

Abstract
An overview of studies on hydrogenative reductive aldol addition is presented. By simply hydrogenating enones in the presence of aldehydes at ambient temperature and pressure, aldol adducts are generated under neutral conditions in the absence of any stoichiometric byproducts. Using cationic rhodium complexes modified by tri(2-furyl)phosphine, highly syn-diastereoselective reductive aldol additions of vinyl ketones are achieved. Finally, using novel monodentate TADDOL-like phosphonite ligands, the first highly diastereo- and enantioselective reductive aldol couplings of vinyl ketones were devised. These studies, along with other works from our laboratory, demonstrate that organometallics arising transiently in the course of catalytic hydrogenation offer byproduct-free alternatives to preformed organometallic reagents employed in classical carbonyl addition processes.
Keywords: hydrogenation, rhodium, green chemistry, aldol, enantioselective catalysis
1 Introduction
The aldol reaction is a gateway to Nature’s vast library of polyketide natural products and a methodological cornerstone of synthetic organic chemistry. Although nearly 150 years have elapsed since the discovery of the aldol reaction,1 regio- and stereoselective methods for aldol addition have begun to emerge only within the last few decades.2 Beyond the considerable stereochemical challenges posed by aldol addition, modern aldolization protocols should satisfy the economic and aesthetic standards evoked by green chemistry, including the concepts of atom-economy,3 step-economy and the ‘ideal synthesis’.4 Hence, an especially significant milestone in aldol chemistry is the advent of metallic5 and organic6 catalysts for ‘direct’ asymmetric aldol addition of unmodified ketones. Such direct aldol additions are stereoselective, byproductfree and step-economic, as they depart from the use of chiral auxiliaries and preformed enol(ate) derivatives.
Withstanding these enormous advances, regioselective enolization in direct asymmetric aldol additions of nonsymmetric ketones remains a significant challenge. Although hydroxymethyl ketones regioselectively engage in direct asymmetric aldol additions to deliver glycolate aldol adducts,7 simple nonsymmetric ketones such as butan-2-one generally favor coupling at the less substituted enolizable position. For example, direct aldol couplings of butan-2-one catalyzed by l-proline8,9 or the heterobimetallic catalyst LaLi3-tris(binaphthoxide) (LLB) furnish linear aldol adducts.10 Such regiochemistry is incompatible with polypropionate construction. For this reason, direct methods for the stereoselective formation of the corresponding branched aldol adducts are highly desirable (Scheme 1).
Scheme 1.
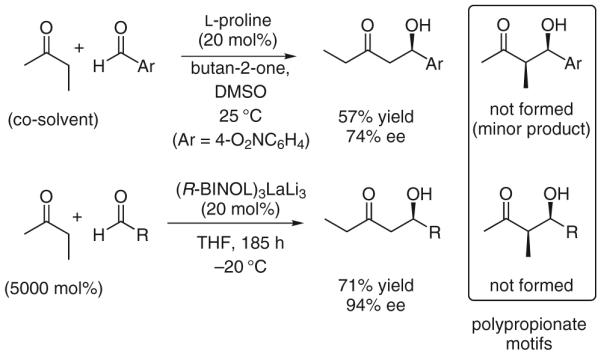
Regiochemistry in direct asymmetric aldol couplings of butan-2-one and the challenge of polypropionate construction
The use of enones as regiocontrol elements enables reductive generation of enolate isomers that cannot be formed selectively upon deprotonation of the parent carbonyl compounds. For example, although it is generally appreciated that regioselective deprotonation of nonsymmetric ketones possessing different degrees of substitution at the α-positions, for example methylcyclohexanone, may be achieved by exploiting kinetic or thermodynamic control,11 regioselective enolization of nonsymmetric ketones that possess identical degrees of α-substitution is often problematic. The deprotonation of cholesteran-3-one is a classic example. The Δ3-enolate is thermodynamically preferred and the Δ2-enolate cannot be formed exclusively via deprotonation under kinetically or thermodynamically controlled conditions. The balance is capricious: introduction of 7,8-unsaturation inverts the thermodynamic preference and now the Δ3-enolate cannot be formed selectively under kinetically or thermodynamically controlled conditions.12,13 As first reported by Stork,14 regiospecific enolate generation may be achieved via enone reduction. Specifically, dissolving metal reduction (Li/NH3) of conjugated enones was found to promote regiospecific enolate formation, enabling access to enolate isomers, and adducts thereof, that cannot be formed by way of base-mediated deprotonation (Scheme 2).
Scheme 2.

Enones as regiocontrol elements in the enolization of nonsymmetric ketones
Following Stork’s seminal work, metal-catalyzed reductive couplings of enones to aldehydes, the so-called ‘reductive aldol reactions’, were developed.15 To date, catalysts for reductive aldol coupling based on rhodium,16,17 cobalt,18 iridium,19 ruthenium,20 palladium,21 copper,22,23 nickel,24 and indium25,26 are known. Such transformations generally employ silanes or stannanes as the terminal reductant, mandating generation of stoichiometric byproducts. We were motivated by the prospect of developing completely byproduct-free regiospecific reductive aldol couplings using elemental hydrogen as terminal reductant. However, prior to our work, hydrogen-mediated carbon–carbon coupling had only been systematically explored in the context of alkene hydroformylation27 and the parent Fischer–Tropsch reaction.28 With the exception of two isolated reports,29 along with the infrequent detection of reductive coupling side-products in catalytic hydrogenation,30 the field of hydrogenative carbon–carbon bond formation was largely undeveloped. As organic molecules, by their very definition, contain carbon and hydrogen, we envisioned a broad new family of hydrogenative carbon–carbon couplings; processes wherein two or more unsaturated reactants combine upon exposure to gaseous hydrogen in the presence of a metallic catalyst to produce a single, more complex product. Considerable progress toward this end has been made (Scheme 3).31
Scheme 3.

Redefining the chemistry of carbonyl addition through catalytic hydrogenation: byproduct-free carbonyl addition in the absence of preformed organometallic reagent
In this review, we present an overview of our efforts to merge the fields of catalytic hydrogenation and aldol addition chemistry. Whereas related silane-mediated reductive aldol additions enable diastereo- and enantioselective coupling of α,β-unsaturated esters, the hydrogenative processes we report represent the first diastereo- and enantioselective reductive aldol couplings of vinyl ketones.
2 Intramolecular Hydrogenative Aldol Addition
Reductive aldol addition under hydrogenation conditions was first observed in the catalytic hydrogenation of enone-aldehydes 1a–g.17a A distinct dichotomy in the reactivity of neutral versus cationic rhodium catalysts was observed. For example, using the neutral complex Rh(PPh3)3Cl, hydrogenation of the enone-aldehyde 1a results in conventional conjugate reduction. However, rhodium salts that embody increased cationic character, such as Rh(cod)2OTf, provide a nearly equal proportion of the aldol product 2a and 1,4-reduction product. Further, when Rh(cod)2OTf is used in conjunction with substoichiometric quantities of a basic additive, potassium acetate, the aldol product 2a is formed as the major reaction product, with nearly complete suppression of the competitive 1,4-reduction manifold. Under these conditions, a range of enone-aldehydes engage in cycloreduction to form five- and six-membered ring products (Table 1).
Table 1.
Reductive Aldol Cyclization of Aldo-Enones under Hydrogenation Conditions
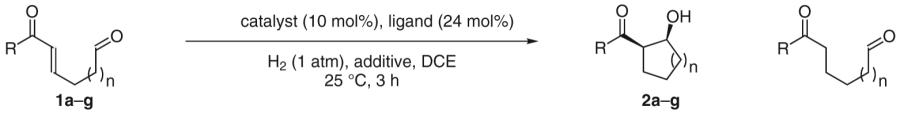 | |||||
|---|---|---|---|---|---|
| Substrate | Catalyst | Ligand | Additive (mol%) |
Yield of aldol (syn/anti) |
Yield of 1,4-reduction |
| 1a, n = 2, R = Ph | Rh(PPh3)3Cl | – | – | 2a, 1% (99:1) | 95% |
| 1a, n = 2, R = Ph | Rh(cod)2OTf | Ph3P | – | 2a, 21% (99:1) | 25% |
| 1a, n = 2, R = Ph | Rh(cod)2OTf | Ph3P | KOAc (30) | 2a, 59% (58:1) | 21% |
| 1a, n = 2, R = Ph | Rh(cod)2OTf | (4-F3CC6H4)3P | – | 2a, 57% (14:1) | 22% |
| 1a, n = 2, R = Ph | Rh(cod)2OTf | (4-F3CC6H4)3P | KOAc (30) | 2a, 89% (10:1) | 0.1% |
| 1b, n = 2, R = 4-MeOC6H4 | Rh(cod)2OTf | (4-F3CC6H4)3P | KOAc (30) | 2b, 74% (5:1) | 3% |
| 1c, n = 2, R = 2-naphthyl | Rh(cod)2OTf | (4-F3CC6H4)3P | KOAc (30) | 2c, 90% (10:1) | 1% |
| 1d, n = 2, R = 2-thienyl | Rh(cod)2OTf | (4-F3CC6H4)3P | KOAc (30) | 2d, 76% (19:1) | 2% |
| 1e, n = 2, R = 2-furyl | Rh(cod)2OTf | (4-F3CC6H4)3P | KOAc (30) | 2e, 70% (6:1) | 10% |
| 1f, n = 1, R = Ph | Rh(cod)2OTf | (4-F3CC6H4)3P | KOAc (30) | 2f, 71% (24:1) | 1% |
| 1g, n = 2, R = Me | Rh(cod)2OTf | (4-F3CC6H4)3P | KOAc (30) | 2g, 65% (1:5) | – |
Given the pronounced effect of basic additives, a mechanism was postulated where partitioning of dihydride and monohydride based catalytic cycles is achieved through the deprotonation of (hydrido)rhodium intermediates. The observance of syn-aldol adducts is consistent with the intervention of a Z-enolate and carbonyl addition through a closed Zimmerman–Traxler-type transition structure.32 Reexposure of the 1,4-reduction product to the reaction conditions does not result in aldolization, nor does reexposure of the aldol product to the reaction conditions result in retro-aldol addition. Finally, exposure of the substrate to standard reaction conditions in the absence of hydrogen does not provide products of Morita–Baylis–Hillman cyclization (Scheme 4).
Scheme 4.

Bifurcated catalytic mechanism accounting for the effect of basic additives on partitioning of conjugate reduction and aldol addition pathways
While mechanisms involving enone-aldehyde oxidative coupling cannot be excluded on the basis of available data, the aforementioned interpretation finds support in studies of olefin hydrogenation by Osborn and Schrock, which involve the use of cationic rhodium precatalysts.33 Basic additives were found to increase the rate of olefin isomerization with respect to the rate of olefin reduction. This observation was rationalized on the basis of a bifurcated catalytic mechanism that partitions dihydride and monohydride cycles by virtue of an acid–base equilibrium between a cationic rhodium(III) dihydride and a neutral rhodium(I) monohydride. Further support for the indicated equilibrium derives from the stoichiometric reaction of a cationic rhodium(I) complex with elemental hydrogen to furnish a neutral rhodium(I) monohydride, representing a formal heterolytic activation of elemental hydrogen (H2 + MX → MH + HX).34 Cationic rhodium hydrides, unlike their neutral counterparts, are especially acidic and, hence, are particularly prone to heterolytic hydrogen activation (Scheme 5).35
Scheme 5.
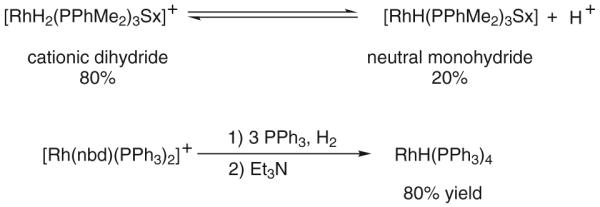
Equilibration of cationic rhodium dihydrides and neutral rhodium monohydrides and stoichiometric formation of the latter through base-mediated heterolytic hydrogen activation
Reductive aldol cyclization of keto-enones 3a–l is promoted under nearly identical hydrogenation conditions.17b Both five- and six-membered ring syn-aldol cyclization products 4a–l form as single diastereomers, accompanied by variable quantities of conjugate reduction product. While aldolization proceeds readily at ambient temperature, more reproducible ratios of aldol and 1,4-reduction product are observed at 80 °C. Products of retro-aldol addition are not observed upon resubmission of the aldol products to the reaction conditions. Aldol cyclization under an atmosphere of deuterium results in deuterium incorporation exclusively at the former enone β-position, but as a distribution of deuterated adducts, suggesting reversible enone hydrometallation in the case of ketone acceptors, where reversible carbonyl addition is anticipated (Scheme 6).
Scheme 6.

Reductive aldol cyclization of keto-enones via catalytic hydrogenation; yield of conjugate reduction product is indicated in parentheses
As suggested by the preceding results, ketones are only marginally less electrophilic than required to fully suppress competitive 1,4-reduction manifolds. 1,3-Diones 5a–f are more electrophilic due to inductive effects and dipole-dipole interactions. Exposure of enone-diones 5a–f to standard conditions for hydrogenative aldol cyclization at ambient temperature and pressure provides the corresponding aldol cyclization products 6a–f as single diastereomers. Only when forming a strained cis-decalone 6f is competitive 1,4-reduction observed (Scheme 7).17b
Scheme 7.

Reductive aldol cyclization of enone-diones via catalytic hydrogenation; yield of conjugate reduction product is indicated in parentheses
A particularly challenging variant of the aldol reaction involves the addition of aldehyde enolates to ketones. A single stoichiometric variant of this transformation is known.36 As aldolization is driven thermodynamically by chelation, intramolecular addition to furnish a robust transition-metal aldolate may serve to bias the enolate–aldolate equilibria toward the latter.37,38 Indeed, upon exposure to standard carbon–carbon bond-forming hydrogenation conditions, keto-enals 7a–d reductively cyclize to deliver the corresponding cycloaldol products 8a–d along withvariable quantities of 1,4-reduction product (Scheme 8).17c
Scheme 8.

Reductive aldol cyclization of enal-ketones via catalytic hydrogenation; yield of conjugate reduction product is indicated in parentheses
3 Intermolecular Hydrogenative Aldol Addition
For hydrogenative aldol addition chemistry to serve as a useful tool for polypropionate construction, stereoselective intermolecular couplings should be developed. Under our initially disclosed hydrogenation conditions employing a catalyst derived from Rh(cod)2OTf and triphenylphosphine, intermolecular reductive aldol coupling of vinyl ketones provides good yields of aldol adducts, but as mixtures of diastereomers.17a In subsequent work, it was found that π-acidic ligands such as tri(2-furyl)phosphine39 promote exceptional syn-diastereoselectivity.17e As for the cyclization processes, intervention of a Z-(O)-enolate and aldolization through a Zimmerman–Traxler-type transition structure is postulated. Tri(2-furyl)phosphine may increase diastereoselectivity by increasing Lewis acidity at the rhodium center, which should tighten the chair-like transition structure and destabilize competitive boat-like transition structures. Enhanced Lewis acidity at rhodium also should mitigate reversible aldol addition, which would erode kinetic syn-diastereoselectivity. The observed levels of syn-diastereoselectivity require kinetic control at the stages of both enolization and aldolization.2c The kinetic pathway for Z-selective enolization likely involves internal hydride delivery to the enone s-cis conformer through a six-centered transition structure.40
As demonstrated by the coupling of methyl vinyl ketone (MVK) and ethyl vinyl ketone (EVK) to aldehydes 9a–d,17e a remarkable feature of the intermolecular hydrogenative aldol addition is that competitive reduction of ‘hydrogen-labile’ functional groups (alkynes, alkenes, benzylic ethers and nitroarenes) is not observed. Divinyl ketones, such as crotyl vinyl ketone (CVK), react chemoselectively with aldehydes 9a–c and 9e at the less substituted vinyl residue to furnish adducts 12a–c and 12e without over-reduction.17f Because the hydrogenative coupling occurs under essentially neutral conditions, sensitive N-Boc-α-aminoaldehydes 9f and 9g are subject to reductive aldol addition without racemization. In the relatively low dielectric medium provided by dichloromethane, an intramolecular hydrogen-bond directs diastereofacial selectivity to provide aldol adducts 13f, 13g, 14f and 14g that embody exceptional levels of syn-aldol diastereoselectivity and anti-Felkin–Ahn control (Scheme 9).17g
Scheme 9.

syn-Diastereoselective hydrogenative aldol coupling catalyzed by cationic rhodium catalysts ligated by tri(2-furyl)phosphine
To probe the mechanism of the diastereoselective reductive coupling, methyl vinyl ketone and p-nitrobenzaldehyde (9a) were coupled under an atmosphere of deuterium. The aldol adduct deuterio-10a incorporates a single deuterium atom at the former enone β-position.17e This result is consistent with a mechanism involving irreversible enone hydrometallation. However, enone-aldehyde oxidative coupling followed by hydrogenolytic cleavage of the resulting oxarhodacycle cannot be excluded. Deuterium incorporation at the α-carbon is not observed, demonstrating that Morita–Baylis–Hillman pathways are not operative (Scheme 10).
Scheme 10.
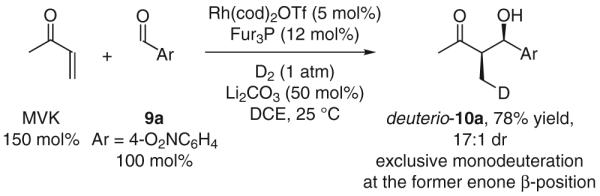
Rhodium-catalyzed hydrogenative aldol coupling under an atmosphere of deuterium
Diastereo- and enantioselective reductive aldol couplings of vinyl ketones, such as methyl vinyl ketone, would enable access to branched aldol adducts, providing a regiochemical complement to direct organocatalyzed and metal-catalyzed aldol couplings of nonsymmetric ketones, such as butan-2-one, which furnish linear aldol adducts.8-10 Enantioselective variants of the hydrogenative aldol couplings are especially challenging for the following reasons: (a) only trace quantities of product are obtained using chelating phosphine ligands, (b) π-acidic ligands such as tri(2-furyl)phosphine are required to enforce high levels of diastereoselection,6 yet (c) commercially available π-acidic chiral monodentate ligands, for example, BINOL-derived phosphites and phosphoramidites, are presumably too π-acidic, and provide only trace quantities of product. Hence, the design, preparation and assay of novel chiral monodentate phosphorus-based ligands were undertaken.
Over several years numerous ligands were assayed, yet those displaying promising levels of asymmetric induction were not amenable to facile and systematic structural variation. Hence, a versatile template enabling well-defined structure–selectivity trends was sought. TADDOL-like phosphonites41 present three structural elements that may be independently optimized: (a) the P-aryl moiety, (b) the ketal substructure, and (c) the groups appended to the tertiary carbinol center. Gratifyingly, by combining the optimal diethyl ketal, dimethylcarbinol, and P-2-benzothienyl substructures, the highly effective TADDOL-like phosphonite ligand AP-I was identified (Scheme 11).17h
Scheme 11.

Optimization of TADDOL-like phosphonite ligand for enantioselective hydrogenative aldol coupling. a Reaction was conducted at 0 °C.
Using the optimal TADDOL-like phosphonite ligand AP-I, the enantioselective hydrogenative aldol coupling of methyl vinyl ketone and ethyl vinyl ketone to aldehydes 9b, 9c, 9h and 9i was explored. Optimal efficiencies and selectivities were observed using the preformed complex [Rh(cod)(AP-I)2]OTf. Uniformly high levels of relative and absolute stereocontrol were observed in additions to α- and β-heteroatom substituted aldehydes 9b and 9c, and α-(hetero)aryl substituted aldehydes 9h and 9i. Diastereo- and enantioselectivities were determined by chiral stationary phase HPLC analyses made in comparison to racemic diastereomeric mixtures. These first-generation catalysts for intermolecular hydrogenative aldol coupling are restricted to vinyl ketones, as acrylates provide conventional hydrogenation. Additionally, slightly activated aldehydes are required, as simple aliphatic aldehydes provide diminished yields of adduct (Scheme 12).
Scheme 12.

syn-Diastereoselective and enantioselective hydrogen-mediated aldol coupling employing novel TADDOL-like phosphonite ligands
Single crystal X-ray diffraction analysis of [Rh(cod)(L)2]OTf (L = the acetonide of AP-I) reveals a C2-symmetric arrangement (Figure 1). A similar metalligand arrangement is likely evident in the stereo-determining event, as the reactants should occupy adjacent co-ordination sites. Based on this hypothesis, second-generation ligands AP-II and AP-IV were designed. For ligands AP-II and AP-IV, the (benzo)thiophene moiety is substituted such that the purported chiral pocket is deepened, potentially conferring heightened levels of enantioselection. The veracity of this analysis is supported by results obtained for the enantioselective hydrogenative aldol coupling of methyl vinyl ketone to aldehyde 9j. Both AP-II and AP-IV are found to induce higher levels of optical enrichment in the formation of aldol product 10j, whereas AP-III, which projects the methyl residue into an inactive volume of space, displays selectivities comparable to those obtained with AP-I (Scheme 13).
Figure 1.
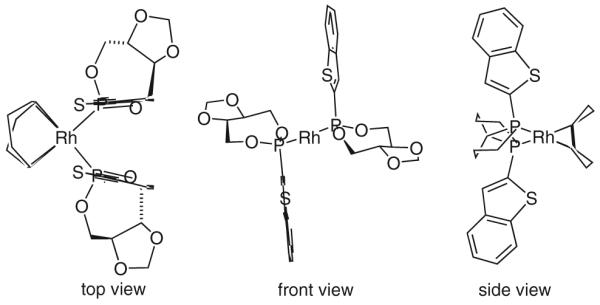
Structure of [Rh(cod)(L)2]OTf (L = the acetonide analogue of AP-I) determined by X-ray diffraction reveals C2-symmetric arrangement. The figure graphics are depictions of crystallographic data imported into ChemDraw Ultra 9.0. For clarity, the following substructures were omitted. Top view: methyl groups and triflate ion. Front view: methyl groups, triflate ion and 1,5-cyclooctadiene. Side view: methyl groups, triflate ion, dioxolane rings and phosphonite oxygen atoms.
Scheme 13.

Enantioselective aldol coupling of methyl vinyl ketone to aldehyde 9j using ligands AP-I, AP-II, AP-III and AP-IV
4 Conclusion and Outlook
Inspired by the enormous socioeconomic impact of the prototypical carbon–carbon bond forming hydrogenations, the Fischer–Tropsch reaction and alkene hydroformylation, we seek to develop diverse hydrogenative carbonyl and imine additions based upon catalytic hydrogenation31,17,42 and catalytic transfer hydrogenation.43 These studies already evoke the question of whether processes that have traditionally employed one or more stoichiometric metallic reagents can now be conducted catalytically under hydrogenative or transfer hydrogenative conditions. As demonstrated in this account, the combination of catalytic hydrogenation and aldol chemistry has brought forth a powerful new catalytic reductive variant of the aldol reaction that circumvents the use of preformed metallo-enolates – an aldol coupling that is regiospecific, diastereo- and enantioselective and byproduct-free. Related hydrogenative enolate couplings, including reductive Mannich additions, are in progress.
Acknowledgment
Acknowledgment is made to Merck, Johnson & Johnson, the Robert A. Welch Foundation, the ACS-GCI Pharmaceutical Roundtable, and the NIH-NIGMS (RO1-GM069445) for partial support of this research. Dr. Oliver Briel of Umicore is thanked for the generous donation of rhodium salts.
Biography

Soo Bong Han was born in Seoul, Korea in 1975. He received his B.S. degree in chemistry from Sogang University in 2002 and his M.S. degree from KAIST under supervision of Professor Suk Bok Chang in 2004. He then worked as a senior researcher for LG Life Science to develop medicine for Alzheimer’s disease. In 2005, he began his Ph.D. studies under Professor Michael J. Krische at University of Texas at Austin, where he contributed to the development of a diastereo- and enantioselective reductive aldol reaction mediated by hydrogen.

Abbas Hassan was born in Mardan, Pakistan in 1980. He obtained his B.S. degree in chemistry from University of Peshawar in 2001, followed by an M.S. degree in chemistry from the same university in 2004. During his M.S. he worked on the synthesis of azaheterocyclic compounds with Professor Muhammad Arfan. He then moved to the HEJ Research Institute of Chemistry, University of Karachi to join the group of Dr. Khalid M. Khan where he worked on the synthesis of medicinally important nitrogen- and sulfur-containing heterocyclic compounds. He now works in the laboratory of Professor Michael J. Krische at University of Texas at Austin, where he has contributed to the development of a novel family of TADDOL-like phosphonite ligands for enantioselective hydrogenative aldol coupling.

Professor Michael J. Krische obtained a B.S. degree in chemistry from the University of California at Berkeley, where he performed research under the tutelage of Professor Henry Rapoport. After one year study abroad as a Fulbright Fellow, he initiated graduate research at Stanford University under the mentorship of Professor Barry Trost as a Veatch Graduate Fellow. Following his Ph.D., he worked under Jean-Marie Lehn at the Université Louis Pasteur as an NIH Post-Doctoral Fellow. In Fall 1999, Professor Krische was appointed Assistant Professor of Chemistry at the University of Texas at Austin. He was promoted directly to Full Professor in Fall 2004 and in Fall 2007 was granted the Robert A. Welch Chair in Science. His research represents the first systematic efforts to exploit catalytic hydrogenation in carbon–carbon couplings beyond hydroformylation. His research group has shown that diverse π-unsaturated reactants couple to carbonyl compounds and imines under hydrogenation conditions, providing byproductfree alternatives to stoichiometrically preformed organometallics in a range of classical C=X (X = O, NR) addition processes. This work has been recognized by the following awards: Novartis Chemistry Lectureship (2008), Presidential Green Chemistry Award (2007), Dowpharma Prize (2007), ACS Elias J. Corey Award (2007), Solvias Ligand Prize (2006), Society of Synthetic Chemistry Japan Lectureship (2005), Johnson & Johnson Focused Giving Award (2005), Dreyfus Teacher Scholar Award (2003), Alfred P. Sloan Research Fellowship (2003), Cottrell Scholar Award (2002), Frasch Foundation Award in Chemistry (2002), Lilly Grantee Award (2002), National Science Foundation-CAREER Award (2000).
References
- (1).Though largely attributed to Wurtz, the aldol reaction was reported first by Borodin: von Richter V. Ber. Dtsch. Chem. Ges. 1869;2:552. Borodin’s earliest results are cited in this article. Wurtz A. Bull. Soc. Chim. Fr. 1872;17:436. Borodin A. Ber. Dtsch. Chem. Ges. 1873;6:982. Kane R. Ann. Phys. Chem., Ser. 2. 1838;44:475.
- (2).For selected reviews on stereoselective aldol additions, see: Heathcock CH. Science. 1981;214:395. doi: 10.1126/science.214.4519.395. Heathcock CH. In: Asymmetric Reactions and Processes in Chemistry, ACS Symposium Series. Eliel EL, Otsuka S, editors. Vol. 185. American Chemical Society; Washington DC: 1982. p. 55. Evans DA, Nelson JV, Taber TR. Top. Stereochem. 1982;13:1. Machajewski TD, Wong C-H. Angew. Chem. Int. Ed. 2000;39:1352. doi: 10.1002/(sici)1521-3773(20000417)39:8<1352::aid-anie1352>3.0.co;2-j. Palomo C, Oiarbide M, García JM. Chem. Soc. Rev. 2004;33:65. doi: 10.1039/b202901d.
- (3).For reviews, see: Trost BM. Science. 1991;254:1471. doi: 10.1126/science.1962206. Trost BM. Angew. Chem., Int. Ed. Engl. 1995;34:259.
- (4).(a) Wender PA, Miller BL. Org. Synth. Theor. Appl. 1993;2:27. [Google Scholar]; (b) Wender PA, Handy S, Wright DL. Chem. Ind. 1997:767. [Google Scholar]
- (5).For recent reviews on the use of organic catalysts for direct enantioselective aldol addition, see: Shibasaki M, Matsunaga S, Kumagai N. In: Modern Aldol Reactions. Mahrwald R, editor. Vol. 2. Wiley-VCH; Weinheim: 2004. p. 197.
- (6).For a recent review on the use of metallic catalysts for direct enantioselective aldol additions, see: List B. In: Modern Aldol Reactions. Mahrwald R, editor. Vol. 1. Wiley-VCH; Weinheim: 2004. p. 161. Notz W, Tanaka F, Barbas CF., III. Acc. Chem. Res. 2004;37:580. doi: 10.1021/ar0300468.
- (7).(a) Notz W, List B. J. Am. Chem. Soc. 2000;122:7386. [Google Scholar]; (b) Yoshikawa N, Kumagai N, Matsunaga S, Moll G, Ohshima T, Suzuki T, Shibasaki M. J. Am. Chem. Soc. 2001;123:2466. doi: 10.1021/ja015580u. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Ito H, Silcoff ER. J. Am. Chem. Soc. 2001;123:3367. doi: 10.1021/ja003871h. [DOI] [PubMed] [Google Scholar]
- (8).Sakthivel K, Notz W, Bui T, Barbas CF., III J. Am. Chem. Soc. 2001;123:5260. doi: 10.1021/ja010037z. [DOI] [PubMed] [Google Scholar]
- (9).Tang Z, Yang Z-H, Chen X-H, Cun L-F, Mi A-Q, Jiang Y-Z, Gong L-Z. J. Am. Chem. Soc. 2005;127:9285. doi: 10.1021/ja0510156. Luo S, Lu H, Li J, Zhang L, Cheng J-P. J. Am. Chem. Soc. 2007;129:3074. doi: 10.1021/ja069372j.
- (10).Yoshikawa N, Yamada YMA, Das J, Sasai H, Shibasaki M. J. Am. Chem. Soc. 1999;121:4168. [Google Scholar]
- (11).(a) House HO, Czuba LJ, Gall M, Olmstead HD. J. Org. Chem. 1969;34:2324. [Google Scholar]; (b) Kraft ME, Holton RA. Tetrahedron Lett. 1983;24:1345. [Google Scholar]
- (12).For a review, see: Velluz L, Valls J, Nomine G. Angew. Chem. Int. Ed. 1965;4:181.
- (13).(a) Corey EJ, Sneen RA. J. Am. Chem. Soc. 1955;77:2505. [Google Scholar]; (b) Berkoz B, Chavez EP, Djerassi C. J. Chem. Soc. 1962:1323. [Google Scholar]; (c) Mazur Y, Sondheimer F. J. Am. Chem. Soc. 1958;80:6296. [Google Scholar]
- (14).(a) Stork G, Rosen P, Goldman NL. J. Am. Chem. Soc. 1961;83:2965. [Google Scholar]; (b) Stork G, Rosen P, Goldman N, Coombs RV, Tsuji J. J. Am. Chem. Soc. 1965;87:275. [Google Scholar]
- (15).For recent reviews on the reductive aldol reaction, see: Huddleston RR, Krische MJ. Synlett. 2003:12. Nishiyama H, Shiomi T. Top. Curr. Chem. 2007;279:105. Garner S, Han SB, Krische MJ. In: Modern Reductions. Andersson P, Munslow I, editors. Wiley-VCH; Weinheim: 2008. pp. 387–408.
- (16).For rhodium-catalyzed reductive aldol reactions mediated by silane, see: Revis A, Hilty TK. Tetrahedron Lett. 1987;28:4809. Matsuda I, Takahashi K, Sato S. Tetrahedron Lett. 1990;31:5331. Taylor SJ, Morken JP. J. Am. Chem. Soc. 1999;121:12202. Taylor SJ, Duffey MO, Morken JP. J. Am. Chem. Soc. 2000;122:4528. Zhao C-X, Bass J, Morken JP. Org. Lett. 2001;3:2839. doi: 10.1021/ol016279l. Emiabata-Smith D, McKillop A, Mills C, Motherwell WB, Whitehead AJ. Synlett. 2001:1302. Freiría M, Whitehead AJ, Tocher DA, Motherwell WB. Tetrahedron. 2004;60:2673. Nishiyama H, Shiomi T, Tsuchiya Y, Matsuda I. J. Am. Chem. Soc. 2005;127:6972. doi: 10.1021/ja050698m. Willis MC, Woodward RL. J. Am. Chem. Soc. 2005;127:18012. doi: 10.1021/ja056130v. Fuller NO, Morken JP. Synlett. 2005:1459. Ito JI, Shiomi T, Nishiyama H. Adv. Synth. Catal. 2006;348:1235. Shiomi T, Ito J-I, Yamamoto Y, Nishiyama H. Eur. J. Org. Chem. 2006:5594. Shiomi T, Nishiyama H. Org. Lett. 2007;9:1651. doi: 10.1021/ol070251d.
- (17).For rhodium-catalyzed reductive aldol reactions mediated by hydrogen, see: Jang HY, Huddleston RR, Krische MJ. J. Am. Chem. Soc. 2002;124:15156. doi: 10.1021/ja021163l. Huddleston RR, Krische MJ. Org. Lett. 2003;5:1143. doi: 10.1021/ol0300219. Koech PK, Krische MJ. Org. Lett. 2004;6:691. doi: 10.1021/ol030136c. Marriner GA, Garner SA, Jang HY, Krische MJ. J. Org. Chem. 2004;69:1380. doi: 10.1021/jo030310a. Jung CK, Garner SA, Krische MJ. Org. Lett. 2006;8:519. doi: 10.1021/ol052859x. Han SB, Krische MJ. Org. Lett. 2006;8:5657. doi: 10.1021/ol0624023. Jung CK, Krische MJ. J. Am. Chem. Soc. 2006;128:17051. doi: 10.1021/ja066198q. Bee C, Han SB, Hassan A, Iida H, Krische MJ. J. Am. Chem. Soc. 2008;130:2747. doi: 10.1021/ja710862u.
- (18).For cobalt-catalyzed reductive aldol reactions, see: Isayama S, Mukaiyama T. Chem. Lett. 1989:2005. Baik TG, Luis AL, Wang LC, Krische MJ. J. Am. Chem. Soc. 2001;123:5112. doi: 10.1021/ja0040971. Wang LC, Jang H-Y, Roh Y, Lynch V, Schultz AJ, Wang X, Krische MJ. J. Am. Chem. Soc. 2002;124:9448. doi: 10.1021/ja020223k. Lam HW, Joensuu PM, Murray GJ, Fordyce EAF, Prieto O, Luebbers T. Org. Lett. 2006;8:3729. doi: 10.1021/ol061329d. Lumby RJR, Joensuu PM, Lam HW. Org. Lett. 2007;9:4367. doi: 10.1021/ol701980e.
- (19).For iridium-catalyzed reductive aldol reaction, see: Zhao CX, Duffey MO, Taylor SJ, Morken JP. Org. Lett. 2001;3:1829. doi: 10.1021/ol015859f.
- (20).For ruthenium-catalyzed reductive aldol reaction, see: Doi T, Fukuyama T, Minamino S, Ryu I. Synlett. 2006:3013.
- (21).For palladium-catalyzed reductive aldol reaction, see: Kiyooka SI, Shimizu A, Torii S. Tetrahedron Lett. 1998;39:5237.
- (22).For copper-promoted reductive aldol reaction, see: Chiu P, Chen B, Cheng KF. Tetrahedron Lett. 1998;39:9229. Chiu P. Synthesis. 2004:2210. For copper-promoted reductive intramolecular Henry reaction, see: Chung WK, Chiu P. Synlett. 2005:55. For copper-promoted and -catalyzed reductive cyclizations of α,β-acetylenic ketones tethered to ketones, see: Chiu P, Leung SK. Chem. Commun. 2004:2308. doi: 10.1039/b407842j.
- (23).For copper-catalyzed reductive aldol reaction, see: Ooi T, Doda K, Sakai D, Maruoka K. Tetrahedron Lett. 1999;40:2133. Lam HW, Joensuu PMA. Org. Lett. 2005;7:4225. doi: 10.1021/ol051649h. Lam HW, Murray GJ, Firth JD. Org. Lett. 2005;7:5743. doi: 10.1021/ol052599j. Deschamp J, Chuzel O, Hannedouche J, Riant O. Angew. Chem. Int. Ed. 2006;45:1292. doi: 10.1002/anie.200503791. Chuzel O, Deschamp J, Chauster C, Riant O. Org. Lett. 2006;8:5943. doi: 10.1021/ol062398v. Zhao D, Oisaki K, Kanai M, Shibasaki M. Tetrahedron Lett. 2006;47:1403. Zhao D, Oisaki K, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2006;128:14440. doi: 10.1021/ja0652565. Welle A, Diez-Gonzalez S, Tinant B, Nolan SP, Riant O. Org. Lett. 2006;8:6059. doi: 10.1021/ol062495o.
- (24).For nickel-catalyzed reductive aldol reaction, see: Chrovian CC, Montgomery J. Org. Lett. 2007;9:537. doi: 10.1021/ol063028+. Joensuu PM, Murray GJ, Fardyce EAF, Luebbers T, Lam HW. J. Am. Chem. Soc. 2008;130:7328. doi: 10.1021/ja0775624.
- (25).For a reductive aldol coupling employing stoichiometric quantities of indium reagent, see: Inoue K, Ishida T, Shibata I, Baba A. Adv. Synth. Catal. 2002;344:283.
- (26).For indium-catalyzed reductive aldol reaction, see: Shibata I, Kato H, Ishida T, Yasuda M, Baba A. Angew. Chem. Int. Ed. 2004;43:711. doi: 10.1002/anie.200352738. Miura K, Yamada Y, Tomita M, Hosomi A. Synlett. 2004:1985.
- (27).Roelen O. (Chemische Verwertungsgesellschaft mbH, Oberhausen) German Patent DE 849548, 1938. Chem. Abstr. 1944;38:5501.
- (28).(a) Fischer F, Tropsch H. Brennst.–Chem. 1923;4:276. [Google Scholar]; (b) Fischer F, Tropsch H. Chem. Ber. 1923;56:2428. [Google Scholar]
- (29).(a) Molander GA, Hoberg JO. J. Am. Chem. Soc. 1992;114:3123. [Google Scholar]; (b) Kokube K, Miura M, Nomura M. Organometallics. 1995;14:4521. [Google Scholar]
- (30).Side products of reductive carbon–carbon bond formation have been observed in catalytic hydrogenation on rare occasion: Moyes RB, Walker DW, Wells PB, Whan DA, Irvine EA. In: Catalysis and Surface Characterization (Special Publication) Dines TJ, Rochester CH, Thomson J, editors. Vol. 114. Royal Society of Chemistry; London: 1992. p. 207. Bianchini C, Meli A, Peruzzini M, Vizzi F, Zanobini F, Frediani P. Organometallics. 1989;8:2080.
- (31).For recent reviews on hydrogen-mediated C–C coupling, see: Ngai M-Y, Krische M. J. Chim. Oggi/Chemistry Today. 2006;24(4):12. chiral technologies supplement. Iida H, Krische M. J. Top. Curr. Chem. 2007;279:77. Ngai M-Y, Kong J-R, Krische MJ. J. Org. Chem. 2007;72:1063. doi: 10.1021/jo061895m. Skucas E, Ngai M-Y, Komanduri V, Krische M. J. Acc. Chem. Res. 2007;40:1394. doi: 10.1021/ar7001123.
- (32).Zimmerman HE, Traxler MD. J. Am. Chem. Soc. 1957;79:1920. [Google Scholar]
- (33).Monohydride catalytic cycles initiated via deprotonation of cationic rhodium dihydrides have been postulated: Schrock RR, Osborn JA. J. Am. Chem. Soc. 1976;98:2134. Schrock RR, Osborn JA. J. Am. Chem. Soc. 1976;98:2143. Schrock RR, Osborn JA. J. Am. Chem. Soc. 1976;98:4450.
- (34).For reviews on the heterolytic activation of elemental hydrogen, see: Brothers PJ. Prog. Inorg. Chem. 1981;28:1. Jeske G, Lauke H, Mauermann H, Schumann H, Marks TJ. J. Am. Chem. Soc. 1985;107:8111.
- (35).For a review of the acidity of metal hydrides, see: Kristjansdottir SS, Norton JR. In: Transition Metal Hydrides. Dedieu A, editor. VCH; Weinheim: 1992. p. 309.
- (36).Yachi K, Shinokubo H, Oshima K. J. Am. Chem. Soc. 1999;121:9465. [Google Scholar]
- (37).Arnett EM, Fisher FJ, Nichols MA, Ribeiro AA. J. Am. Chem. Soc. 1989;111:748. [Google Scholar]
- (38).The failure of tris(dialkylamino)sulfonium enolates to react with aldehydes is attributed to unfavorable enolate–aldolate equilibria: Noyori R, Sakata J, Nishizawa M. J. Am. Chem. Soc. 1980;102:1223. Noyori R, Nishida I, Sakata J. J. Am. Chem. Soc. 1981;103:2106. Noyori R, Nishida I, Sakata J. J. Am. Chem. Soc. 1983;105:1598.
- (39).For tri(2-furyl)phosphine and triphenylarsine effects in metal-catalyzed reactions, see: Farina V, Krishnan B. J. Am. Chem. Soc. 1991;113:9585. Farina V. Pure Appl. Chem. 1996;68:73. Anderson NG, Keay BA. Chem. Rev. 2001;101:997. doi: 10.1021/cr000024o.
- (40).Consistent with internal hydride delivery to the enone s-cis conformer through a six-centered transition structure, enones constrained in the s-trans configuration, such as cyclohexenone, do not participate in hydrogenative reductive aldol coupling.
- (41).For TADDOL-derived phosphonites, see: Seebach D, Hayakawa M, Sakaki J-I, Schweizer WB. Tetrahedron. 1993;49:1711. Sakaki J-I, Schweizer WB, Seebach D. Helv. Chim. Acta. 1993;76:2654. Haag D, Runsink J, Scharf H-D. Organometallics. 1998;17:398.
- (42).For recent examples of hydrogenative C–C couplings developed in our laboratory, see: Kong J-R, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2006;128:718. doi: 10.1021/ja056474l. Skucas E, Kong J-R, Krische MJ. J. Am. Chem. Soc. 2007;129:7242. doi: 10.1021/ja0715896. Barchuk A, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2007;129:8432. doi: 10.1021/ja073018j. Barchuk A, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2007;129:12644. doi: 10.1021/ja075438e. Skucas E, Bower JF, Krische MJ. J. Am. Chem. Soc. 2007;129:12678. doi: 10.1021/ja075971u.
- (43).For recent examples of transfer hydrogenative C–C couplings developed in our laboratory, see: Bower JF, Skucas E, Patman RL, Krische MJ. J. Am. Chem. Soc. 2007;129:15134. doi: 10.1021/ja077389b. Bower JF, Patman RL, Krische MJ. Org. Lett. 2008;10:1033. doi: 10.1021/ol800159w. Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:6338. doi: 10.1021/ja801213x. Kim IS, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2008;130:6340. doi: 10.1021/ja802001b.