

Abstract
Silymarin is a hepatoprotective agent, having poor water solubility and oral absorption of about 23 – 47%, leading to low bioavailability of the drug. The aim of the present study is to improve the solubility and dissolution rate and in turn the hepatoprotective activity of the drug, by formulating its inclusion complex with beta (β)-cyclodextrin, using different methods. The phase solubility analysis indicates the formation of 1:1 molar inclusion complex of the drug with beta cyclodextrin. Apparent stability constant for Silymarin (Kc) was 722 K-1 with β-cyclodextrin complex. The inclusion complexes were prepared by four different methods, namely, physical mixing, kneading, co-precipitation, and solvent evaporation. The prepared complexes were characterized using differential scanning colorimetry, scanning electron microscopy, and x-ray diffractometry. The inclusion complex prepared by the co-precipitation methods exhibits an overall best result, with respect to the formulation of sustained release formulations.
Keywords: Bioavailability, inclusion complexes, silymarin, β-cyclodextrin
INTRODUCTION
Silymarin (482.43) is not a good water soluble hepatoprotective agent. The oral absorption of silymarin is only about 23 – 47%, leading to low bioavailability of the drug, which limits its use. It is a mixture of mainly three flavanolignans, namely, silybin, silydianin, and silicristine, with silybin being most active. silymarin has been used medicinally to treat liver disorders, including acute and chronic liver hepatitis, toxin –/drug-induced hepatitis and cirrhosis, and alcoholic liver diseases. It is also reported to be effective in certain cancers.[1–5] The poor aqueous solubility of the drug may lead to dissolution-related bioavailability problems. Many approaches such as solubilization with surfactant systems, formation of water soluble complexes, and use of pro-drugs and soluble salt formation have been reported for improving the solubility and dissolution, and in turn the bioavailability of the drug.[6–8] Cyclodextrin and its derivatives play an important role in the formulation development due to their effect on solubility, dissolution rate, chemical stability, and absorption of a drug.[9] Nagarsenkar et al. reported faster dissolution and better bioavailability of ketorolac solid dispersion with HP-β-CD.[10] Reddy et al. reported enhanced solubility and dissolution rate of Celecoxib by complexation with β-Cyclodextrin.[11] The objective of the present study is to investigate the possibility of improving the solubility and dissolution rate of silymarin by complexation with β-cyclodextrin and also to compare the different complexation methods with respect to their dissolution study. In addition, the physiochemical characteristics of solid inclusion complexes were also investigated.
MATERIALS AND METHODS
A gift sample of silymarin was obtained from AGAPE pharmaceuticals, Majhitar, East Sikkim; β-cyclodextrin was obtained from Zim Laboratories Ltd., Nagpur. HCl, methanol potassium dihydrogen phosphate, and sodium hydroxide were of analytical grade (Merck, Mumbai). All other chemicals and reagents used were of analytical grade.
Phase solubility analysis for silymarin
Phase solubility studies were performed to determine the stoichiometric proportions of silymarin with β-cyclodextrin.[12,13] The data was used to determined the stability constant of the complexes. For this, the stock solution of 0.01 M β-cyclodextrin was prepared using distilled water. These stock solutions were diluted with distilled water to give molar solutions in the range of 0.002 to 0.01 M β-cyclodextrin. Five ml of each molar solution was filled in screw capped vials and the excess quantity of the drug was added to each vial separately.[7,14] The vials were shaken at an ambient temperature, for 48 hours, using a laboratory shaker (Remi). The supernatant solutions were collected carefully and filtered using Whatman filter paper (No. 41). The concentration of the drug in filtered solutions was determined using a UV visible spectrophotometer. No changes in λ max of the drug were found after complexation with cyclodextrin, hence absorbance of the resultant solutions were recorded at 286 nm, which was λ max of the drug. From the slope and intercept value (S0) of the phase solubility curve, a stability constant (Kc) was determined.
KC = Slope/[S0 (1 – Slope)]
Preparation of physical mixture and inclusion complexes
Physical mixture method
The required molar (1 : 1) quantities of the drug and cyclodextrin were weighted accurately and mixed together thoroughly in a mortar, with vigorous trituration, for about three hours. These mixtures were then passed through sieve No. 44, and finally stored in airtight containers till further use.[7,15,16]
Kneading method
The required quantities of the drug (Silymarin) and β-cyclodextrin were weighed accurately in a ratio of 1:1. A homogenous paste of cyclodextrin was prepared in a mortar by adding water : Methanol mixture (1 : 1) in small quantities. Silymarin powder was then added to this paste in portions, with continuous kneading, for three hours. An appropriate quantity of water : Methanol mixture (1:1) was added further to maintain suitable consistency of the paste. This paste was dried in a hot air oven at 45°–50° for 24 hours. The dried complexes were then powdered and passed through sieve No. 44 and stored in airtight containers till further use.[15]
Co-precipitation method
Quantities of drug and cyclodextrin, in the required molar ratio (1 : 1), were dissolved in methanol : Water, respectively. The solution of the drug was added dropwise into cyclodextrin solution. The contents were continuously stirred for 6 hours and finally were dried at 45°– 50° for 48 hours, collected, and stored in airtight containers till further use.[16]
Solvent evaporation method
In this technique, silymarin along with solubilizsing additives such as acetone were dissolved at 25°C temperature. Next, the required moles of β-cyclodextrin in hot distilled water were added dropwise into this solution, with continuous stirring, for one hour. The complexes formed were filtered and dried under a vacuum. Then the prepared solid mass was stored in a desiccator under vacuum to a constant weight. The dried products were removed, pulverized, and passed through sieve No. 100 and finally stored in a closed airtight container.
Characterization of silymarin inclusion complexes
Drug content estimation
The quantities of inclusion complex equivalent to 70 mg of silymarin were dissolved in water: Methanol mixture (1:1). Appropriate dilutions were made and the drug content of each complex was calculated from UV absorbance recorded at λ max 286 nm.
Scanning electron microscopy
The morphology of the inclusion complexes by physical mixture, kneading method, and co-precipitation method was studied using a scanning electron microscope (JSM-5610 LV Jeol, Japan). The samples were coated with platinum to provide a conductive layer for observing images at 15 kV.
IR spectrum analysis
Infra red (IR) spectra of the drug and inclusion complexes were recorded using the KBr method using Fourier Transform Infrared Spectrophotometer (FT IR-8400 S). A baseline correction was made using dried potassium bromide, and then the spectra of the dried mixtures of drug and inclusion complexes with potassium bromide were recorded.
Differential scanning calorimetric analysis
This scanning was performed using DSC model (Perkin Elmer). The samples were placed in a closed platinum crucible and DSC thermograms were recorded at a heating rate of 10°/minute in the range of 20° to 310°C.[16]
X-ray diffraction study
The X-ray diffraction pattern of the selected inclusion complex was compared with that of pure silymarin. This was performed by measuring 2θ in the range of 4° to 50° with reproducibility of ±0°–001° on an X-ray diffractometer (Phillips).
Dissolution study of silymarin and its inclusion complexes
Dissolution of inclusion complexes (equivalent to 70 mg of silymarin) was studied using the USP XXII eight station dissolution apparatus (Electrolab TDT-08L). The dissolution was carried out in 900 ml of Phosphate buffer, pH 6.8, at a speed of 75 rpm. Aliquots of 10 mL were withdrawn periodically and replaced with 10 mL of fresh dissolution medium. The concentrations of the drug in the samples were determined by measuring their absorbance at 286 nm, using Shimadzu 1700 UV-visible spectrophotometer. Cumulative percent of the drug released was determined at every point of time. The pure drug was used as a control. The T90 (time required for 90% dissolution of drug) of various solid dispersions were calculated.[17,18]
RESULTS AND DISCUSSION
Phase solubility analysis of silymarin
The phase solubility study was done to determine the stoichiometric proportion of silymarin with complexing agent β-cyclodextrin. The solubility analysis indicated the formation of a 1 : 1 molar inclusion complex of the drug with β-cyclodextrin. The apparent stability constant for silymarin (Kc) was 722 K–1 with the β-cyclodextrin complex.
Characterization of silymarin inclusion complexes
All the inclusion complexes prepared using different methods, such as, physical mixture, kneading method, co-precipitation, and solvent evaporation method were found to be slightly brown, free-flowing powders.
Estimation of drug content
Inclusion complexes prepared by co-precipitation showed nearly 100% drug content. The drug content of the inclusion complex prepared by kneading, physical mixture, and solvent evaporation shows slightly less drug content as compared to that prepared by using co-precipitation [Table 1].
Table 1.
Comparison of drug content and T90 value of silymarin inclusion complex with β-cyclodextrin
Scanning electron microscopy
Scanning electron microscopy (SEM) of the physical mixture method, inclusion complex by kneading method, and co-precipitation method were studied. The representative photographs are shown in Figures 1–3. Pure drug particles in the physical mixture method were very small in size with reduced effective surface area, due to agglomeration. They remained dispersed and physically adsorbed on the surface of β-cyclodextrin. Figures 2 and 3, show drug particles distributed on the surface of β-cyclodextrin. Both kneaded and the co-precipitated systems show a homogeneity, signifying the inclusion complex formation. The kneaded system is of poor crystal structure, lacks distinct crystal faces, and has numerous cracks and fissures. This may also have contributed to faster dissolution compared to the co-precipitation system.
Figure 1.

Silymarin with β-cyclodextrin (Physical mixture)
Figure 3.

Silymarin with β-cyclodextrin (Co-precipitation method)
Figure 2.

Silymarin with β-cyclodextrin (Kneading method)
IR spectral analysis
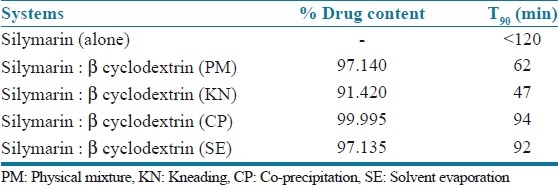
Infra red spectra of the pure drug and inclusion of silymarin with β-cyclodextrin, by different methods, are given in Figure 4 (a - e). The guest molecules within the cyclodextrin cavity show in their peaks or show peaks of less intensity. Basically peaks that lie in the fingerprint, and peaks due to C-O or O-H stretching are affected (shifted or intensity is changed).
Figure 4.
IR spectra of inclusion complexes of silymarin with β-cyclodextrin (a) FT IR study of silymarin (b) Physical mixture method (c) Kneading method (d) Co-precipitation method (e) Solvent evaporation
Differential scanning calorimetric analysis
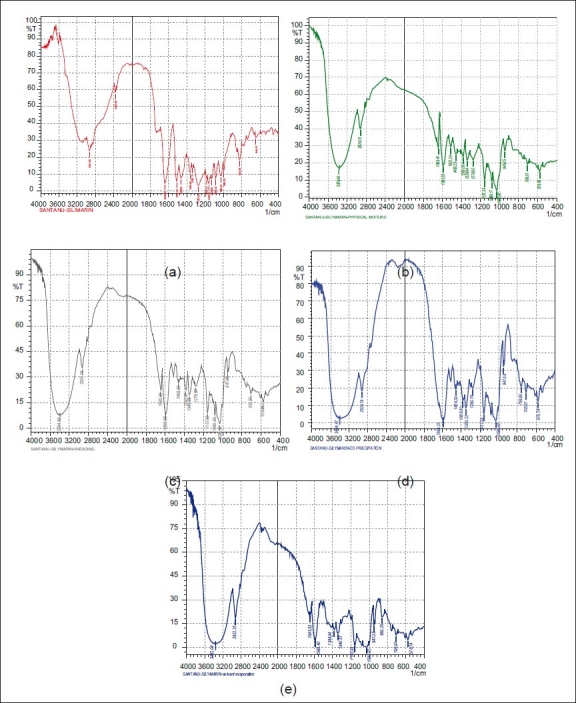
Silymarin, β-cyclodextrin, and the complex prepared by the co-precipitation method were subjected to Differential scanning calorimetric (DSC) analysis. The thermal curves are given in Figure 5. The DSC curve of silymarin shows one characteristic sharp endothermic peak at around 150°C, indicating the melting point of the drug. The DSC curve shows that the sharp endothermic peak at around 150°C, which is observed for silymarin, decreases in the inclusion complex (1:1). Furthermore, the wide peak at 90°C, which is observed for β-cyclodextrin, shifts to 60°C in the inclusion complex, indicating that the inclusion complex does not contain much residue of silymarin or β-cyclodextrin, thus suggesting that the drug is well dispersed in the β-cyclodextrin cavity.
Figure 5.
Differential scanning calorimetry study of silymarin, β-cyclodextrin and silymarin-β-cyclodextrin complex
X-ray diffraction study
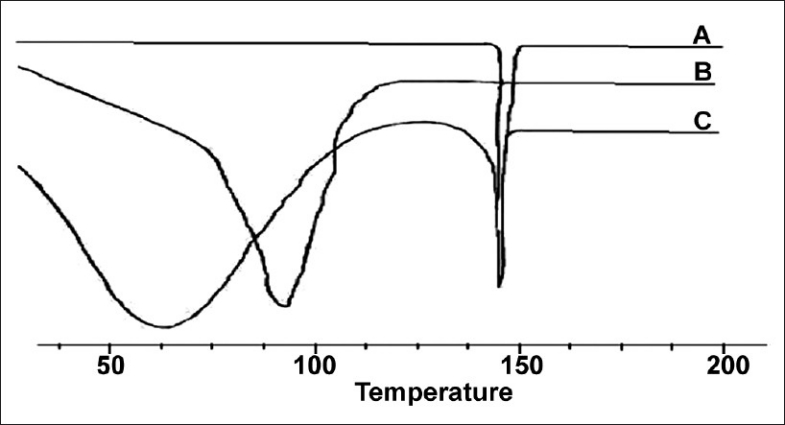
The inclusion complex of the drug prepared with β-cyclodextrin, using the co-precipitation method, which showed a good result overall, was characterized further by an XRD study The X-ray diffraction patterns of pure silymarin, as well as the silymarin–β-Cyclodextrin complex obtained by using the co-precipitation method are represented in Figure 6. The peak position (angle of diffraction) is an indication of the amorphous nature of the sample. The diffractogram of pure silymarin shows some intense peaks, which are indicative of crystallinity. However, in case of silymarin complexed with β-Cyclodextrin diffractogram, it attributes to a new solid phase with low crystallinity, indicating inclusion complex formation (more water soluble). A reduced number of signals, of markedly low intensity, are noticeable in the complex, indicating the greater amorphous nature of the inclusion complex compared to the free molecules.
Figure 6.
X-ray diffraction study on silymarin and silymarin complexed with β-cyclodextrin
Dissolution study of silymarin and its inclusion complexes
The inclusion complexes of silymarin with β-cyclodextrin produce pronounced enhancement in its dissolution as shown in Table 2 and Figure 7. From Table 1-T90 value for kneading shows the quickest rate of dissolution as compared to the other inclusion complexes, prepared by other inclusion methods. Inclusion complexes prepared by the co-precipitation methods show their T90 in about 94 minutes.
Table 2.
Percentage cumulative release of inclusion complexes of silymarin and β-cyclodextrin prepared by different methods
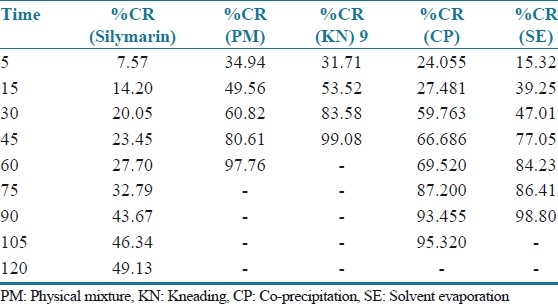
Figure 7.
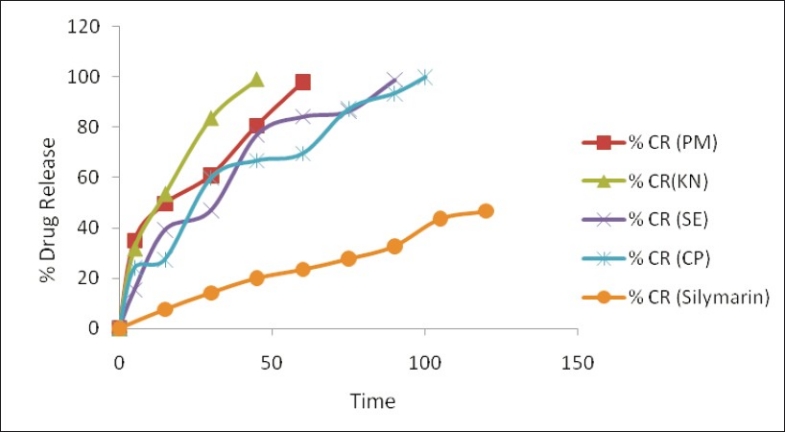
Dissolution profiles of inclusion complexes of silymarin and β-cyclodextrin prepared using different methods. PM = Physical mixture, KN = kneading, CP = co-precipitation, SE = solvent evaporation
CONCLUSION
The phase solubility data suggest a 1 : 1 complex formation with β-cyclodextrin. All inclusion complexes show increase in dissolution than in the drug alone. The inclusion complex of the drug prepared by the co-precipitation method shows the best results overall, in terms of drug content and dissolution profile, for preparation of sustained release formulations. However, it should be noted that this is a result of a preliminary study of β-cyclodextrin inclusion complexation with silymarin.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Luper SA. Review of plants used in the treatment of liver disease, Part 1. Altern Med Rev. 1998;3:410–21. [PubMed] [Google Scholar]
- 2.Pradhan SC, Girish C. Hepatoprotective herbal drug, Silymarin from experimental pharmacology to clinical medicine. Indian J Med Res. 2006;124:491–504. [PubMed] [Google Scholar]
- 3.Reynolds JE. London: Pharmaceutical Press; 2002. Martindale the Extra Pharmacopoeia. [Google Scholar]
- 4.Pepping J. Milk thistle: Silybum marianum. Am J Health Sys Pharma. 1999;56:1195–7. doi: 10.1093/ajhp/56.12.1195. [DOI] [PubMed] [Google Scholar]
- 5.Saller R, Meier R, Brignoli R. The use of Silymarin in the treatment of liver diseases. Drugs. 2001;61:2035–63. doi: 10.2165/00003495-200161140-00003. [DOI] [PubMed] [Google Scholar]
- 6.Pinnamaneni S, Das N, Das SK. Formulation approaches for orally administered poorly soluble drugs. Pharmazie. 2002;57:291–300. [PubMed] [Google Scholar]
- 7.Deshmukh SS, Potnis VV, Shelar DB, Mahaparale PR. Studies on inclusion complexes of ziprasone hydrochloride with β cyclodextrin and hydroxypropyl βcyclodextrin. Indian Drugs. 2007;44:677–82. [Google Scholar]
- 8.Patil JS, Pandya NR, Marapur SC, Shiralashetti SS. Influence of method of preparation on physiochemical properties and in vitro drug release profile of nimodipine–cyclodextrin inclusion complexes: A comparative study. Int J Pharmacy Pharmaceutical Sci. 2010;2:71–81. [Google Scholar]
- 9.Loftsson T, Brewster ME, Masson M. Role of cyclodextrins in improving oral drug delivery. Am J Drug Deliv. 2004;2:1–15. [Google Scholar]
- 10.Nagarsenkar MS, Meshram R, Ramprakash G. Solid dispersion of hydroxypropyl a cyclodextrin and Ketorolac: Enhancement of in vitro dissolution rates, improvement in anti-inflammatory activity and reduction in ulcerogenecity in rats. J Pharm Pharmacol. 2000;52:949–56. doi: 10.1211/0022357001774831. [DOI] [PubMed] [Google Scholar]
- 11.Reddy MN, Chowdary KP, Diwan PV. β-Cyclodextrin complexes of celecoxib: Molecular modeling, characterization and dissolution studies. AAPS Pharm Sci. 2004;6:E7. doi: 10.1208/ps060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loftsson T, Brewster ME. Pharmaceutical application of cyclodextrin in drug solubilisation and stabilization. J Pharm Sci. 1996;85:1017–25. doi: 10.1021/js950534b. [DOI] [PubMed] [Google Scholar]
- 13.Kamal D, Ramana MD, Himaja M, Agarwal A, Garg V. Investigation of enhancement of solubility of norfloxacin beta cyclodextrin in presence of acidic solubilizing additives. Curr Drug Deliv. 2007;4:21–5. doi: 10.2174/156720107779314776. [DOI] [PubMed] [Google Scholar]
- 14.Attma AA, Ndibe ON, Nnamani PO. Studies on diclofenac-β-Cyclodextrin inclusion complexes. J Pharm Res. 2004;3:47–9. [Google Scholar]
- 15.Soniwala MM, Patel PR, Ansuri MS, Parikh RK, Goel MC. Various approaches in dissolution enhancement of Rofecoxib. Indian J Pharm Sci. 2005;67:61–3. [Google Scholar]
- 16.Aries MJ, Moyano JR, Munoz P, Justo A. Study of Omeprazole-g-cyclodextrin complexation in the solid state. Drug Dev Ind Pharm. 2000;26:253–9. doi: 10.1081/ddc-100100353. [DOI] [PubMed] [Google Scholar]
- 17.Becket G, Schep LJ, Tan MY. Improvement of the in vitro dissolution of praziquantel by complexation with α-, β-, and γ-cyclodextrin. Int J Pharm. 1999;179:65–71. doi: 10.1016/s0378-5173(98)00382-2. [DOI] [PubMed] [Google Scholar]
- 18.Bekers O, Uijtendaal EV, Beijnen JH, Bult A, Underberg WJ. Cyclodextrins in the pharmaceutical field. Drug Dev Ind Pharm. 1991;17:1503–49. [Google Scholar]