based on speculation and yield possibly limited results.
Influence of chronic morphine exposure on brain and spinal cord μ receptors
Both [3H]naloxone and DAMGO-stimulated [35S]GTPγS binding levels were measured in the brains and spinal cords of P17 rat pups The μ receptor was expressed in numerous brain regions, as demonstrated using [3H]naloxone binding (Table 1). Furthermore, the widespread distribution of DAMGO-stimulated [35S]GTPγS binding indicates that the μ receptor was functionally active by P17. Finally, autoradiographic examination of [3H]naloxone and [35S]GTPγS binding indicate that the μ receptor was distributed anatomically in a fashion similar to adult rats [22,23].
Yet, [35S]GTPγS binding revealed that chronic infusion of P17 rats with morphine failed to desensitize μ receptors in any region of the brain or spinal cord (Table 1). Adult rats treated chronically with morphine or heroin resulted in μ receptor desensitization in the thalamus, periaqueductal gray, and brain stem [12,21,23]. One major difference with the previous studies was that desensitization in adults occurred after 12 days of morphine infusion and 34 to 40 days of heroin self-administration. A longer morphine exposure might be needed to see desensitization in rat pups. However, μ receptor desensitization cannot explain the decrease in morphine’s antinociceptive efficacy in tolerant P17 rat pups.
In the present study, the μ receptor was not downregulated based on the [3H]naloxone binding experiment. The issue has been more clouded in adult animals, probably reflecting differences in species, animal strains, and in the methods of infusing and injecting animals with morphine. Chronic infusion of morphine at 40 mg/kg/day or repeated bolus dosing failed to downregulate the μ receptor in some studies [18,20,26], while other groups reported a significant up-regulation of the μ receptor [4,40]. Petruzzi and colleagues concluded that genetic traits contribute to tolerance when chronic morphine treatment downregulated the μ receptor in inbred C57BL/6J mice, but not DBA/2J mice [17]. However, μ receptor downregulation cannot account for the profound decrease in the antinociceptive efficacy of morphine in tolerant P17 rat pups.
In summary, this study provides evidence that high doses of morphine stimulate κ opioid receptors to elicit antinociception in tolerant rat pups. Yet the loss of morphine’s efficacy cannot be attributed to either μ receptor desensitization or downregulation, suggesting that non-opioid mechanisms might be involved. This and other laboratories have demonstrated that the adenylyl cyclase and phosphatidylinositol cascades, as well as various Ca++ and K+ channels, contribute to the expression of morphine tolerance [6,9,24,25,34]. These and other mechanisms could be investigated to determine their possible contribution mediating the decrease in morphine’s efficacy in tolerant neonatal and infant rats.
4. Experimental Procedure
Source of rat pups
Nulliparous female Sprague Dawley rat dams were purchased from Zivic Miller (Zedianople, PA, U.S.A.). Litter sizes were culled to 10 pups per dam (five males and five females). The animals were allowed food and water ad libitum and were housed at the Virginia Commonwealth University Medical Center animal care facilities with a 12-h light-dark cycle (0700-light, 1900-dark). The Institutional Animal Care and Use Committee at Virginia Commonwealth University approved the experiments. The laboratory and IACUC committee at VCU follows the NIH guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978) to minimize pain and distress in laboratory animals.
Surgical implantation of osmotic minipumps
Alzet® osmotic minipumps were surgically implanted as previously described [32]. P14 rat pups, weighing 30 g, were anesthetized with isoflurane USP (Henry Schein, Inc. Melville, NY, U.S.A.) for implantation of Alzet 1003D osmotic minipumps with a delivery rate of 1μL/h. The rat pups received either saline-filled or morphine-filled pumps. The surgical incisions were sealed with Vetbond Tissue Adhesive (3M Animal Care Products, St. Paul, MN, U.S.A.) and swabbed with 10% povidone iodine (General Medical, Prichard, WV, U.S.A.). Morphine sulfate concentrations were adjusted to a delivery rate of 2 mg/kg/h [27].
Warm Water Tail-Immersion Test
Seventy-two hours after pump implantation, the rats were assessed for morphine antinociception using the warm water tail immersion test. The rats were gently restrained, and the distal third of the tail was immersed in a warm water bath. Varying temperatures of water were tested, including 52°C and 49°C. Base-line (control) latencies of 2 to 4 s were used, while a 10 s maximum (test) cut-off time was imposed to prevent tissue damage of the tail. Antinociception was quantified as the percentage of maximum possible effect (MPE), as represented by the formula
Morphine Antinocicepetive Testing
Saline- and morphine-infused rats were injected with morphine 30-min before testing in the warm water tail withdrawal assay. Morphine dose response curves were generated using 4 to 6 rats per dose. For the studies assessing the role of the δ opioid receptor, the δ antagonist naltrindole was dissolved in deionized H2O and was injected (3 mg/kg; s.c.) 5 min prior to morphine injections. For the studies assessing the role of the κ opioid receptor, the κ antagonist nor-binaltorphimine (nor-BNI) was dissolved in deionized H2O and was injected (10 mg/kg; s.c.) 3 h prior to morphine injections. For cross-tolerance studies, the κ receptor agoinist, U50,488 was dissolved in deionized H2O and was injected i.p. 15 min prior to the tail immersion test.
Re-establishment of morphine’s efficacy
P14 rats were implanted with saline-filled or morphine-filled pumps as previously described. Seventy-two hours later, the pumps were surgically removed to allow the rats, now P17, to undergo spontaneous morphine withdrawal. The rats were anesthetized with isoflurane USP and the initial incision was swabbed with 10% povidone iodine, washed with 70% ethanol USP, and reopened with sterile scissors. The minipumps were removed from the subcutaneous space using sterile forceps. The incision was resealed with Vetbond Tissue Adhesive and washed with 10% povidone iodine. After five days of spontaneous withdrawal, the antinociceptive efficacy of morphine was determined using the warm-water tail-withdrawal test as described previously.
Tissue preparation for autoradiography
To assess the activity of the μ receptor during morphine tolerance, P14 rats were implanted with vehicle- or morphine-filled pumps as previously described. After seventy-two hours, the rats were sacrificed by decapitation. The brains were removed and immersed in isopentane at -35 °C, and then stored frozen at -80 °C to be used for autoradiography. Coronal sections (20 μm) were cut on a cryostat at -20 °C and thaw-mounted onto gelatin-coated slides. Sections were cut at five levels to include the following regions: 1) caudate putamen 2) thalamus/amygdala, 3) periaqueductal gray 4) parabrachial nucleus, 5) cervical spinal cord. Slides were dried under a vacuum and stored at -80 °C until use.
Agonist-stimulated [35S]GTPγS binding autoradiography
Slides were incubated in assay buffer (50 mM Tris-HCl, 3 mM MgCl2, 0.2 mM EGTA, 100 mM NaCl, pH 7.4) at 25 °C for 10 min. Slides were then incubated in assay buffer with GDP (2 mM) and adenosine deaminase (9.5 mU/ml) for 20 min at 25 °C. Sections were then incubated in assay buffer with 2 mM GDP, 9.5 mU/ml adenosine deaminase, 0.04 nM [35S] GTPγS, and 10 μM DAMGO at 25 °C for 2 h. Basal [35S]GTPγS binding was assessed in the absence of DAMGO. Slides were rinsed in twice for 2 min each in cold Tris buffer (50 mM Tris-HCl, pH 7.4) and once in deionized H2O for 30 s. Slides were dried overnight and exposed to Kodak MR film for 24 h in film cassettes containing [14C] microscales (Amersham, Arlington Heights, IL) for densitometric analysis.
[3H]Naloxone autoradiography
Sections were equilibrated in assay buffer for 15 min at 25 °C. Slides were then incubated in 2 nM [3H]naloxone in assay buffer for 1 h at 25 °C. Slides were rinsed three times for 2 min each in 50 mM Tris (pH 7.4) buffer at 4 °C, then for 30 s in deionized H2O at 4 °C. Nonspecific binding was assessed in the presence of 10 μM naltrexone. Slides were dried and exposed to Kodak MS film for approximately 10 weeks. All film cassettes included a [3H] microscale (Amersham) for calibration of results.
Analysis of data
Dose-response curves were analyzed by least-squares linear regression analysis followed by calculation of 95% confidence limits (CL) by the method of Bliss [3]. %MPE values for the morphine-tolerant rats were compared to those of tolerant rats treated with naltrindole or nor-BNI by two-factor ANOVA followed by the Tukey’s post hoc test. Autoradiographic films were digitized with a Sony XC-77 video camera and analyzed densitometrically using the NIH IMAGE program for Macintosh computers. For [35S]GTPγS autoradiography, resulting values were expressed as nanocuries of [35S] per gram of tissue and were corrected for [35S] from [14C] standards based on incorporation of [35S] into sections of frozen brain paste [21]. Net agonist-stimulated [35S]GTPγS binding was calculated by subtracting basal binding (obtained in the absence of agonist) from agonist-stimulated binding. For [3H]naloxone binding, nonspecific binding was subtracted from total binding, and resulting values represent specific picocuries of [3H]naloxone binding per gram of tissue. Data are reported as mean values ± SE of triplicate sections of brains from ten treated and control animals. For the statistical comparisons between vehicle- and morphine-treated rats, an ANOVA was performed and followed by the Tukey’s post hoc test.
Acknowledgments
This research was funded by the National Institute on Drug Abuse grants DA-01647-28 and K05-DA-00480; D.C.S. was supported by T32-DA-07027.
Abvbreviations
- %MPE
percent maximum possible effect
- P14
postnatal day 14
- P17
postnatal day 17
- P21
postnatal day 21
- ED50
50% effective dose
- s.c
subcutaneous
- CL
confidence limits
- Nor-BNI
nor-binaltophamine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alberti I, Fernandez B, Alguacil LF, Aguilar A, Caamano M, Romero EM, Viveros MP. Preweanling naltrindole administration differentially affects clonidine induced antinociception and plasma adrenaline levels in male and female neonatal rats. Br J Pharmacol. 1999;128:953–60. doi: 10.1038/sj.bjp.0702852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barr GA, Wang S. Tolerance and withdrawal to chronic morphine treatment in the week-old rat pup. Eur J Pharmacol. 1992;215:35–42. doi: 10.1016/0014-2999(92)90605-4. [DOI] [PubMed] [Google Scholar]
- 3.Bliss C. Statistics in Biology. McGraw Hill; New York: 1967. p. 439. [Google Scholar]
- 4.Brady LS, Herkenham M, Long JB, Rothman RB. Chronic morphine increases mu-opiate receptor binding in rat brain: a quantitative autoradiographic study. Brain Res. 1989;477:382–6. doi: 10.1016/0006-8993(89)91432-7. [DOI] [PubMed] [Google Scholar]
- 5.Corbett ADPS, Kosterlitz HW. Selectivity of Ligands for Opioid Receptors. Springer-Verlag; Berlin: 1993. [Google Scholar]
- 6.Dalton GD, Smith FL, Smith PA, Dewey WL. Protein Kinase A activity is increased in mouse lumbar spinal cord but not brain following morphine antinociceptive tolerance for 15 days. Pharmacol Res. 2005;52:204–10. doi: 10.1016/j.phrs.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 7.Deng HB, Yu Y, Wang H, Guang W, Wang JB. Agonist-induced mu opioid receptor phosphorylation and functional desensitization in rat thalamus. Brain Res. 2001;898:204–14. doi: 10.1016/s0006-8993(01)02179-5. [DOI] [PubMed] [Google Scholar]
- 8.Fernandez B, Antelo MT, Kitchen I, Viveros MP. Effects of neonatal naltrindole treatment on antinociceptive and behavioral responses to mu and kappa agonists in rats. Pharmacol Biochem Behav. 1999;62:145–9. doi: 10.1016/s0091-3057(98)00138-5. [DOI] [PubMed] [Google Scholar]
- 9.Granados-Soto V, Kalcheva I, Hua X, Newton A, Yaksh TL. Spinal PKC activity and expression: role in tolerance produced by continuous spinal morphine infusion. Pain. 2000;85:395–404. doi: 10.1016/S0304-3959(99)00281-X. [DOI] [PubMed] [Google Scholar]
- 10.Kim YI, Na HS, Han JS, Hong SK. Critical role of the capsaicin-sensitive nerve fibers in the development of the causalgic symptoms produced by transecting some but not all of the nerves innervating the rat tail. J Neurosci. 1995;15:4133–9. doi: 10.1523/JNEUROSCI.15-06-04133.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitchen I, Pinker SR. Antagonism of swim-stress-induced antinociception by the delta-opioid receptor antagonist naltrindole in adult and young rats. Br J Pharmacol. 1990;100:685–8. doi: 10.1111/j.1476-5381.1990.tb14076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kruzich PJ, Chen AC, Unterwald EM, Kreek MJ. Subject-regulated dosing alters morphine self-administration behavior and morphine-stimulated [35S]GTPgammaS binding. Synapse. 2003;47:243–9. doi: 10.1002/syn.10173. [DOI] [PubMed] [Google Scholar]
- 13.Nemmani KV, Mogil JS. Serotonin-GABA interactions in the modulation of mu- and kappa-opioid analgesia. Neuropharmacology. 2003;44:304–10. doi: 10.1016/s0028-3908(02)00374-x. [DOI] [PubMed] [Google Scholar]
- 14.O'Callaghan JP, Holtzman SG. Prenatal administration of morphine to the rat: tolerance to the analgesic effect of morphine in the offspring. J Pharmacol Exp Ther. 1976;197:533–44. [PubMed] [Google Scholar]
- 15.Paronis CA, Holtzman SG. Development of tolerance to the analgesic activity of mu agonists after continuous infusion of morphine, meperidine or fentanyl in rats. J Pharmacol Exp Ther. 1992;262:1–9. [PubMed] [Google Scholar]
- 16.Petrillo P, Tavani A, Verotta D, Robson LE, Kosterlitz HW. Differential postnatal development of mu-, delta- and kappa-opioid binding sites in rat brain. Brain Res. 1987;428:53–8. doi: 10.1016/0165-3806(87)90082-4. [DOI] [PubMed] [Google Scholar]
- 17.Petruzzi R, Ferraro TN, Kurschner VC, Golden GT, Berrettini WH. The effects of repeated morphine exposure on mu opioid receptor number and affinity in C57BL/6J and DBA/2J mice. Life Sci. 1997;61:2057–64. doi: 10.1016/s0024-3205(97)00864-3. [DOI] [PubMed] [Google Scholar]
- 18.Polastron J, Meunier JC, Jauzac P. Chronic morphine induces tolerance and desensitization of mu-opioid receptor but not down-regulation in rabbit. Eur J Pharmacol. 1994;266:139–46. doi: 10.1016/0922-4106(94)90103-1. [DOI] [PubMed] [Google Scholar]
- 19.Przewlocki R, Przewlocka B. Opioids in chronic pain. Eur J Pharmacol. 2001;429:79–91. doi: 10.1016/s0014-2999(01)01308-5. [DOI] [PubMed] [Google Scholar]
- 20.Shen J, Benedict Gomes A, Gallagher A, Stafford K, Yoburn BC. Role of cAMP-dependent protein kinase (PKA) in opioid agonist-induced mu-opioid receptor downregulation and tolerance in mice. Synapse. 2000;38:322–7. doi: 10.1002/1098-2396(20001201)38:3<322::AID-SYN11>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 21.Sim-Selley LJ, Selley DE, Vogt LJ, Childers SR, Martin TJ. Chronic heroin self-administration desensitizes mu opioid receptor-activated G-proteins in specific regions of rat brain. J Neurosci. 2000;20:4555–62. doi: 10.1523/JNEUROSCI.20-12-04555.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sim-Selley LJ, Xiao R, Childers SR. Anatomical distribution of sodium-dependent [(3)H]naloxone binding sites in rat brain. Synapse. 2000;35:256–64. doi: 10.1002/(SICI)1098-2396(20000315)35:4<256::AID-SYN3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 23.Sim LJ, Selley DE, Dworkin SI, Childers SR. Effects of chronic morphine administration on mu opioid receptor-stimulated [35S]GTPgammaS autoradiography in rat brain. J Neurosci. 1996;16:2684–92. doi: 10.1523/JNEUROSCI.16-08-02684.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith FL, Dombrowski DS, Dewey WL. Involvement of intracellular calcium in morphine tolerance in mice. Pharmacol Biochem Behav. 1999;62:381–8. doi: 10.1016/s0091-3057(98)00168-3. [DOI] [PubMed] [Google Scholar]
- 25.Smith FL, Lohmann AB, Dewey WL. Involvement of phospholipid signal transduction pathways in morphine tolerance in mice. Br J Pharmacol. 1999;128:220–6. doi: 10.1038/sj.bjp.0702771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stafford K, Gomes AB, Shen J, Yoburn BC. mu-Opioid receptor downregulation contributes to opioid tolerance in vivo. Pharmacol Biochem Behav. 2001;69:233–7. doi: 10.1016/s0091-3057(01)00525-1. [DOI] [PubMed] [Google Scholar]
- 27.Stoller DC, Smith FL. Buprenorphine blocks withdrawal in morphine-dependent rat pups. Paediatr Anaesth. 2004;14:642–9. doi: 10.1111/j.1460-9592.2004.01264.x. [DOI] [PubMed] [Google Scholar]
- 28.Stoller DC, Thornton SR, Smith FL. Loss of antinociceptive efficacy in rat pups infused with morphine from osmotic minipumps. Pharmacology. 2002;66:11–8. doi: 10.1159/000063250. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki T, Mori T, Tsuji M, Misawa M, Nagase H. The role of delta-opioid receptors in the discriminative stimulus properties of a low dose of methamphetamine. Eur J Pharmacol. 1997;331:1–8. doi: 10.1016/s0014-2999(97)01020-0. [DOI] [PubMed] [Google Scholar]
- 30.Takemori AE, Ho BY, Naeseth JS, Portoghese PS. Nor-binaltorphimine, a highly selective kappa-opioid antagonist in analgesic and receptor binding assays. J Pharmacol Exp Ther. 1988;246:255–8. [PubMed] [Google Scholar]
- 31.Tempel A, Espinoza K. Morphine-induced downregulation of mu-opioid receptors and peptide synthesis in neonatal rat brain. Ann N Y Acad Sci. 1992;654:529–30. doi: 10.1111/j.1749-6632.1992.tb26021.x. [DOI] [PubMed] [Google Scholar]
- 32.Thornton SR, Smith FL. Long-term alterations in opiate antinociception resulting from infant fentanyl tolerance and dependence. Eur J Pharmacol. 1998;363:113–9. doi: 10.1016/s0014-2999(98)00783-3. [DOI] [PubMed] [Google Scholar]
- 33.Thornton SR, Wang AF, Smith FL. Characterization of neonatal rat morphine tolerance and dependence. Eur J Pharmacol. 1997;340:161–7. doi: 10.1016/s0014-2999(97)01434-9. [DOI] [PubMed] [Google Scholar]
- 34.Welch SP, Smith FL, Dewey WL. Morphine tolerance-induced modulation of [3H]glyburide binding to mouse brain and spinal cord. Drug Alcohol Depend. 1997;45:47–53. doi: 10.1016/s0376-8716(97)01343-4. [DOI] [PubMed] [Google Scholar]
- 35.Wilson SG, Smith SB, Chesler EJ, Melton KA, Haas JJ, Mitton B, Strasburg K, Hubert L, Rodriguez-Zas SL, Mogil JS. The heritability of antinociception: common pharmacogenetic mediation of five neurochemically distinct analgesics. J Pharmacol Exp Ther. 2003;304:547–59. doi: 10.1124/jpet.102.041889. [DOI] [PubMed] [Google Scholar]
- 36.Windh RT, Little PJ, Kuhn CM. The ontogeny of mu opiate tolerance and dependence in the rat: antinociceptive and biochemical studies. J Pharmacol Exp Ther. 1995;273:1361–74. [PubMed] [Google Scholar]
- 37.Yamada H, Shimoyama N, Sora I, Uhl GR, Fukuda Y, Moriya H, Shimoyama M. Morphine can produce analgesia via spinal kappa opioid receptors in the absence of mu opioid receptors. Brain Res. 2006;1083:61–9. doi: 10.1016/j.brainres.2006.01.095. [DOI] [PubMed] [Google Scholar]
- 38.Yashpal K, Pitcher GM, Parent A, Quirion R, Coderre TJ. Noxious thermal and chemical stimulation induce increases in 3H-phorbol 12,13-dibutyrate binding in spinal cord dorsal horn as well as persistent pain and hyperalgesia, which is reduced by inhibition of protein kinase C. J Neurosci. 1995;15:3263–72. doi: 10.1523/JNEUROSCI.15-05-03263.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yi DK, Barr GA. The induction of Fos-like immunoreactivity by noxious thermal, mechanical and chemical stimuli in the lumbar spinal cord of infant rats. Pain. 1995;60:257–65. doi: 10.1016/0304-3959(94)00119-y. [DOI] [PubMed] [Google Scholar]
- 40.Yoburn BC, Billings B, Duttaroy A. Opioid receptor regulation in mice. J Pharmacol Exp Ther. 1993;265:314–20. [PubMed] [Google Scholar]