

Abstract
For years, a vast majority of clinical trial industry has followed the tenet of 100% source data verification (SDV). This has been driven partly by the overcautious approach to linking quality of data to the extent of monitoring and SDV and partly by being on the safer side of regulations. The regulations however, do not state any upper or lower limits of SDV. What it expects from researchers and the sponsors is methodologies which ensure data quality. How the industry does it is open to innovation and application of statistical methods, targeted and remote monitoring, real time reporting, adaptive monitoring schedules, etc. In short, hybrid approaches to monitoring. Coupled with concepts of optimum monitoring and SDV at site and off-site monitoring techniques, it should be possible to save time required to conduct SDV leading to more available time for other productive activities. Organizations stand to gain directly or indirectly from such savings, whether by diverting the funds back to the R&D pipeline; investing more in technology infrastructure to support large trials; or simply increasing sample size of trials. Whether it also affects the work-life balance of monitors who may then need to travel with a less hectic schedule for the same level of quality and productivity can be predicted only when there is more evidence from field.
Keywords: 100% source data verification, cost savings, hybrid monitoring, on-site monitoring, statistical methods and adaptive monitoring, targeted and remote monitoring techniques
INTRODUCTION
Monitoring has always been considered an important activity and one which always lends itself to the interpretation of “more the better.”
Let us have a look at the ICH E6 definition of Monitoring:[1]
“The act of overseeing the progress of a clinical trial, and of ensuring that it is conducted, recorded, and reported in accordance with the protocol, Standard Operating Procedures (SOPs), Good Clinical Practice (GCP), and the applicable regulatory requirement(s).”
The definition above is a well-rounded one; however it does not mandate any specifics of how much or how little monitoring would ensure quality, nor does it say how and when to conduct visits. The global definition also affirms that the specifics of how and when to conduct monitoring and hence SDV should be laid down on a case-to-case basis depending on several criteria.
Section 5.18.3 of Guideline E6 states: “The sponsor should ensure that trials are adequately monitored. The sponsor should determine the appropriate extent and nature of monitoring. The determination of the extent and nature of monitoring should be based on considerations such as the objective, purpose, design, complexity, blinding, size and endpoints of the trial. In general there is a need for on-site monitoring, before, during, and after the trial; however, in exceptional circumstances, the sponsor may determine that central monitoring in conjunction with procedures such as investigators′ training and meetings, and extensive written guidance can assure appropriate conduct of the trial in accordance with GCP. Statistically controlled sampling may be an acceptable method for selecting the data to be verified.”
So why the industry is still focused on elaborate site monitoring visit schedules and 100% or near 100% SDV in most trials?
CURRENT POINTS OF VIEW
Traditional view of “more SDV leads to better quality”
Researchers and sponsors find it hard to let go of the comfort feel of looking at all the data all over again to “catch” mistakes. The focus is still on catching the mistakes rather than preventing them from happening. Since, prevention is proactive, it need not happen at the site or during the monitoring visit. It can be a continuous process even before a site visit has started. The temptation of reviewing all the data also means that the time that should be spent on the critical data points such as inclusion-exclusion criteria, primary secondary endpoints, informed consent, etc., is divided into other noncritical data points which are enmeshed in the voluminous patient records and CRF binders.
Being on the safer side of regulations
It has been largely believed that more SDV means better quality and since none of the current regulations speak explicitly and quantitatively about the nature and extent of monitoring activities, researchers, and monitors err on the side of caution. In fact, FDA clearly recommends a review of a “representative” number of study volunteer records, and not all records.[2] How to choose the representative population of records is dependent on a set of predefined SDV criteria or is flexible as per the quality trends seen in the data that have already been monitored at a particular site or throughout the trial. Instead, if a cookie cutter approach is taken toward SDV and monitoring visits, the monitoring plans would remain overtly conservative and forever seek the wasteful path of 100% SDV.
Perhaps the regulators realize that although driven by the same overarching concepts of GCP, all clinical trials are different in terms of operational conduct and intended outcome. Within the ambit of the set guidelines, the conduct (including nature and extent of SDV) of the trial is largely left to the sponsors, so that there is scope of flexibility and innovation in how monitoring is done.
Considering one of the findings from Clinical Trials Transformation Initiatives′ (CTTI) Survey of Current Monitoring Practices, on-site monitoring visits are conducted for only about 31% of studies sponsored by NIH.[2,3]
DIFFERENT TYPES OF MONITORING
Monitoring contrary to the popular belief is an activity that can start even before the trial sites are chosen and/or active. This means that the preparation of monitoring and the actual act of monitoring can begin and happen at any point of the trial value chain. Like most of the trial activities, phases of monitoring can also be classified under the “preinitiation phase;” “on-site monitoring/data generation phase;” and “post close-out phase.”
We can thus broadly classify study monitoring into the following three categories:[4,5]
Trial oversight committees
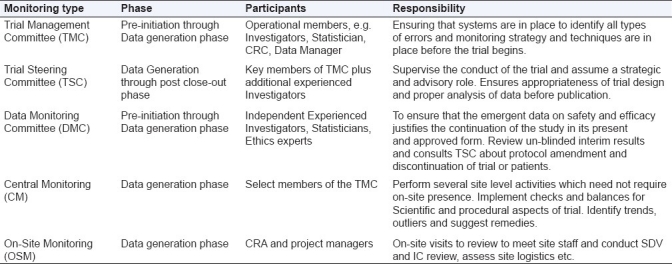
Trial Management Committee (TMC)
Trial Steering Committee (TSC)
Data Monitoring Committee (DMC)
The subtypes largely represent the different phases when these committees are applicable, e.g., TMC is relevant before the trial begins; TSC is more strategic and provides advisory throughout the trial progress and posttrial representation of the data. DMC is an independent committee which is active during the entire data acquisition phase and is responsible for ensuring that the emergent data are reviewed and meaningful conclusions about continuation of the study on grounds of safety and efficacy may be drawn [Table 1].
Table 1.
Different types of monitoring committees, their composition and responsibilities across various phases of study conduct
Central monitoring
Think of it as central lab versus local lab assessment of protocol required lab tests.
Similar to central labs, central monitoring techniques have several advantages. It allows a view of all the sites in a trial almost on a real time basis. The assessment of the trial parameters (scientific as well as procedural) are done consistently as it is done centrally. This can help in spotting trends in some of the parameters and can set off a trial level or site level remedial action. The study progress across sites can be evaluated by looking at several parameters:
Scientific aspects: Adherence to enrolment criteria; protocol deviations; primary efficacy outcomes (e.g., radiological assessments); safety trends, etc.
Procedural aspects: Assessment of drug storage and accountability; document life cycle management at site level; audit readiness checks; deviations in the patient follow-up schedules, etc.
Since via central monitoring, study data are available real time (through EDC) it is possible to use programmed statistical methods to trace discrepancies in the monitored/unmonitored data and identify outliers. This could be done at the study level, site level, patient level as well as CRA level. Since this could be automated and real time, it is a timely opportunity to investigate the root causes and implement remedies, which in rare cases could lead to protocol amendments or termination of sites.
For example, in the Second European Stroke Prevention Study, data on 438 patients were fabricated at one site. This was first detected by statistical anomalies in the data and later confirmed by a central review of blood results: A visit to the site had failed to identify any transgression.[5,6]
In most cases though, it leads to a better understanding of the training needs and procedural/logistic gaps that the trial has. In turn, it could define the level of on-site monitoring and extent of SDV required on a case-to-case basis. This does not exclude the possibility of even recommending additional on-site monitoring visit for sites where no OSM was planned initially.
On-site monitoring
As the name suggests, this is the variant of monitoring that is most easily recognizable and traditionally has been considered the safest and only way monitoring can and should be done. On-site monitoring as a concept has not been challenged enough. Although it is well accepted that OSM is needed to an extent for performing SDV and informed consent review, widespread attempt has not been made to proactively define the threshold of OSM by implementing analytics and statistical methods to tailor SDV and thus impact OSM duration and extent.
There is more than one reason why OSM needs to be optimized. OSM is expensive both in terms of the money spent on travel/stay logistics as well as the time spent on-site by numerous CRA across all sites though the life of the trial.
While OSM should ideally be used as an important interface between the investigator and the industry staff, it turns out that more time is spent on activities which could have easily been done from a remote location or in doing activities in excess of what is needed.
More often than not, the content and duration of on-site monitoring is defined by the volume of data and the extent of SDV that is planned for the site/study. It is reported that almost 46% of on-site monitoring effort goes into SDV which translates to about 34% of the total phase III study budget[7,8,2][Figure 1].
Figure 1.
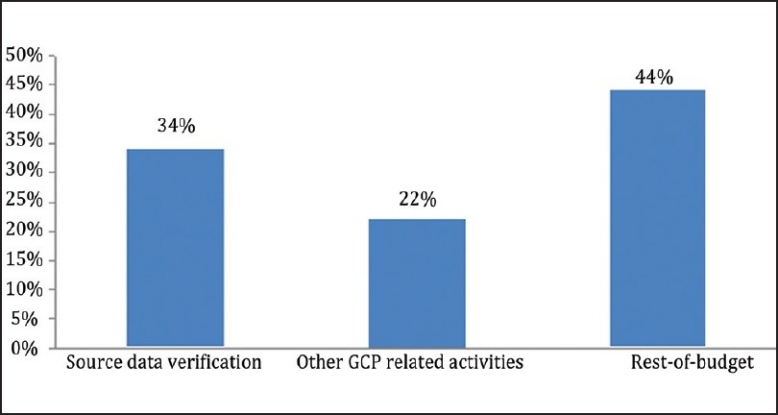
SDV costs companies about one-third of the total phase III study budget. [Sources: Funning et al., Quality Assurance Journal, 2008(10)]
So what are the alternatives to 100% SDV and extensive OSM?
Alternative monitoring approaches
There are several approaches that have been theorized and practised which help in optimizing the content and nature of SDV and prioritizing the data points to be SD verified. The strategies mentioned below could be used as stand-alone methods or as combination of several methods depending on the ease of implementation and value.
Targeted SDV
This could be defined as the SDV strategy where programmed statistical methods are utilized to select data points that should be verified against source during an OSM visit. There are two basic ways in which targeted SDV could be planned.[7]
Fixed field approach
Random field approach
In fixed field approach, consideration is given to the data points which are high risk (prone to errors) and critical (efficacy endpoints, consent, safety). Armed with this knowledge, the data points that need maximum attention are prioritized and 100% is mandated for only those fields. Depending on the total sample size of subjects in the trial, a number could be decided for whom 100% SDV for the fixed fields need to be done.
While the fixed field monitoring is in progress, central monitoring techniques can be used hand in hand on the incoming data to trace discrepancies in the monitored/un-monitored data and identify outliers. These departures from expected pattern may necessitate additional on-site visit, retraining, stepping up of monitoring efforts (including 100% SDV). In any case a combination of fixed field monitoring and central monitoring gives more information about the data quality much ahead of time and possibly much less on-site visit efforts.
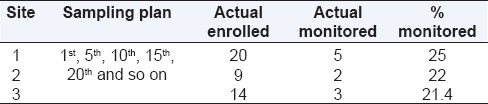
The other type of targeted SDV is the random field approach. This uses a random sampling technique to select the specific subjects and data points to be monitored during an OSM visit. However, even with the random sampling approach, a bit of caution could be employed. The first one or two patients could be subjected to 100% SDV including the inclusion-exclusion criteria, efficacy endpoints, safety, informed consent, etc. Thereafter, several techniques could be used to produce a random sample, e.g., choosing patient numbers at particular intervals; 3rd; 6th; 9th, and so on. In case the first one or two patients were not subjected to 100% SDV, it might better to have a sampling strategy like 1st, 4th, 7th, and so on. This will ensure that data quality can be reviewed right from beginning of enrolment at a site. One risk of having a random sampling plan like this could be ending up monitoring lesser percentage of patients from sites which are low enrolling or which do not enroll enough patients to touch the upper limit of the sample interval. Consider the simple scenario below.
As it is evident from above [Table 2], there could be different scenarios at different sites or at the same site based on actual enrolment. Therefore it is advisable to also specify a minimum SDV % so that the subjects who have not been monitored could be grouped and sampled again till the overall SDV target of the site is met.[7] So in the above case if the minimum SDV target is set at 25% of enrolled subjects, then for site/scenario 3, the recommendation would be to SDV an additional patient, and the % SDV would then be close to 29%.
Table 2.
Hypothetical sampling plans and %SDV in each case
Risk-based SDV
Risk-based monitoring approach focuses on the high risk data points (data points which are prone to mistakes or difference in interpretation or transcription and which have a high impact on the quality of the data and the outcome of the study). The SDV and monitoring strategies are customized to focus on these areas. In addition to the specific study details, several other factors are kept into account while formulating a risk-based monitoring plan, including the data quality from similar studies, experience level of site in handling such studies.[2]
Adaptive monitoring
In this approach the initial monitoring plan suggests a certain level of SDV based on the considerations discussed. However, as actual visits happen and data are generated, the frequency and extent of SDV may be increased or decreased depending on the quality of data assessed by predefined benchmarks. This adaptation in SDV requirement over the duration of the trial has a direct impact on the frequency of monitoring visits also.
Remote monitoring
This is an approach where activities that are traditionally considered to be on-site activities are carefully analyzed and new ways of conducting those from an off-site/remote location wre suggested, e.g., a simple activity of checking temperature logs at site could become challenging if there are discrepancies or temperature excursions noted during an OSM visit. One way of preventing this could be a remote monitoring technique where the site scans and emails a copy of the temperature log on a routine basis. This helps the CRA and the sponsor to discuss and resolve any issues even before a visit; leading to time saving and allowing temperature excursion discussions promptly.
If customized for each study and site, remote monitoring can take care of several on-site activities either wholly or partly. Initial findings from an in-house assessment indicate that there could be a potential time saving of 15% to 25% in the select range of activities if remote monitoring concepts are used.
Hybrid monitoring
Hybrid monitoring stresses upon the importance of moving away from a fixed concept of on-Site monitoring and replace it with a sustainable mix of targeted SDV (based on statistical sampling, risk based approach and flexible SDV %) and remote monitoring from an off-site location by optimally utilizing technology.
CONCLUSIONS
In conclusion, would it be premature to say that the industry should now segregate the concept of monitoring and the necessity for an on-site monitoring visit?
From the time when monitoring was necessarily done at the site with paper CRFs, the industry has come a long way with eClinical and remote data capture solutions. What it needs to continue is to implement methods and techniques to replace full-fledged 100% SDV.
By doing so, precious time and resources would be saved and directed back to the plethora of clinical research pursuits. CRAs who travel endlessly and seem to be always on-site could probably travel lesser but for more meaningful reasons and for activities which score the highest in the clinical monitoring value chain.
Acknowledgments
I wish to thank Mr. Arvind Kumar Pathak (Six Sigma Master Black Belt, General Manager-Quality) and Mr. Ashok Patel (Six Sigma Black Belt, Service Delivery Manager-Quality), Cognizant Technology Solutions for all the in-house assessments of remote monitoring concepts.
Footnotes
Source of Support: Nil,
Conflict of Interest: None declared.
REFERENCES
- 1.Rockville, MD: FDA; 1996. Food and Drug Administration, ICH E6 Good Clinical Practice Consolidated Guidance. [Google Scholar]
- 2.Getz KA. Low Hanging Fruit in the Fight Against Inefficiency. Appl Clin Trials. 2011;20:30–2. [Google Scholar]
- 3.Morrison BW, Cochran CJ, Giangrande J, Harley J, Kleppinger CF, Mitchel JT, et al. A CTTI Survey of Current Monitoring Practices.Society for Clinical Trials, 31st Annual Meeting Poster Presentation. 2010 May 17; [Google Scholar]
- 4.Buyse M, George SL, Evans S, Geller NL, Ranstam J, Scherrer B, et al. The role of biostatistics in the prevention, detection and treatment of fraud in clinical trials. Stat Med. 1999;18:3435–51. doi: 10.1002/(sici)1097-0258(19991230)18:24<3435::aid-sim365>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 5.Baigent C, Harrell FE, Buyse M, Emberson RJ, Altman DG. Ensuring Trial Validity by Data Quality Assurance and Diversification of Monitoring Methods. Clin Trials. 2008;5:49–55. doi: 10.1177/1740774507087554. [DOI] [PubMed] [Google Scholar]
- 6.The ESPS2 Group. European Stroke Prevention Study 2. Efficacy and safety data. J Neurol Sci. 1997;151 Suppl:S1–77. doi: 10.1016/s0022-510x(97)86566-5. [DOI] [PubMed] [Google Scholar]
- 7.Hines S. Targeting Source Document Verification. Available via Applied Clinical Trials Online. Search word: Sandra Hines. [Last accessed on 2011 March 10]. Available from: http://appliedclinicaltrialsonline.findpharma.com/appliedclinicaltrials/
- 8.Breslauer C. Could Source Document Verification Become a Risk in a Fixed-Unit Price Environment. The Monitor. 2006 Dec;:43–7. [Google Scholar]