

Abstract
Intensive research in recent years has begun to unlock the mysteries surrounding the molecular pathogenesis of melanoma, the deadliest of skin cancers. The high-penetrance, low-frequency susceptibility gene CDKN2A produces tumor suppressor proteins that function in concert with p53 and retinoblastoma protein to thwart melanomagenesis. Aberrant CDKN2A gene products have been implicated in a great many cases of familial cutaneous melanoma. Sporadic cases, on the other hand, often involve constitutive signal transduction along the mitogen-activated protein kinase (MAPK) pathway, with particular focus falling upon mutated RAS and RAF protooncogenes. The proliferative effects of the MAPK pathway may be complemented by the antiapoptotic signals of the PI3K/AKT pathway. After skin, melanoma most commonly affects the eye. Data for the constitutive activation of the MAPK pathway in uveal melanoma exists as well, however, not through mutations of RAS and RAF. Rather, evidence implicates the proto-oncogene GNAQ. In the following discussion, we review the major molecular pathways implicated in both familial and sporadic cutaneous melanomagenesis, the former accounting for approximately 10% of cases. Additionally, we discuss the molecular pathways for which preliminary evidence suggests a role in uveal melanomagenesis.
1. Introduction
Melanoma remains a disproportionate cause of death among skin cancers [1, 2]. Currently, early diagnosis followed by complete surgical removal of the tumor offers the best hope for cure [3]. Once advanced, melanoma is notoriously resistant to medical interventions [3]. Thus, great interest lies in the discovery of new therapeutic options that may improve the prognoses of those afflicted with this unforgiving disease.
New insights into the development and/or progression of cutaneous melanoma have been achieved through the study of its molecular pathogenesis. Key molecules at crucial junctions have been identified and have begun serving as potential targets for clinicians tasked with containing this lethal disease.
After skin, primary melanoma most commonly affects the eye [4]. The two most commonly employed modalities for the treatment of uveal melanoma, the most lethal of ocular melanomas, are radiation therapy and enucleation [5]. Despite these valiant efforts at local disease control, up to 50% of patients succumb to their disease, and impact on patient survival remains questionable at best [6]. Thus, a great need for improved therapy exists for the treatment of uveal melanoma.
In the following discussion, we review the major molecular pathways implicated in both familial and sporadic cutaneous melanomagenesis, the former accounting for approximately 10% of cases [7]. Additionally, we discuss the molecular pathways for which preliminary evidence suggests a role in uveal melanomagenesis.
2. Familial Cutaneous Melanoma
Knowledge of some of the earliest molecular pathways involved in melanomagenesis derived from investigations of familial cutaneous melanoma. In affected individuals, a complex network of interrelated pathways functions to promote cellular proliferation and cellular survival.
2.1. CDKN2A
The best-characterized high-penetrance susceptibility gene predisposing to cutaneous melanoma is CDKN2A [3, 8–12]. This gene is located on chromosome 9p21 and encodes two distinct tumor-suppressor proteins—p14/ARF and p16/INK4a—implicated in the pathogenesis of 25–40% of familial cutaneous melanomas (Figure 1) [3, 13]. The former deters melanomagenesis through its indirect effect on p53, a tumor-suppressor protein also known as “the guardian of the genome.” Upon sensing DNA damage, p53 promotes the transcription of numerous genes involved in cell cycle arrest and/or apoptosis. Simply stated, if DNA damage can be repaired during cell cycle arrest, the cell returns to its normal functional state. If damage is irreparable, however, p53 stimulates the transcription of microRNAs (miRNAs), specifically the mir34 family of miRNAs, which silence the translation of proproliferative and antiapoptotic transcripts resulting in either quiescence/senescence or apoptosis, respectively.
Figure 1.
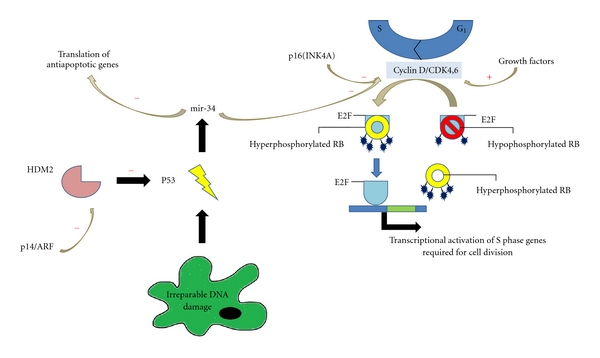
Roles of p14/ARF, p16(INK4A), and cyclin-dependent kinase 4 protein in cellular proliferation and survival. Loss of function of any of these molecules has been implicated in the pathogenesis of Familial Cutaneous Melanoma.
Under homeostatic conditions, p53 maintains a relatively short half-life due to the function of human homolog of murine Mdm2 (HDM2), a protein that ubiquitinates other proteins for destruction. When the cell is stressed, however, p14/ARF binds to and inhibits the function of HDM2 allowing p53 to escape ubiquitination. Mutated p14/ARF, on the other hand, is unable to bind and suppress HDM2, allowing it to mark p53 for destruction. With less p53 available to identify damaged DNA, genomic instability results, predisposing the afflicted individual to the development of cutaneous melanoma.
p16/INK4a functions in concert with retinoblastoma protein (RBp), another tumor-suppressor protein, to regulate the Gap 1 (G1) phase of the cell cycle. During this phase, cells can exit the cell cycle into quiescence or senescence, or make the necessary preparations to progress onward into the S phase of the cycle. Specifically, phosphorylation of RBp, which is partly dependent upon the cyclin D—CDK4/6 complex, is necessary for the transcription of genes encoding cyclin E, a protein that is required for the initiation of DNA replication in the S phase. p16/INK4a inhibits the phosphorylation of RBp by inactivating the cyclin D—CDK4/6 complex and consequently prevents the cell progression through the cell cycle. When p16 expression is compromised, and so is RBp's regulatory control on the cell cycle.
2.2. CDK4
Linkage studies have allowed the identification of another high-penetrance, low-frequency melanoma susceptibility gene, CDK4, which is mutated in three cutaneous melanoma kindreds worldwide [3, 9–11]. Located on chromosome 12q14, CDK4 encodes cyclin-dependent kinase 4 protein, a constituent of the CDK4/6 complex discussed above (Figure 1). Germline mutations that activate this gene occur at codon 24 (Arg24Cys and Arg24His) and render the CDK4/6 complex resistant to p16 inhibition. Similar to the case of aberrations in the CDKN2A gene, mutations in CDK4 lend to an increased risk for cutaneous melanomagenesis.
3. Sporadic Cutaneous Melanoma
3.1. RAS/RAF/MEK/ERK Signaling Pathway
Constitutive stimulation of the mitogen-activated protein kinase (MAPK) pathway, which regulates cellular proliferation, has been implicated in up to 90% of cutaneous melanomas [3, 9, 11, 12, 14, 15]. The MAPK pathway exerts its effects through signal transduction along the cascade of RAS, RAF, MAPK extracellular signal-regulated kinase (MEK), and extracellular signal-regulated kinase (ERK) (Figure 2). MAPK signaling initiates when a receptor tyrosine kinase in the cell membrane binds its respective ligand—an event that results in the activation of RAS, a membrane-bound protein with GTPase activity. Activated RAS then recruits RAF, a cytosolic serine-threonine-specific protein kinase, to the plasma membrane. Through phosphorylation, RAS activates RAF, which in turn phosphorylates and activates MEK. Activated MEK activates ERK, which induces several proliferative and survival processes, one of which is activation of the cyclin D-CDK4/6 complex (discussed above) upon translocation to the nucleus.
Figure 2.
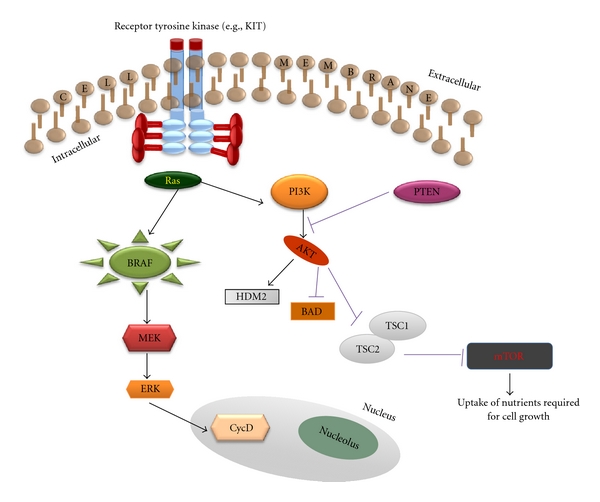
MAPK and PI3K/AKT pathways regulate cellular proliferation and survival. Aberrant functioning of these pathways has been implicated in the pathogenesis of sporadic cutaneous melanoma.
3.1.1. c-KIT
c-KIT, a protooncogene that encodes the type III receptor tyrosine kinase KIT, was first identified in 1987 as a result of sequence similarity to the Hardy-Zuckerman 4 feline sarcoma virus oncogene, v-KIT [16]. Upon binding its ligand, stem cell factor (SCF), KIT undergoes receptor dimerization, autophosphorylation, and activation of its intracellular tyrosine kinase domain [8, 17]. Once activated, KIT is capable of stimulating downstream signaling pathways, such as MAPK [8, 17, 18].
Activating mutations and/or gene amplification of KIT are now being described in significant subsets of melanomas [19–23]. One study, in particular, recognized such aberrations in 39% of mucosal melanomas, 36% of acral melanomas, and 28% of melanomas arising in chronically sun-damaged skin (as defined by the presence of solar elastosis on review of histopathology)—anatomic sites at which BRAF mutations occur far less frequently [22].
The most prevalent KIT mutations are L576P (exon 11), K642E (exon 13), V559A (exon 11), and D816H (exon 17) [17, 24]. These mutations are thought to promote the constitutive activation of KIT either through precluding the protein from assuming its default autoinhibited conformation, or by promoting its dimerization in the absence of SCF [25, 26]. The precise manner in which constitutive KIT activation promotes melanomagenesis remains unclear.
3.1.2. NRAS
First implicated in sarcoma in rats, HRAS and KRAS—members of the RAS family of protooncogenes—were recognized for their ability to undergo activating transformation under the influence of the Harvey and Kirsten sarcoma viruses. A third member of the RAS family, NRAS, was subsequently identified in human neuroblastoma cells. Of these three protooncogenes, activating mutations in NRAS occur most frequently in melanocytes and have been identified in nearly one-third of all melanomas [3, 12, 15]. The most commonly documented NRAS aberration in melanoma is a missense mutation at codon 61 (Q61R), which results in the substitution of arginine in place of glutamine and impairs GTP hydrolysis locking the protein in a state of constitutive activation [27]. In comparison to other solid tumors, mutations in RAS do not occur with as high as a frequency in melanoma [3]. Nonetheless, in the absence of the inhibitory effects of p16/INK4a, oncogenic RAS has been shown to induce melanoma and appears to have an integral function in tumor maintenance and progression [28, 29].
3.1.3. BRAF
Each of the three RAF protooncogenes—ARAF, BRAF, and CRAF—have been identified in mammals. While ARAF and CRAF mutations are rare in human cancers, a significant percentage of human malignancies have been shown to harbor activating mutations in BRAF, with the highest rate occurring in melanoma [3, 9, 11, 12]. BRAF mutations in melanoma tend to occur at anatomic sites exposed to intermittent, rather than chronic, sun damage. With approximately 70% of cases harboring such a mutation, BRAF is the most commonly mutated protooncogene in cutaneous melanoma. Furthermore, a significant proportion of both benign and dysplastic melanocytic nevi have been shown to harbor mutation of BRAF as well, suggesting a relatively early event in melanomagenesis [3, 11, 12, 30].
Greater than 90% of BRAF mutations in melanoma result from a single base missense mutation (T→A) at codon 1799 that leads to the substitution of valine in favor of glutamic acid at position 600 of the BRAF protein [31]. This alteration introduces a conformational change in BRAF's kinase domain, which can lead to a 480-fold increase in kinase activity when compared to that of wild-type BRAF [32]. However, the story is not as simple as this may suggest. While mutated BRAF induces uncontrolled proliferation in melanoma, it lends to senescence in benign melanocytic nevi. Furthermore, a subset of melanocytes is able to bypass the senescent response and undergo malignant transformation upon the accumulation of additional insults.
3.2. PI3K/AKT Signaling Pathway
Activated RAS also triggers the phosphatidylinositol 3-kinase (PI3K)/AKT pathway, which conducts antiapoptotic signals that complement the proliferative effects of the MAPK pathway (Figure 2) [3, 11, 12, 15]. PI3K phosphorylates phosphatidylinositols of the cell membrane to produce phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3 then recruits the serine/threonine kinase AKT, also known as protein kinase B, to the cell membrane. Activation of AKT requires phosphorylation by PDK1,2. AKT then promotes cell survival by phosphorylating several substrates, such as BAD (a member of the bcl-2 family of apoptosis regulator proteins) and HDM2 (discussed above). AKT also inactivates the complex formed by TSC1 and TSC2, tumor-suppressor proteins known to be mutated in tuberous sclerosis. Inactivation of the TSC1/TSC2 complex stimulates activity of mammalian target of rapamycin (mTOR), a kinase that promotes the uptake of nutrients necessary for cellular growth.
3.2.1. PTEN
The phosphatase and tensin homolog (PTEN) gene is located on chromosome 10 and encodes a tumor-suppressor protein that functions as both a lipid and protein phosphatase [3, 11, 12, 15]. Specifically, the PTEN protein degrades the products of PI3K, thereby antagonizing the activity of the PI3K/AKT pathway. This results in increased apoptosis and decreased tumorigenesis. Loss of functional PTEN protein has been demonstrated in approximately 20% of primary cutaneous melanomas and has been shown to positively correlate with increasing Breslow thickness [33]. Frequency of mutations and deletions only partly account for PTEN loss in melanoma samples, suggesting epigenetic mechanisms [34]. PTEN losses occur with greater frequency in primary melanomas when compared with benign melanocytic nevi, but with lesser frequency when compared with metastatic melanomas [34, 35]. Thus, PTEN losses appear to occur in later stages of melanomagenesis and may contribute to the transformation of a benign melanocytic proliferation into an invasive melanoma.
4. Ocular Melanoma
Melanomas of the eye are categorized as either conjunctival or uveal, with the latter being further divided between anterior uveal melanomas (iris) and posterior uveal melanomas (ciliary and choroidal). Not surprisingly, differences in the histopathology and/or clinical presentation of these neoplasms exist, and the most striking distinction is in prognosis [36, 37]. Melanomas of the conjunctiva and iris have relatively good outcomes, while survival rates of those with posterior uveal melanoma markedly decline. Specifically, the 5-year mortality rate due to metastasis of ciliary body or choroidal melanomas is approximately 30%, compared to about 3% for iris melanomas [37]. Thus, a greater urgency in exploring the molecular mechanisms of posterior uveal melanomagenesis in the hopes of developing new and effective therapies is understandable.
5. Uveal Melanoma
The identification of susceptibility genes through linkage analysis has not proved fruitful due to the low occurrence of uveal melanoma in the familial setting. Furthermore, while CDKN2A is implicated in 25–40% of familial cutaneous melanomas, the germline mutation infrequently occurs in the rare uveal melanoma-prone families studied [38]. Finally, data implicating the protooncogenes NRAS and BRAF is not as convincing in uveal melanomagenesis as it is in sporadic cutaneous melanomagenesis [39–41]. Nonetheless, evidence for the constitutive activation of the MAPK pathway in uveal melanoma exists, suggesting involvement of a disparate protooncogene [41].
5.1. GNAQ
Screening of potential oncogenes that may activate the MAPK pathway has led to the discovery of mutations in GNAQ, a stimulatory αq subunit of heterotrimeric G proteins (Gαβγ) [42–44]. Upon the binding of an extracellular signal molecule to a G-protein-coupled receptor on the cell surface, GDP bound to the Gα subunit of Gαβγ is replaced with GTP, allowing for the dissociation of Gα from Gβγ. No longer tethered to Gβγ, Gαq stimulates phospholipase Cβ, until its intrinsic GTPase activity renders it unable to do so. Phospholipase Cβ hydrolyzes phosphatidylinositol 4,5-bisphosphonate (PIP2) into two second messengers: inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). IP3 functions to increase cytosol calcium levels by releasing the cation from endoplasmic reticulum stores. Calcium then activates conventional isoforms of the serine/threonine kinase, protein kinase C, while DAG activates both conventional and novel isoforms of the kinase. Finally, protein kinase C stimulates the RAF/MEK/ERK pathway, which induces several proliferative and survival processes, as discussed above.
Mutation of GNAQ at codon 209 prevents the hydrolysis of GTP and locks GNAQ in its active, GTP-bound state. Constitutive activation of GNAQ is tantamount to oncogenic activation of the MAPK cascade and accounts for approximately 50% of uveal melanomas [43]. While inhibitors of GNAQ, phospholipase Cβ, or protein kinase C isoforms have yet to be developed, small, preliminary studies involving the downstream inhibition of MEK have encouraged investigation through formal clinical trials [45]. The results of these larger studies may offer much needed hope to those afflicted with this aggressive disease.
6. Conclusion
Many of the pivotal discoveries regarding the key molecular players discussed in this paper are highlighted on the timeline pictured in Figure 3 [22, 42, 46–50]. While much progress has been made in unlocking the mysteries surrounding the molecular pathogenesis of cutaneous melanoma, the story is far from complete. Just as researchers are beginning to understand the mechanisms by which activating mutations in the RAS and RAF protooncogenes lead to proliferative and antiapoptotic effects, evidence is mounting for the role of constitutive MAPK activity in tumor evasion of immune surveillance, suppression of immune response, tumor angiogenesis, and metastatic dissemination [51–62]. Furthermore, approximately 40% of melanoma kindreds harbor CDKN2A mutations, and significantly less perpetuate CDK4 mutations, thus the genetic basis of a substantial proportion of cases of familial cutaneous melanoma clearly remains unresolved [7, 63]. Genome-wide association studies have enabled the identification of several low-penetrance, high-frequency susceptibility genes [64, 65]. Analyses of these genes will likely reveal additional molecular pathways involved in melanoma formation and/or progression.
Figure 3.
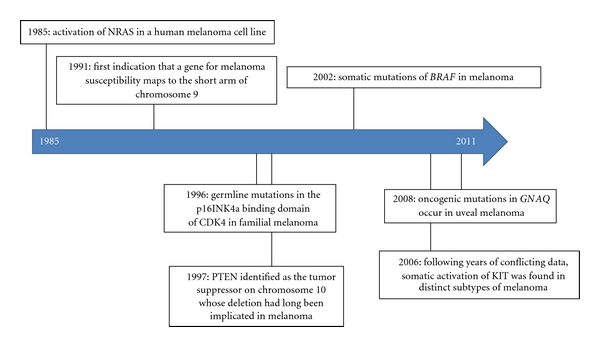
Timeline highlighting pivotal discoveries of key molecular players discussed in this paper on melanomagenesis.
While the battle against uveal melanoma may not be as far along as that against its cutaneous counterpart, lines have been drawn in the sand, and war is under way. In addition to taking aim at the constitutive activation of the MAPK pathway, researchers have discovered potential targets in the PI3/AKT and insulin growth factor (IGF) signaling pathways, mTOR, protooncogene c-MET, and tumor-suppressor breast cancer-1 (BRCA1)-associated protein-1 (BAP1), early onset [43]. While clinical trials are under way to determine if aberrations in the aforementioned molecules and pathways can be manipulated to stifle and/or reverse uveal melanomagenesis, the need for intervention at more than just one critical junction will likely be needed.
Despite the plethora of questions that remain, the potential of this early research to benefit disease-stricken patients is already being realized. Preliminary studies employing selective inhibitors against key players detailed in the aforementioned discussion have demonstrated encouraging results [43, 66, 67]. Whether these particular treatments prove effective in rigorous clinical trials remains to be seen. Regardless, researchers are reassured in seeking a molecular basis for the treatment of melanoma.
Acknowledgments
The authors would like to thank Keiran Smalley, Ph.D. and Raymond M. Fertig, B. S., M. B. A. for their kind contributions of the graphics used in this paper.
References
- 1.National Cancer Institute SEER Stat Fact Sheets. Melanoma of the skin. 2010. http://seer.cancer.gov/statfacts/html/melan.html.
- 2.National Cancer Institute Skin Cancer. 2010. http://www.cancer.gov/cancertopics/types/skin.
- 3.Ibrahim N, Haluska FG. Molecular pathogenesis of cutaneous melanocytic neoplasms. Annual Review of Pathology. 2009;4:551–579. doi: 10.1146/annurev.pathol.3.121806.151541. [DOI] [PubMed] [Google Scholar]
- 4.Chang AE, Karnell LH, Menck HR. The national cancer data base report on cutaneous and noncutaneous melanoma: a summary of 84,836 cases from the past decade: the American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer. 1998;83(8):1664–1678. doi: 10.1002/(sici)1097-0142(19981015)83:8<1664::aid-cncr23>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 5.Laver NV, McLaughlin ME, Duker JS. Ocular melanoma. Archives of Pathology and Laboratory Medicine. 2010;134(12):1778–1784. doi: 10.5858/2009-0441-RAR.1. [DOI] [PubMed] [Google Scholar]
- 6.Damato B. Does ocular treatment of uveal melanoma influence survival. British Journal of Cancer. 2010;103(3):285–290. doi: 10.1038/sj.bjc.6605765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeh I, Bastian BC. Genome-wide associations studies for melanoma and nevi. Pigment Cell and Melanoma Research. 2009;22(5):527–528. doi: 10.1111/j.1755-148X.2009.00622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calder KB, Morgan MB. Carcinogenic pathway of malignant melanoma. In: Coppola D, editor. Mechanisms of Oncogenesis: An Update on Tumorigenesis . Springer; 2010. pp. 149–157. [Google Scholar]
- 9.Meyle KD, Guldberg P. Genetic risk factors for melanoma. Human Genetics. 2009;126(4):499–510. doi: 10.1007/s00439-009-0715-9. [DOI] [PubMed] [Google Scholar]
- 10.Nelson AA, Tsao H. Melanoma and genetics. Clinics in Dermatology. 2009;27(1):46–52. doi: 10.1016/j.clindermatol.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 11.Palmieri G, Capone M, Ascierto ML, et al. Main roads to melanoma. Journal of Translational Medicine. 2009;7 doi: 10.1186/1479-5876-7-86. Article ID 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sekulic A, Haluska P, Miller AJ, et al. Malignant melanoma in the 21st century: the emerging molecular landscape. Mayo Clinic Proceedings. 2008;83(7):825–846. doi: 10.4065/83.7.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chin L. The genetics of malignant melanoma: lessons from mouse and man. Nature Reviews Cancer. 2003;3(8):559–570. doi: 10.1038/nrc1145. [DOI] [PubMed] [Google Scholar]
- 14.Smalley KS. Understanding melanoma signaling networks as the basis for molecular targeted therapy. Journal of Investigative Dermatology. 2010;130(1):28–37. doi: 10.1038/jid.2009.177. [DOI] [PubMed] [Google Scholar]
- 15.Kong Y, Kumar SM, Xu X. Molecular pathogenesis of sporadic melanoma and melanoma-initiating cells. Archives of Pathology and Laboratory Medicine. 2010;134(12):1740–1749. doi: 10.1043/2009-0418-RAR.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yarden Y, Kuang WJ, Yang-Feng T, et al. Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO Journal. 1987;6(11):3341–3351. doi: 10.1002/j.1460-2075.1987.tb02655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woodman SE, Davies MA. Targeting KIT in melanoma: a paradigm of molecular medicine and targeted therapeutics. Biochemical Pharmacology. 2010;80(5):568–574. doi: 10.1016/j.bcp.2010.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flaherty KT, Hodi FS, Bastian BC. Mutation-driven drug development in melanoma. Current Opinion in Oncology. 2010;22(3):178–183. doi: 10.1097/cco.0b013e32833888ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Antonescu CR, Busam KJ, Francone TD, et al. L576P KIT mutation in anal melanomas correlates with KIT protein expression and is sensitive to specific kinase inhibition. International Journal of Cancer. 2007;121(2):257–264. doi: 10.1002/ijc.22681. [DOI] [PubMed] [Google Scholar]
- 20.Ashida A, Takata M, Murata H, Kido K, Saida T. Pathological activation of KIT In metastatic tumors of acral and mucosal melanomas. International Journal of Cancer. 2009;124(4):863–868. doi: 10.1002/ijc.24048. [DOI] [PubMed] [Google Scholar]
- 21.Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clinical Cancer Research. 2008;14(21):6821–6828. doi: 10.1158/1078-0432.CCR-08-0575. [DOI] [PubMed] [Google Scholar]
- 22.Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. Journal of Clinical Oncology. 2006;24(26):4340–4346. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 23.Rivera RS, Nagatsuka H, Gunduz M, et al. C-kit protein expression correlated with activating mutations in KIT gene in oral mucosal melanoma. Virchows Archiv. 2008;452(1):27–32. doi: 10.1007/s00428-007-0524-2. [DOI] [PubMed] [Google Scholar]
- 24.Smalley KS, Sondak VK, Weber JS. C-KIT signaling as the driving oncogenic event in sub-groups of melanomas. Histology and Histopathology. 2009;24(5):643–650. [Google Scholar]
- 25.Lennartsson J, Blume-Jensen P, Hermanson M, Pontén E, Carlberg M, Rönnstrand L. Phosphorylation of Shc by Src family kinases is necessary for stem cell factor receptor/c-kit mediated activation of the Ras/MAP kinase pathway and c-fos induction. Oncogene. 1999;18(40):5546–5553. doi: 10.1038/sj.onc.1202929. [DOI] [PubMed] [Google Scholar]
- 26.Mol CD, Dougan DR, Schneider TR, et al. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. Journal of Biological Chemistry. 2004;279(30):31655–31663. doi: 10.1074/jbc.M403319200. [DOI] [PubMed] [Google Scholar]
- 27.Platz A, Egyhazi S, Ringborg U, Hansson J. Human cutaneous melanoma; a review of NRAS and BRAF mutation frequencies in relation to histogenetic subclass and body site. Molecular Oncology. 2008;1(4):395–405. doi: 10.1016/j.molonc.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ackermann J, Frutschi M, Kaloulis K, McKee T, Trumpp A, Beermann F. Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Research. 2005;65(10):4005–4011. doi: 10.1158/0008-5472.CAN-04-2970. [DOI] [PubMed] [Google Scholar]
- 29.Chin L, Tam A, Pomerantz J, et al. Essential role for oncogenic ras in tumour maintenance. Nature. 1999;400(6743):468–472. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- 30.Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nature Genetics. 2003;33(1):19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 31.Puzanov I, Flaherty KT. Targeted molecular therapy in melanoma. Seminars in Cutaneous Medicine and Surgery. 2010;29(3):196–201. doi: 10.1016/j.sder.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 32.Wan PTC, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 33.Goel VK, Lazar AJF, Warneke CL, Redston MS, Haluska FG. Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. Journal of Investigative Dermatology. 2006;126(1):154–160. doi: 10.1038/sj.jid.5700026. [DOI] [PubMed] [Google Scholar]
- 34.Mirmohammadsadegh A, Marini A, Nambiar S, et al. Epigenetic silencing of the PTEN gene in melanoma. Cancer Research. 2006;66(13):6546–6552. doi: 10.1158/0008-5472.CAN-06-0384. [DOI] [PubMed] [Google Scholar]
- 35.Tsao H, Goel VK, Wu H, Yang G, Haluska FG. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. Journal of Investigative Dermatology. 2004;122(2):337–341. doi: 10.1046/j.0022-202X.2004.22243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zembowicz A, Mandal RV, Choopong P. Melanocytic lesions of the conjunctiva. Archives of Pathology and Laboratory Medicine. 2010;134(12):1785–1792. doi: 10.5858/2009-0522-RAR.1. [DOI] [PubMed] [Google Scholar]
- 37.Intraocular (Eye) melanoma treatment. 2007. http://www.cancer.gov/cancertopics/pdq/treatment/intraocularmelanoma/HealthProfessional.
- 38.Goldstein AM, Stacey SN, Olafsson JH, et al. CDKN2A mutations and melanoma risk in the Icelandic population. Journal of Medical Genetics. 2008;45(5):284–289. doi: 10.1136/jmg.2007.055376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cruz F, Rubin BP, Wilson D, et al. Absence of BRAF and NRAS mutations in uveal melanoma. Cancer Research. 2003;63(18):5761–5766. [PubMed] [Google Scholar]
- 40.Rimoldi D, Salvi S, Liénard D, et al. Lack of BRAF mutations in uveal melanoma. Cancer Research. 2003;63(18):5712–5715. [PubMed] [Google Scholar]
- 41.Zuidervaart W, Van Nieuwpoort F, Stark M, et al. Activation of the MAPK pathway is a common event in uveal melanomas although it rarely occurs through mutation of BRAF or RAS. British Journal of Cancer. 2005;92(11):2032–2038. doi: 10.1038/sj.bjc.6602598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Onken MD, Worley LA, Long MD, et al. Oncogenic mutations in GNAQ occur early in uveal melanoma. Investigative Ophthalmology and Visual Science. 2008;49(12):5230–5234. doi: 10.1167/iovs.08-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patel M, Smyth E, Chapman PB, et al. Therapeutic implications of the emerging molecular biology of uveal melanoma. Clinical Cancer Research. 2011;17(8):2087–2100. doi: 10.1158/1078-0432.CCR-10-3169. [DOI] [PubMed] [Google Scholar]
- 44.Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457(7229):599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phase II trial of temozolomide versus AZD6244 in patients with metastatic uveal melanoma. 2011. http://www.mskcc.org/mskcc/html/2270.cfm?IRBNO=10-053.
- 46.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 47.Nancarrow DJ, Mann GJ, Holland EA, et al. Confirmation of chromosome 9p linkage in familial melanoma. American Journal of Human Genetics. 1993;53(4):936–942. [PMC free article] [PubMed] [Google Scholar]
- 48.Padua RA, Barrass NC, Currie GA. Activation of N-ras in a human melanoma cell line. Molecular and Cellular Biology. 1985;5(3):582–585. doi: 10.1128/mcb.5.3.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nature Genetics. 1997;15(4):356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 50.Zuo L, Weger J, Yang Q, et al. Germline mutations in the p16(INK4a) binding domain of CDK4 in familial melanoma. Nature Genetics. 1996;12(1):97–99. doi: 10.1038/ng0196-97. [DOI] [PubMed] [Google Scholar]
- 51.Benbow U, Tower GB, Wyatt CA, Buttice G, Brinckerhoff CE. High levels of MMP-1 expression in the absence of the 2G single nucleotide polymorphism is mediated by p38 and ERK1/2 mitogen-activated protein kinases in VMM5 melanoma cells. Journal of Cellular Biochemistry. 2002;86(2):307–319. doi: 10.1002/jcb.10225. [DOI] [PubMed] [Google Scholar]
- 52.Cartlidge RA, Thomas GR, Cagnol S, et al. Oncogenic BRAFV600E inhibits BIM expression to promote melanoma cell survival. Pigment Cell and Melanoma Research. 2008;21(5):534–544. doi: 10.1111/j.1755-148X.2008.00491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eisenmann KM, VanBrocklin MW, Staffend NA, Kitchen SM, Koo HM. Mitogen-activated protein kinase pathway-dependent tumor-specific survival signaling in melanoma cells through inactivation of the proapoptotic protein bad. Cancer Research. 2003;63(23):8330–8337. [PubMed] [Google Scholar]
- 54.Huntington JT, Shields JM, Der CJ, et al. Overexpression of collagenase 1 (MMP-1) is mediated by the ERK pathway in invasive melanoma cells: role of BRAF mutation and fibroblast growth factor signaling. Journal of Biological Chemistry. 2004;279(32):33168–33176. doi: 10.1074/jbc.M405102200. [DOI] [PubMed] [Google Scholar]
- 55.Kono M, Dunn IS, Durda PJ, et al. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Molecular Cancer Research. 2006;4(10):779–792. doi: 10.1158/1541-7786.MCR-06-0077. [DOI] [PubMed] [Google Scholar]
- 56.Kumar SM, Yu H, Edwards R, et al. Mutant V600E BRAF increases hypoxia inducible factor-1α expression in melanoma. Cancer Research. 2007;67(7):3177–3184. doi: 10.1158/0008-5472.CAN-06-3312. [DOI] [PubMed] [Google Scholar]
- 57.Sharma A, Tran MA, Liang S, et al. Targeting mitogen-activated protein kinase/extracellular signal-regulated kinase kinase in the mutant (V600E) B-Raf signaling cascade effectively inhibits melanoma lung metastases. Cancer Research. 2006;66(16):8200–8209. doi: 10.1158/0008-5472.CAN-06-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Research. 2005;65(6):2412–2421. doi: 10.1158/0008-5472.CAN-04-2423. [DOI] [PubMed] [Google Scholar]
- 59.Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene. 2003;22(20):3138–3151. doi: 10.1038/sj.onc.1206454. [DOI] [PubMed] [Google Scholar]
- 60.Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. Journal of Experimental Medicine. 2006;203(7):1651–1656. doi: 10.1084/jem.20051848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woods D, Cherwinski H, Venetsanakos E, et al. Induction of β3-integrin gene expression by sustained activation of the Ras-regulated Raf-MEK-extracellular signal-regulated kinase signaling pathway. Molecular and Cellular Biology. 2001;21(9):3192–3205. doi: 10.1128/MCB.21.9.3192-3205.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang XD, Borrow JM, Zhang XY, Nguyen T, Hersey P. Activation of ERK1/2 protects melanoma cells from TRAIL-induced apoptosis by inhibiting Smac/DIABLO release from mitochondria. Oncogene. 2003;22(19):2869–2881. doi: 10.1038/sj.onc.1206427. [DOI] [PubMed] [Google Scholar]
- 63.Newton-Bishop J, Gruis N. Melanoma susceptibility genes. Melanoma Research. 2010;20(3):161–162. doi: 10.1097/CMR.0b013e328336b000. [DOI] [PubMed] [Google Scholar]
- 64.Bishop DT, Demenais F, Iles MM, et al. Genome-wide association study identifies three loci associated with melanoma risk. Nature Genetics. 2009;41(8):920–925. doi: 10.1038/ng.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Falchi M, Bataille V, Hayward NK, et al. Genome-wide association study identifies variants at 9p21 and 22q13 associated with development of cutaneous nevi. Nature Genetics. 2009;41(8):915–919. doi: 10.1038/ng.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Puzanov I, Nathanson KL, Chapman PB, et al. PLX4032, a highly selective V600EBRAF kinase inhibitor: clinical correlation of activity with pharmacokinetic and pharmacodynamic parameters in a phase I trial. Journal of Clinical Oncology. 2009;27(15s):p. 9021. [Google Scholar]
- 67.Hodi FS, Friedlander P, Corless CL, et al. Major response to imatinib mesylate in KIT-mutated melanoma. Journal of Clinical Oncology. 2008;26(12):2046–2051. doi: 10.1200/JCO.2007.14.0707. [DOI] [PubMed] [Google Scholar]