

Abstract
Understanding how genetic variants impact the regulation and expression of genes is important for forging mechanistic links between variants and phenotypes in personal genomics studies. In this work, we investigate statistical interactions among variants that alter gene expression and identify 79 genes showing highly significant interaction effects consistent with genetic heterogeneity. Of the 79 genes, 28 have been linked to phenotypes through previous genomic studies. We characterize the structural and statistical nature of these 79 cis-epistasis models, and show that interacting regulatory SNPs often lie far apart from each other and can be quite distant from the gene they regulate. By using cis-epistasis models that account for more variance in gene expression, investigators may improve the power and replicability of their genomics studies, and more accurately estimate an individual's gene expression level, improving phenotype prediction.
1. Introduction
Epistasis, or gene-gene interaction, is thought to be an important component of complex, multifactorial diseases due to the monumental complexity of biological systems [1]. Over the past 10 years, a wealth of data from model organisms has supported a role for epistasis [2, 3]. Furthermore, epistasis is one way to account for the problem of “missing heritability”, where the analysis of single SNPs (single nucleotide polymorphisms) has explained very little of the heritability estimated from twin and adoption studies for complex traits [4, 5]. Accounting for interactions among SNPs may explain a larger portion of this heritability [6], expanding our understanding of the genomics of human disease and personalized medicine.
One often cited potentially causal mechanism of gene-gene interaction is due to variation in multiple genes in similar pathways, protein families, or genes with similar or redundant biological function [7, 8]. This generally implies that interaction occurs between genes scattered throughout the genome due to a trans-epistasis effect. Several approaches have been applied to investigate these effects in genome-wide association studies [9–12].
The occurrence of epistatic interactions, however, is not restricted to variation between distant genes. Epistatic interactions could also occur between genetic variants in close proximity which may impact transcriptional regulation. Recent work investigating the transcriptome of HapMap-based cell lines has led to the identification of expression quantitative trait loci (eQTLs) - genetic variants that influence the expression of a gene [13, 14]. Veyrieras et al. published an analysis of gene expression for 11,446 genes from HapMap-based lymphoblastoid cell lines leveraging genotypes for roughly 3 million single nucleotide polymorphisms (SNPs) to identify eQTL SNPs in a 500 kilobase (kb) window both upstream of the transcription start site and downstream of the transcription end site [15]. This work discovered 744 genes containing at least one significant eQTL SNP (p<7×10−6). The single-SNP analysis, however, does not assess the variance in gene expression that can be explained by the interaction of multiple SNPs in regulatory regions of the gene. It has been shown that the underlying mechanisms of gene expression are incredibly complex, involving the binding of multiple factors to DNA to facilitate transcription and mRNA stability [16]. Furthermore, polymorphisms within the binding sites of multiple factors may alter binding affinities to various degrees, exerting a non-linear influence on gene expression due to synergistic effects [17, 18]. This principle has been demonstrated with multiple sclerosis where severity is impacted by functional effects of two alleles in close proximity in the MHC region [19]. Despite the known complexity of gene regulation, multi-SNP interaction analysis has been previously examined only for genes having highly heritable expression but lacking single SNP associations [20]. As a secondary analysis of eQTLs using lymphoblastoid lines isolated from children with asthma, the authors successfully explain some of the missing heritability from single SNP analysis using interactions. From this limited assessment, the authors conclude that genetic interactions may have an important role in the regulation of gene expression. From these points, we hypothesize that combinations of SNPs within the 500 kb window of potential transcriptional influence will alter gene expression in humans in a non-linear fashion, here dubbed cis-epistasis.
An analysis of gene expression phenotypes provides a unique opportunity to systematically assess the degree to which epistasis, or nonlinear interactions between genetic variants, might influence human traits. Linking the HapMap cell line expression data from [15] with publicly available genotype data on the same cell lines gives us a dense collection of genetic variants in regions with strong biological plausibility for non-linear multi-SNP interaction within 11,466 quantitative expression outcomes with established main effects. Here we leverage this data to investigate the nature and degree to which cis-epistasis affects gene expression in humans. Furthermore, if epistasis plays an important role in influencing gene regulation, then it logically follows that epistasis is an important part of more complex downstream human disease phenotypes, as these traits are often associated to SNPs that alter gene expression [21]. Finally, investigators could prioritize established combinations of eQTL SNPs to inform a SNP-SNP interaction analysis in complex human traits to reduce both the computational and multiple testing burdens that plague epistasis analysis in high-throughput genetic analysis. This would also motivate reanalysis of existing datasets for multi-SNP interactions that influence complex disease, many of which are publicly available at the database of genotypes and phenotypes (dbGaP) [22]. Put simply, if a study design which considers cis-epistasis can explain more heritability in gene expression, then personal genomics studies that account for cis-epistasis should be more fruitful.
2. Methods
2.1. Genotype and Gene Expression Data
As a starting point for these analyses, we retrieved the full eQTL results database and normalized gene expression data from the Veryrieras et al. analysis (available online: http://eqtnminer.sourceforge.net/), containing 11,966,533 results (significant and non-significant) from 2,437,821 distinct SNPs and 11,466 distinct microarray probes [15]. These results establish a mapping between eQTL SNPs and the genes they regulate using a 500kb window both upstream and downstream of the regulated gene. We limited all analyses to these SNPs and microarray probes. Genotype data for these SNPs was retrieved from release #23 of the International HapMap project for 210 unrelated individuals, including 60 Yoruba (YRI) and 60 CEPH (CEU) parents, and 90 unrelated Chinese (CHB) and Japanese (JPT) samples [23]. Processed gene expression data was retrieved from (http://eqtnminer.sourceforge.net/) that had been normalized first by quantile normalization within replicates and then median normalized across all HapMap individuals. We then applied the normalization procedure from [15], which is a Gaussian quantile normalization for each gene within each population separately to avoid results confounded by population stratification (the distribution of expression values within each population is now the same).
2.2. Statistical Analysis
From the Veryrieras et al. analysis results database, we extracted all SNPs with eQTL p-values <0.05 and their associated microarray probe - that is, all nominally significant SNPs falling within 500 kb upstream of the transcription start site and 500 kb downstream of the transcription end site. Based on this data we generated all possible pair-wise combinations of associated SNPs for each microarray probe, constructing 12,107,627 two-SNP models in total. For each model, we performed a multiple linear regression analysis fitting a model with additive main effect terms (AA = 0, Aa = 1, aa = 2) for the two individual SNPs and a multiplicative interaction term. We tested for significance of interaction via a student's T-test of the interaction term coefficient. All regression analyses were conducted using the `rms' package for the R statistical computing environment [24]. Statistical significance was determined by controlling the false discovery rate (FDR) at 0.20, using the `qvalue' package available for R [25]. Linkage disequilibrium was computed using PLINK software, analyzing the combined set of 210 HapMap samples without phasing using the `--r2' option [26].
2.3. Annotation of Results Using GWAS Catalog
The National Human Genome Research Institute (NHGRI) actively maintains a catalog of all significant (p<10−5) findings from published Genome-Wide Association Studies (GWAS) [27](accessed March, 2010). The National Heart, Lung, and Blood Institute (NHLBI) also recently released comprehensive open access database of 118 GWAS studies containing 56,411 significant SNP-phenotype associations [28]. Illumina expression probe IDs were matched to transcripts within the Ensembl database (Release 49). Transcripts were matched to Ensembl Genes which have associated gene symbols within the Ensembl database. These symbols were matched to the “gene” fields in the GWAS catalogs to assess the number of matches. We also referenced the SNPs from our most significant results against these catalogs to determine if any single SNPs in the regions around our findings were known to influence any complex human phenotypes.
3. Results
3.1. Gene Expression in Humans is Influenced by Cis-Epistasis
After exhaustively fitting two-SNP models between known eQTL SNPs surrounding each microarray probe (12,107,627 two-SNP models in total), we examined the distribution of the p-values from the interaction term. The full results catalog from this analysis is available online at http://chgr.mc.vanderbilt.edu/bushlab/. Figure 1 is a quantile-quantile plot showing that the distribution of interaction term p-values deviates highly from the expected uniform distribution under the null hypothesis of no epistasis (diagonal line). This indicates that multi-SNP interaction may be common among eQTL SNPs that influence gene expression in humans.
Fig. 1.
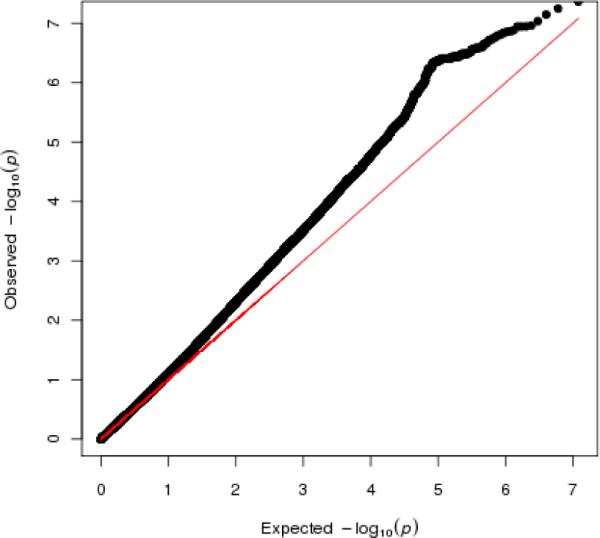
Quantile-quantile plot showing the distribution of observed –log10(p-values) against the expected –log10(p-values) for the interaction term among 12,107,627 cis-epistasis models. Deviation from the expected uniform distribution of p-values under the null hypothesis (indicated by the red line) indicates an abundance of significant cis-epistatic interactions.
Because a large number of statistical tests were performed, we corrected for multiple testing using the false discovery rate (FDR) method described in the methods section. Of the ~12 million two-SNP interaction models tested with multiple linear regression, 706 were still significant after correcting for multiple testing. It is of note that our multiple testing correction is extremely conservative because our tests of interaction are not independent of each other. The deviation from the null hypothesis of no interaction shown in figure 1 suggests that there may be many more than 706 SNP-SNP interactions truly influencing gene expression that we are insufficiently powered to detect when applying our FDR correction. These 706 significant SNP-SNP interaction models influenced the expression of 79 unique probes, representative of 79 unique genes. 706 SNP-SNP interactions reduce to 79 genes because multiple SNP-SNP pairs are associated with the same gene. This redundancy is due to LD between SNPs across models, for example when SNP 1 of model 1 and SNP 1 of model 2 show strong correlation. However, there was relatively weak LD between the two SNPs participating within the interaction; i.e. SNP 1 and SNP 2 of model 1. The distribution of LD statistics (measured by r2) between the SNPs in each interacting pair is shown in Figure 2. The median r2 was 0.043, with a median distance between each pair of 108 kb. Taken together, this suggests that the majority of the most significant results are indeed epistatic effects between independent SNPs, not simple haplotype effects.
Fig. 2.
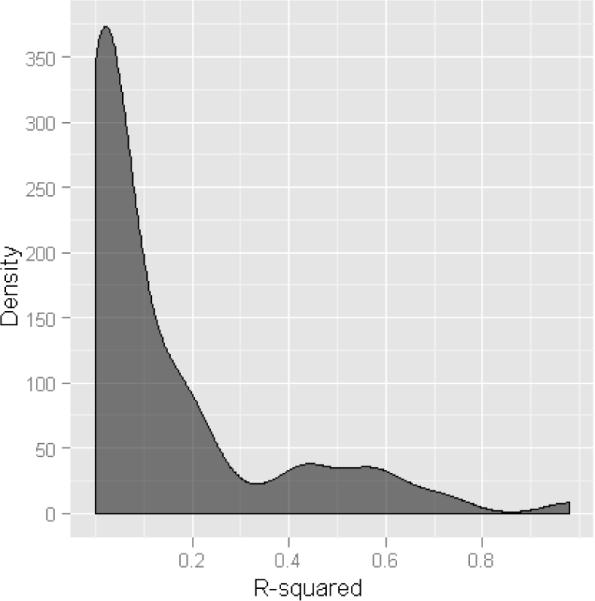
Density histogram showing distribution of linkage disequilibrium (LD) values (r2) between the most significant interacting SNP pair influencing expression of 79 genes after correcting for multiple testing. r2 was calculated using genotype data from the combined set of 210 HapMap samples.
Of the 706 interactions significant after FDR correction, we examined one interaction with the most significant model fit statistic for each of these 79 genes, referencing each regulated gene to the GWAS results catalog described in the methods section. The GWAS results catalog contains SNPs that have been previously associated to a human phenotype, and the associated gene reported by the original GWAS publication. We matched the significant cis-epistatic interactions to the GWAS results catalog in two ways: matching the 79 genes being regulated to the gene reported in the GWAS study, and matching SNPs participating in the 706 interactions to a SNP associated in a GWAS study. When matching by gene, we found that 20 of the 79 genes regulated by cis-epistasis have been previously reported in studies of approximately 20 human disease and morphological phenotypes (Table 1a). When matching by SNP, we found 10 additional cis-interactions where one of the specific SNPs has been associated to one or more disease or morphological phenotypes in humans (Table 1b). These data indicate that genes regulated by cis-epistasis are implicated in human phenotypes.
For the majority the genes in Table 1, examining single SNP effects on expression only resulted in a nominal level of statistical significance (Table 1, columns “eQTL[1/2] P-value”). Examining the cis-epistasis interaction between the two SNPs allowed us to achieve a much greater degree of statistical significance (Table 1, columns “INT P-value” and “Model P-value”). Furthermore, accounting for cis-epistasis allows us to explain a much larger proportion of the heritability (variance) in gene expression (Table 1, column “R2diff”, which is the difference in variance explained by the full model accounting for the interaction, “R2full”, and the reduced model with main effects only, “R2redu”).
3.2. Structural Characterization of Significant Two-SNP Interactions
3.2.1. Genomic Structure
Next we examined the genomic structural characteristics of the single most significant two-SNP epistatic interaction that impact the expression for each of these 79 genes. Specifically, we examined the location of the two eQTL SNPs relative to each other and relative to the transcription start site (TSS) and transcription end site (TES) of the regulated gene. Based on structural characteristics, we defined four distinct classes of regulatory epistatic interactions: upstream, where both eQTL SNPs lie upstream of the TSS of the gene; downstream, where both eQTL SNPs lie downstream of the TES; spanning, where one eQTL SNP is upstream of the TSS and one eQTL SNP is downstream of the TES; and intragenic, where at least one eQTL SNP lies within the genic region, and the other may be either upstream, downstream, or also in the genic region.
We observed 25 upstream interactions (32%), 18 downstream interactions (23%), 17 spanning interactions (21%), and 19 intragenic interactions (24%). Interestingly, all our significant results were evenly distributed among the four structural classes, as a z-test for population proportions revealed no significant difference from 25%. However, this test does not account for gene size or SNP density in the surrounding region. Small genes are less likely to harbor spanning or intragenic interactions, and perhaps the fact that we observe an even distribution of genomic structural classes is meaningful. Figure 3 shows that the four structural classes are distributed evenly among these most significant 79 cis-epistatic interactions. Figure 3 also reveals that the distribution of structural class does not correlate with gene size, organized vertically along the figure.
Fig. 3.
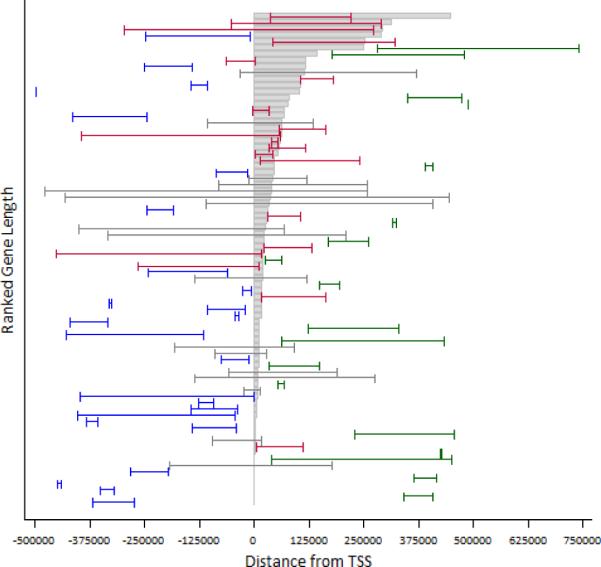
Transcribed regions of these 79 genes (gray boxes) are aligned by transcription start site, ordered by gene size. Epistatically interacting SNPs that influence the gene's expression are shown as connected hash marks, color coded by class: upstream (blue), downstream (green), spanning (gray) and intragenic (red). Analysis of the genomic structure of cis-epistatic interactions reveals that all four structural classes are evenly represented among the most significant cis-epistatic interactions, and that structural class does not correlate with gene size.
3.2.2. Structure of the Statistical Model
Statistical epistasis is classically defined as the deviation from additivity in a linear model [29]. We have shown that there are significant nonlinear effects impacting gene expression throughout the genome. Next we examined the structure of the statistical models of the most significant interactions impacting the 79 unique genes discussed above. Specifically, we examined the direction of the coefficients of both main effect terms and the interaction term in each statistical model.
We found that of these 79 significant cis-epistasis interactions, the main effect coefficients in 75 of these models were in the same direction. That is, if inheriting one copy of the minor allele of a single variant caused an increase in expression, the main effect of the other SNP also resulted in an increase in expression. Recall that we only tested for SNP-SNP interactions among eQTL SNPs that had an established main effect. Interestingly, of these 75 cis-epistasis models where both main effects were in the same direction, the statistically significant interaction term coefficient was in the opposite direction. That is, if the main effect of each variant alone caused an increase in expression by x units, inheriting both variants resulted in an expression level that is significantly lower than the expected 2x increase. Of the remaining four significant cis-epistasis interactions, the main effects were in opposing directions. For three of these four, the main effect coefficient of one SNP in the model approached zero after accounting for the interaction. This suggests a classical modifier effect, where one variant only exerts an effect in the presence of another. In all three of these models, the presence of the “modifier SNP” (β ≈ 0) results in a mitigation of the main effect of the other SNP.
The pattern of coefficients can be seen by examining β1, β2, and βint for the models presented in Table 1 (showing only models related to a human phenotype from a GWAS). These results indicate that the overwhelming majority of significant non-additive two-SNP interactions influencing gene expression represent epistatic genetic heterogeneity rather than multiplicative effects. We consider this in greater detail in the discussion section below.
We also investigated the possibility that aspects of the genomic structure of the model might impact the statistical nature of the interaction. However these analyses revealed no significant relationships between genomic structure characteristics (such as class or the physical distance between the two SNPs) to the variance explained (R2) or magnitude of the interaction coefficient.
4. Discussion
In this work we examined eQTL SNPs known to impact gene expression in humans for non-additive epistatic effects by combining transcriptome-wide expression data from HapMap lymphoblastoid cell lines with genome-wide SNP data from the same cell lines. Specifically, we analyzed over 12 million potential two-SNP interactions for cis-epistasis among SNPs known to regulate transcription of a nearby gene, and found that multiple independent eQTL SNPs may often interact to influence gene expression non-additively. After correcting for multiple testing, we found 706 highly significant cis-epistasis interactions that influence the expression of 79 unique genes.
We characterized the genomic and statistical structure of the most significant cis-epistasis model corresponding to each of these 79 genes. Here we discovered that in the vast majority of cis-epistasis interactions (1) the main effects are in the same direction, and (2) the interaction was in the opposite direction. While still considered a nonlinear epistatic interaction, the structure of this type of model is referred to as a heterogeneity model [30, 31] rather than a multiplicative model. While we observe primarily heterogeneity-type models, our particular approach using linear regression may be underpowered to detect models of other statistical structures. Genetic heterogeneity is a serious concern with large-scale genetic studies, and is often cited as a reason for the widespread lack of replication in GWAS studies [32, 33]. Because epistatic genetic heterogeneity may commonly impact regulation of gene expression, and since SNPs associated to complex human phenotypes often result in changes of gene expression [21], it follows that cis-epistatic genetic heterogeneity could exert a significant influence over complex human traits and should be investigated as such. Others have recently argued that epistatic genetic heterogeneity should be considered when analyzing genomic data for association to disease [34]. Despite the fact that statistical tools have been available for some time now to accomplish this [35, 36], analyses of genome-wide datasets accounting for the possibility of cis-epistasis is a task rarely undertaken. Accounting for genetic heterogeneity in gene expression may improve the replicability of existing personal genomics studies.
By matching cis-epistasis interactions to the GWAS results catalogs by SNP, we discovered that of the 79 significant cis-epistasis interactions, 10 contained one SNP previously associated to a human phenotype via GWAS studies. Nearly all of these associations fall short of “genome-wide” statistical significance [37] and thus would not be reported in the literature as a relevant gene for the phenotype. Furthermore, the statistical significance of each single SNP on the expression of a gene is weak. However, when we consider the joint effect of both SNPs involved in the cis-epistasis interaction, we see a dramatic improvement in the variance of gene expression explained. As such, we hypothesize that some of these reported associations from the GWAS catalog would show stronger associations to the phenotype if modeled with their cis-epistasis partner SNP. In light of the prevalence of cis-epistatic interactions, these examples provide motivation to re-examine existing datasets for cis-epistatic effects on human phenotypes. Our models provide a compelling set of specific regulatory hypotheses to examine in existing data.
Many new approaches have been recently used to examine epistasis in GWAS data [9–12]. All of these approaches focus on interactions among SNPs within genes related to a common biological mechanism, such as pathways, and structural or functional similarity. With these approaches, interaction models consist of SNPs from each of two distant genes – a trans-epistasis effect. In most cases, this precludes the possibility of capturing cis-epstasis effects. While trans-epistasis effects are likely to be important for complex disease etiology, we argue that cis-epistasis may be of equal or greater importance, and coupling cis- and trans-epistasis analysis methods may be more successful.
Furthermore, the collection of available tools for the analysis of multi-locus interactions in personal genomics studies is not likely to discover the cis-epstasis effects we describe here. Knowledge-based approaches generally test models of trans-epistasis (as discussed above). Sliding window-based haplotype association approaches typically use window sizes based on a fixed physical distance or number of SNPs [38]. These approaches would likely not discover cis-epstasis effects due to the variable and often large distances between the pairs of regulatory SNPs within the model (see Figure 3).
Moreover, any gene-based analysis approach that uses SNP data requires mapping SNPs to genes. This is exclusively done using either physical distance (base-pair proximity) or genetic distance (linkage disequilibrium). The genomic window generated using these approaches is typically conservative, including a small region upstream and downstream of the gene region. Others have shown in model organisms that regulatory elements exert effects from extremely long distances [39]. Likewise, the many of the single SNP eQTLs used in the examination of this study illustrate long range regulatory effects [15]. From our analysis, we provide additional evidence that SNPs can influence the regulation of a gene at great distances from the transcription start site, and existing SNP-to-gene mapping approaches should take this into account.
There are several possible molecular phenomena that may underlie these statistical observations. SNPs upstream or downstream of the gene may alter transcription factor binding sites or otherwise affect the efficiency of the transcriptional machinery. SNPs may also alter the binding of micro RNA molecules known to regulate gene transcripts. SNPs in untranslated regions may affect the stability of mRNA molecules. The impact of common variation on these processes is, however, still largely unknown.
We therefore suggest that the re-analysis of existing datasets and the development of new analysis approaches take into account the possibility that long range regulatory interactions could alter gene expression and thus influence human phenotypes. By accounting for more variance in gene expression (thus increasing statistical power), this will improve performance of analytical methods and potentially improve the replicability of GWAS findings. One basic approach would be to use the models we have generated as templates for the analysis of cis-epistasis in existing and future personal genomics studies. The 79 genes we identified after multiple testing correction suggest the most compelling cases of cis-epistasis. However, interaction models with less significant p-values may explain sufficient variance in a gene's expression to resolve an association with a phenotype.
One limitation of this study is that whole-transcriptome data was available for only 210 HapMap samples. However dense genome-wide SNP data is available for 1397 individuals in 11 diverse human sub-populations through the HapMap project [23], so if additional gene expression data were collected we could improve the statistical power of this analysis to detect cis-epistasis effects. Also, we only considered interactions among eQTL SNPs with a known regulatory effect (p < 0.05). A reanalysis of this data including all SNPs, (even those without a known regulatory effect) would be straightforward, perhaps revealing additional cis-epistasis effects; however this would cause a power loss from the increased burden of multiple testing correction.
In summary, we have shown that cis-epistasis is an important phenomenon regulating gene expression in humans. Using this information, we suggest ways in which the performance of existing and future analysis approaches can be improved, and how additional insights into human biology and disease pathogenesis could be gained from personal genomics studies.
Acknowledgments
Work partially supported by National Research Service Award F31-NS066638.
References
- 1.Tyler AL, Asselbergs FW, Williams SM, Moore JH. Bioessays. 2009;31:220. doi: 10.1002/bies.200800022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shao H, et al. Proc. Natl. Acad. Sci. U. S. A. 2008;105:19910. doi: 10.1073/pnas.0810388105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He X, Qian W, Wang Z, Li Y, Zhang J. Nat. Genet. 2010;42:272. doi: 10.1038/ng.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eichler EE, et al. Nat. Rev. Genet. 2010;11:446. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manolio TA, et al. Nature. 2009;461:747. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maher B. Nature. 2008;456:18. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 7.Aguilar PS, et al. Nat. Struct. Mol. Biol. 2010;17:901. doi: 10.1038/nsmb.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costanzo M, et al. Science. 2010;327:425. [Google Scholar]
- 9.Baranzini SE, et al. Hum. Mol. Genet. 2009;18:2078. doi: 10.1093/hmg/ddp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bush WS, Dudek SM, Ritchie MD. Pac Symp Biocomput. 2009;14:368. [PMC free article] [PubMed] [Google Scholar]
- 11.Peng G, et al. Eur. J Hum. Genet. 2010;18:111. doi: 10.1038/ejhg.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruano D, et al. Am. J Hum. Genet. 2010;86:113. doi: 10.1016/j.ajhg.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pickrell JK, et al. Nature. 2010;464:768. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stranger BE, et al. Science. 2007;315:848. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Veyrieras JB, et al. PLoS. Genet. 2008;4:e1000214. doi: 10.1371/journal.pgen.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ravasi T, et al. Cell. 2010;140:744. doi: 10.1016/j.cell.2010.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boj SF, Petrov D, Ferrer J. PLoS. Genet. 2010;6:e1000970. doi: 10.1371/journal.pgen.1000970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du W, Thanos D, Maniatis T. Cell. 1993;74:887. doi: 10.1016/0092-8674(93)90468-6. [DOI] [PubMed] [Google Scholar]
- 19.Gregersen JW, et al. Nature. 2006;443:574. doi: 10.1038/nature05133. [DOI] [PubMed] [Google Scholar]
- 20.Dixon AL, et al. Nat. Genet. 2007;39:1202. doi: 10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- 21.Gamazon ER, et al. Bioinformatics. 2010;26:259. doi: 10.1093/bioinformatics/btp644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mailman MD, et al. Nat. Genet. 2007;39:1181. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.International hapmap consortium Nature. 2007;449:851. [Google Scholar]
- 24.R Development Core Team R: A language and environment for statistical computing. ISBN 3900051070, URL http://www.R-project.org.
- 25.Storey JD, Taylor JE, Siegmund D. Journal of the Royal Statistical Society Series B-Statistical Methodology. 2004;66:187. [Google Scholar]
- 26.Purcell S, et al. Am. J. Hum. Genet. 2007;81:559. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hindorff LA, et al. Proc. Natl. Acad. Sci. U. S. A. 2009;106:9362. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson AD, O'Donnell CJ. BMC Med. Genet. 2009;10:6. doi: 10.1186/1471-2350-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fisher RA. Trans. R. Soc. Edinb. 1918;52:399. [Google Scholar]
- 30.Cordell HJ. Hum. Mol. Genet. 2002;11:2463. doi: 10.1093/hmg/11.20.2463. [DOI] [PubMed] [Google Scholar]
- 31.Neuman RJ, Rice JP. Genet. Epidemiol. 1992;9:347. doi: 10.1002/gepi.1370090506. [DOI] [PubMed] [Google Scholar]
- 32.McClellan J, King MC. Cell. 2010;141:210. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 33.Sillanpaa MJ, Auranen K. Ann. Hum. Genet. 2004;68:646. doi: 10.1046/j.1529-8817.2004.00122.x. [DOI] [PubMed] [Google Scholar]
- 34.Moore JH, Asselbergs FW, Williams SM. Bioinformatics. 2010;26:445. doi: 10.1093/bioinformatics/btp713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lunetta KL, Hayward LB, Segal J, Van EP. BMC Genet. 2004;5:32. doi: 10.1186/1471-2156-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thornton-Wells TA, Moore JH, Haines JL. Trends Genet. 2004;20:640. doi: 10.1016/j.tig.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 37.Pe'er I, Yelensky R, Altshuler D, Daly MJ. Genet. Epidemiol. 2008;32:381. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- 38.Lin S, Chakravarti A, Cutler DJ. Nat. Genet. 2004;36:1181. doi: 10.1038/ng1457. [DOI] [PubMed] [Google Scholar]
- 39.Chandler RL, Chandler KJ, McFarland KA, Mortlock DP. Mol. Cell Biol. 2007;27:2934. doi: 10.1128/MCB.01609-06. [DOI] [PMC free article] [PubMed] [Google Scholar]