

Abstract
Aspergillus fumigatus is an opportunistic human pathogenic fungus responsible for deadly lung infections in immunocompromised individuals. Galactofuranose (Galf) residues are essential components of the cell wall and play an important role in A. fumigatus virulence. The flavoenzyme UDP-galactopyranose mutase (UGM) catalyzes the isomerization of UDP-galactopyranose to UDP-galactofuranose, the biosynthetic precursor of Galf. Thus, inhibitors of UGM that block the biosynthesis of Galf can lead to novel chemotherapeutics for treating A. fumigatus-related diseases. Here, we describe the synthesis of fluorescently labeled UDP analogs and the development of a fluorescence polarization (FP) binding assay for A. fumigatus UGM (AfUGM). High-affinity binding to AfUGM was only obtained with the chromophore TAMRA, linked to UDP by either 2 or 6 carbons with Kd values of 2.6 ± 0.2 μM and 3.0 ± 0.7 μM, respectively. These values were ~6 times lower than when UDP was linked to fluorescein. The FP assay was validated against several known ligands and displayed an excellent Z′ factor (0.79 ± 0.02) and good tolerance to dimethyl sulfoxide.
1. Introduction
Aspergillus fumigatus is an opportunistic human pathogen responsible for diseases such as allergic reactions and lung infections, including bronchopulmonary aspergillosis (ABPA) and invasive pulmonary aspergillosis (IPA) [1, 2]. This fungus is a significant health threat to immunocompromised patients, such as organ transplant recipients and people with AIDS or leukemia [3, 4]. It has been reported that IPA infections are typically accompanied by a mortality rate of 50–70% [5]. Thus, identification of novel and effective drug targets is essential in the fight against fungal infections.
Recently, the biosynthetic pathway of galactofuranose (Galf), the 5-membered ring form of galactose, has been described in A. fumigatus. Galf is a component of the cell wall of A. fumigatus and plays an important role in virulence [6–8]. In A. fumigatus, Galf was first identified as a component of galactomannan by immunodetection in IPA patients [9]. Later, it was found that Galf is also a major component of saccharide structures in membrane lipids and glycosyl phosphoinositol (GPI-)anchored lipophospholipids [10, 11]. UDP-galactopyranose mutase (UGM) is a flavoenzyme that catalyzes the conversion of UDP-galactopyranose (UDP-Galp) to UDP-galactofuranose (UDP-Galf, Figure 1), the biosynthetic precursor of Galf [7, 12]. Deletion of the A. fumigatus UGM (AfUGM) gene results in mutant fungi with attenuated virulence, a decrease in cell wall thickness, and an increase in the sensitivity to antifungal agents [8, 13]. Moreover, Galf is absent in humans [12]. Thus, inhibitors of AfUGM that block the biosynthesis of Galf represent attractive drug targets for the identification of novel therapeutics against A. fumigatus.
Figure 1.
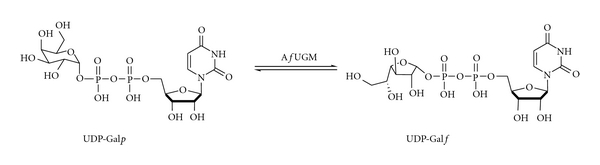
Reaction catalyzed by AfUGM.
Here, we describe the development of a fluorescence polarization (FP) binding assay to identify specific AfUGM inhibitors. Four fluorescently labeled UDP derivatives including two known UDP-fluorescein analogs (1 and 2, Figure 2) and two novel UDP-TAMRA analogs (3 and 4, Figure 2) were synthesized to be used as fluorescent probes in the FP assay. Their concentrations were optimized to obtain a stable FP signal with minimal standard deviation, and their K d values were determined by measuring the anisotropy changes as a function of AfUGM concentration. We found that the UDP-TAMRA analogs bind to AfUGM 6-fold tighter than the UDP-fluorescein analogs, suggesting that UDP-TAMRA analogs are better fluorescent probes for this enzyme. UDP-TAMRA probes could be competed out by UDP, a known ligand of UGMs, and the K d value of UDP was in good agreement with the value determined previously in a fluorescence assay [7]. Furthermore, the FP assay was validated using several known ligands and displayed an excellent Z′ factor (0.79 ± 0.02) and good tolerance to DMSO. Therefore, this fast convenient one-step FP assay is suitable for a high-throughput screening to identify AfUGM inhibitors.
Figure 2.
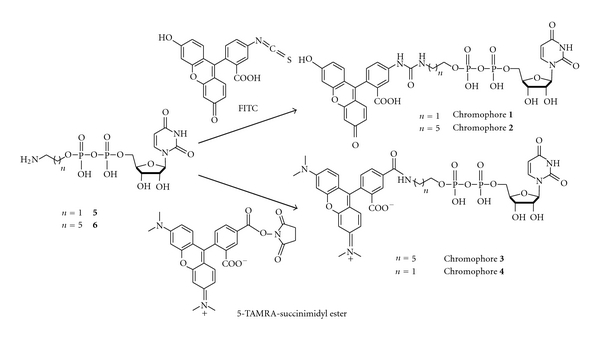
Synthetic scheme of the chromophores used as ligands to AfUGM for application in FP assays.
2. Materials and Methods
2.1. Materials
All chemicals were obtained from commercial sources and were used without further purification. Anhydrous reactions were performed under argon. All solvents were either reagent grade or HPLC grade. NMR spectral data were obtained using a JEOL Eclipse spectrometer at 500 MHz, or a Varian Inova spectrometer at 400 MHz. Chemical shifts were reported as δ-values relative to known solvent residue peaks. High-resolution mass spectra (HRMS) were obtained in the Mass Spec Incubator, Department of Biochemistry, Virginia Tech. High-performance liquid chromatography (HPLC) was performed on a C18 reverse phase column (Phenomenex Luna C18 column, 250 × 21.20 mm, 5 microns) using water and acetonitrile as the elution solvents. All compounds were more than 95% pure as judged by HPLC and 1H NMR.
2.2. Protein Expression and Purification
AfUGM and MtUGM were expressed and purified with the same protocol as described by Oppenheimer et al. [7]. A large quantity of highly pure AfUGM was obtained, which was confirmed by UV-visible spectrophotometry and SDS-PAGE (see Figure S1 Supplementary Material available online at doi:10.4061/2011/513905).
2.3. Synthesis of UDP-Fluorescein Chromophore 1 and 2
The synthesis of chromophore 1 was accomplished by reacting 4 mg of compound 5, which was synthesized following a previously published procedure [15], with 6 mg of fluorescein-5-isothiocyanate (FITC) in 0.1 M pH 9.0 NaHCO3 buffer (50 μL) and DMF (100 μL) (Figure 2). After stirring at room temperature for 2 hours, the yellow solution was concentrated and loaded onto a preparative silica gel TLC plate. The isolated crude product was dissolved in water, injected onto reverse-phase HPLC (Phenomenex Luna C18 column, 250 × 21.20 mm, 5 microns), and purified at a flow rate of 5.0 mL/min with linear gradient elution of 5% to 95% acetonitrile in H2O over 20 min to afford chromophore 1 (4 mg, 52%). 1H NMR (500 MHz, 6 : 1 D2O: d 7-DMF): δ 7.96 (d, J = 8.2, 1H), 7.78 (s, 1H), 7.70 (d, J = 8.1, 1H), 7.30 (dd, J = 8.2, 1.5, 1H), 7.27-7.27 (m, 2H) (t, J = 8.7, 2H), 6.65–6.61 (m, 2H), 6.61–6.58 (m, 2H), 5.91 (s, 1H), 5.91 (s, 1H), 4.36–4.30 (m, 2H), 4.24–4.21 (m, 3H), 4.19–4.16 (m, 2H), 3.88 (s, 2H); HRMS (MALDI) calcd for C32H29N4O17P2S (M-H)−: 835.0729, found 835.0759 (Figure S2).
Chromophore 2 (7.1 mg, 55%) was synthesized from the reaction of compound 6 and FITC by the same procedure as above (Figure 2) and was purified by preparative TLC and reverse-phase HPLC (Phenomenex Luna C18 column, 250 × 21.20 mm, 5 micron). 1H NMR (500 MHz, 6 : 1 D2O: d 7-DMF): δ 8.00 (d, J = 8.0, 1H), 7.73 (s, 1H), 7.61 (d, J = 8.3, 1H), 7.31 (d, J = 8.3, 1H), 7.27-7.27 (m, 2H), 6.66–6.60 (m, 4H), 5.96 (s, 1H), 4.38–4.34 (m, 2H), 4.27–4.23 (m, 2H), 4.23–4.18 (m, 2H), 3.97–3.92 (m, 2H), 3.58 (s, 1H), 1.66–1.61 (m, 4H), 1.42–1.36 (m, 4H); HRMS (MALDI) calcd for C36H37N4O17P2S (M-H)−: 891.1350, found 891.1348 (Figure S3).
2.4. Synthesis of UDP-TAMRA Chromophore 3 and 4
The synthesis of chromophore 3 was accomplished by a reaction of 4 mg of compound 6, which was synthesized following a previously published procedure [15], with 0.8 mg of 5-carboxytetramethylrhodamine, succinimidyl ester (5-TAMRA, SE) in 0.1 M pH 8.3 NaHCO3 buffer (50 μL) and DMF (50 μL) (Figure 2). After stirring at room temperature for 2 hours, the pink solution was concentrated and loaded onto a preparative TLC plate. The isolated crude product was dissolved in water, injected onto reverse-phase HPLC (Phenomenex Luna C18 column, 250 × 21.20 mm, 5 microns), and purified at a flow rate of 5.0 mL/min with linear gradient elution of 5% to 95% acetonitrile in H2O over 20 min to afford chromophore 3 (1.1 mg, 80%). 1H NMR (500 MHz, D2O) δ 8.22 (s, 1H), 8.08 (d, J = 7.7, 1H), 7.89 (d, J = 7.3, 1H), 7.60 (d, J = 8.8, 1H), 7.28–7.22 (m, 1H), 6.91–6.88 (m, 2H), 6.61 (s, 1H), 6.59 (s, 1H), 5.89–5.83 (m, 2H), 4.35–4.31 (m, 1H), 4.30–4.26 (m, 1H), 4.18–4.15 (m, 3H), 4.00 (dd, J = 13.3, 6.4, 2H), 3.49 (t, J = 6.8, 2H), 3.19 (s, 3H), 3.18 (s, 3H), 1.75 – 1.67 (m, 4H), 1.51–1.45 (m, 4H), 1.34 (s, 3H), 1.32 (s, 3H); HRMS (MALDI) calcd for C40H46N5O16P2 (M-H)−: 914.2415, found 914.2431 (Figure S4). The above synthetic approach was also used to synthesize and purify chromophore 4 (1.5 mg, 77%). HRMS (MALDI) calcd for C36H38N5O16P2 (M-H)−: 858.1789, found 858.1851 (Figure S5).
2.5. Optimization of Chromophore Concentration
Solutions containing various concentrations of chromophore in 0.05 M sodium phosphate buffer (pH 7.0) were added to 12 wells in a 96-well half area black bottom plate (Corning) with final volumes of 25 μL. FP was analyzed by a SpectraMax M5 plate reader (Molecular Devices). The parallel fluorescence emission (F=) and perpendicular fluorescence emission (F⊥) at 524 nm (for compounds 1 and 2, excitation at 492 nm) or at 584 nm (for compounds 3 and 4, excitation at 544 nm) were measured by a SpectraMax M5 plate reader (Molecular Devices), and the anisotropy (r) was calculated using (1), the minimal concentration at which stable FP signals with minimal standard deviations were chosen as the optimal concentration for the chromophore.
| (1) |
| (2) |
2.6. FP Binding Assay to Determine the Chromophore Binding Affinities
Solutions containing serially diluted AfUGM and 15 nM of chromophore in 0.05 M sodium phosphate buffer (pH 7.0) were incubated at room temperature for 5 minutes. Each experiment was done in triplicate in a 96-well black bottom plate at final volumes of 25 μL. Fluorescence anisotropy was measured as indicated above, and the K d values were obtained by fitting the anisotropy data to (2), where m 1 and m 2 are the minimum and maximum anisotropy values, respectively; m 3 is the K d value, and the total concentration of UDP-chromophore is represented by C t.
2.7. Determination of the Assay Z′ Factor
Solutions containing 2 μM of AfUGM and 15 nM of chromophore 3 in the absence (negative control) and presence (positive control) of 300 μM of UDP were incubated at room temperature for 5 minutes. Each solution was added to octuplicate wells in a 96-well half area black bottom plate with final volumes of 25 μL. The Z′ factors were calculated using (3), where μ − represents the mean anisotropy value of the negative control, and μ + is the mean anisotropy value of the positive control; σ − represents the standard deviation of the negative control, and σ + is the standard deviation of the positive control. A Z′ factor of 0.79 ± 0.02 was obtained for chromophore 3.
| (3) |
2.8. Optimization of AfUGM Concentration
To determine the optimal concentration of AfUGM in the FP assay, solutions containing 15 nM of chromophore 3 and AfUGM at various concentrations in the absence (negative control) and presence (positive control) of 300 μM of UDP were incubated at room temperature for 5 minutes. Each was added to octuplicate wells at a final volume of 25 μL. FP was analyzed as indicated previously, and Z′ factors were calculated from (3).
2.9. Competitive Binding Experiments Using FP Inhibition Assay
Solutions (25 μL) containing 2 μM of AfUGM and 15 nM of chromophore 3 in 0.05 M sodium phosphate buffer (pH 7.0) were mixed with various concentrations of UDP, UDP-Galp, 7, or 8 (Figure 8), and the reactions incubated at room temperature for 5 minutes. Each solution was done in triplicate. Anisotropy values were measured and the IC50 values obtained by fitting the data to (4), where m 1 and m 2 are the minimum and maximum anisotropy, respectively; m 3 is the slope, and m4 is the IC50. The K d values were obtained using (5), where K i is the binding affinity of chromophore 3 on AfUGM (2.6 ± 0.6 μM), and I is the concentration of the chromophore (15 nM).
Figure 8.
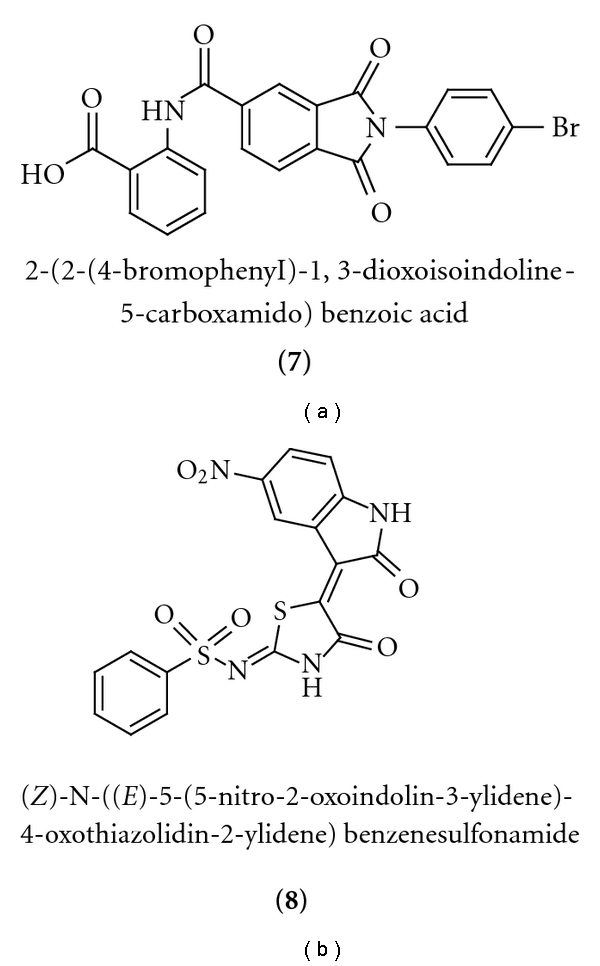
Structures of known inhibitors of bacterial UGM [14].
| (4) |
| (5) |
2.10. AfUGM Activity Assay
The AfUGM activity assay was performed by monitoring the formation of UDP-Galp from UDP-Galf by HPLC. A 20 μL reaction containing 20 mM dithiothreitol, 0.5 mM UDP-Galf in 25 mM HEPES, 125 mM NaCl buffer, pH 7.5 in the absence of 7 or 8 was initiated by the addition of AfUGM at a final concentration of 50 nM. After incubation at 37°C for 10 min, the reaction was quenched by heat denaturation (95°C for 5 min) in a DNA engine thermocycler (BioRad, Hercules, Calif, USA). The same reaction was also performed in the presence of 7 (500 μM) or 8 (50 μM). The suspension was centrifuged and the supernatant was injected onto a CarboPac PA100 (Dionex) anion-exchange column. The sample was eluted isocratically with 75 mM KH2PO4 (pH 4.5), and the absorbance at 262 nm was monitored to identify fractions of substrate and product. The substrate UDP-Galf was eluted at 36.5 min, and the product UDP-Galp was eluted at 28.3 min. The inhibition of AfUGM activity was indicated by the extent of conversion of UDP-Galf to UDP-Galp.
2.11. Tolerance to DMSO
To determine the tolerance of the assay to DMSO, solutions containing 2 μM of AfUGM, 15 nM of chromophore 3, and DMSO at various concentrations in the absence (negative control) and presence (positive control) of 300 μM of UDP were incubated at room temperature for 5 minutes. Fluorescence anisotropy values and Z′ factors were calculated as indicated previously.
3. Results and Discussion
3.1. Assay Design and Optimization
In this study, we report the development of an FP assay that can be used in a high-throughput format for the identification of inhibitors of AfUGM, which we believe will lead to the development of new therapeutics against A. fumigatus-related diseases. The FP assay was designed as shown in Figure 3. If the UDP fluorescent probe binds to AfUGM and is excited with plane-polarized light, the resulting enzyme-ligand complex tumbles slowly in solution, and thus, the fluorescence emission remains polarized (Figure 3(a)). Otherwise, the emission will be depolarized as the free chromophore will rotate rapidly. The change in the rotational motion between the bound and free chromophore can be used as a signal for detection of the binding of small molecules to the active site of AfUGM because, as the small molecule replaces the bound fluorescent probe, the free probe will rapidly rotate increasing the amount of depolarized fluorescence (Figure 3(b)).
Figure 3.
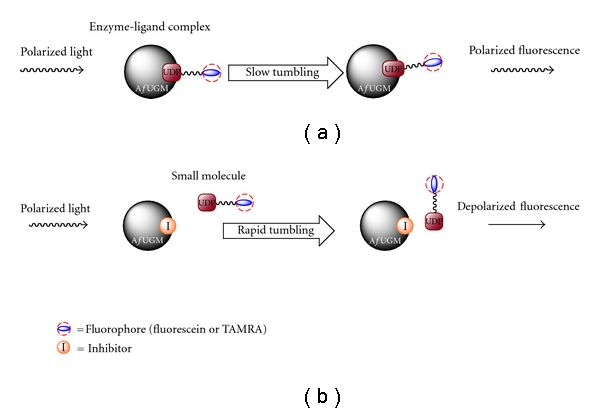
FP assay design. (a) Binding of the FP probe to AfUGM leads to polarized fluorescence. (b) Displacement of the FP probe from AfUGM by inhibitor results in depolarized fluorescence.
An essential component of an FP assay is a fluorescent probe that specifically binds to the enzyme or protein of interest. To design the fluorescent probe, we reasoned that the incorporation of the UDP moiety into the structure would target binding to the AfUGM active site since it is a major part of the UGM substrate. The fluorophore we first selected was fluorescein because UDP-fluorescein derivatives have been found to bind to prokaryotic UGMs from Klebsiella pneumoniae and Mycobacterium tuberculosis [15]. To minimize the steric hindrance of fluorescein with AfUGM binding site residues, UDP and fluorescein were connected with alkyl linkers of different lengths, which resulted in two UDP-fluorescein analogs (1 and 2, Figure 2). We also designed a UDP bound to the chromophore, commercially known as TAMRA (Figure 2). This chromophore offers several advantages over fluorescein. First, TAMRA is more resistant to photobleaching compared to fluorescein [16]. Second, the fluorescence emission of TAMRA does not overlap with that of the flavin cofactor in AfUGM. Fluorescein is typically excited at 494 nm and emits at 520 nm, which significantly overlaps with the absorbance and fluorescence emission of the flavin. In contrast, TAMRA's absorbance and fluorescence maxima is at 546 nm and 580 nm, respectively [16]. This is significantly different from the flavin absorbance/emission properties and improves signal-to-noise ratio. Finally, in comparison with fluorescein, TAMRA has one extra positive charge, which we believe increases the interaction between TAMRA and flavin and helps improve binding of the probe to AfUGM. Alkyl linkers of different lengths were also included to minimize the steric interaction of TAMRA with the binding site residues, giving two novel UDP-TAMRA analogs (3 and 4, Figure 2).
In order to increase the signal-to-noise ratio, stable FP values are necessary. Therefore, we varied the concentration of UDP-chromophores to determine the optimal concentration (Figure 4). Stable FP values with minimal standard deviation were obtained at concentrations higher than 15 nM. Therefore, we chose the 15 nM UDP-chromophore as the minimal concentration to use for further characterization.
Figure 4.
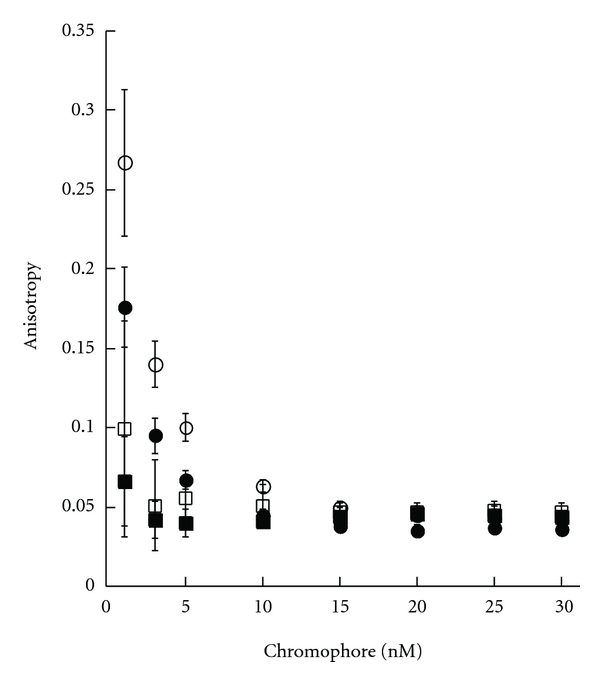
Determination of optimal concentration of fluorescent probe for FP binding assay. Conditions are described in Material and Methods sections. Chromophore 1 (○), 2 (⬤) (excitation at 492 nm and emission at 524 nm), 3 (■), and 4 (□) (excitation at 544 nm and emission at 584 nm).
3.2. AfUGM Specific UDP-Chromophore for HTS Assay Application
Binding of the UDP-chromophore to AfUGM was determined by varying the concentration of the enzyme at a constant concentration of the UDP-chromophores (15 nM) (Figure 5). Binding assays with the UDP-fluorescein probes (chromophores 1 and 2) show that these ligands bind weakly to AfUGM, with K d values of ~15 μM (Figure 5(a)). This relatively low affinity impedes the utilization of these chromophores for a high-throughput FP binding assay, as it will require high quantities of enzyme. Interestingly, we tested the binding of these chromophores to bacterial UGM from M. tuberculosis, and the K d value of chromophore 2 was 0.10 ± 0.01 μM, consistent with previously published values (Table 1) [15]. This tighter binding suggests differences in the active-site architecture between the prokaryotic and A. fumigatus UGM enzymes. This is also consistent with our recent report on binding assays monitoring flavin fluorescence that showed that AfUGM binds UDP-glucose 5 times tighter than K. pneumoniae UGM. Similarly, binding of UDP-Galp to AfUGM was not detected although UDP-Galp binds to the bacterial enzyme with a K d value of 220 μM [7, 17]. These differences in ligand binding might originate from the low amino acid identity between the bacterial and eukaryotic UGMs (<18%). Furthermore, we have shown that the quaternary structure between these enzymes is not conserved as the bacterial enzymes have been shown to function as homodimers, while AfUGM functions as a homotetramer [7].
Figure 5.
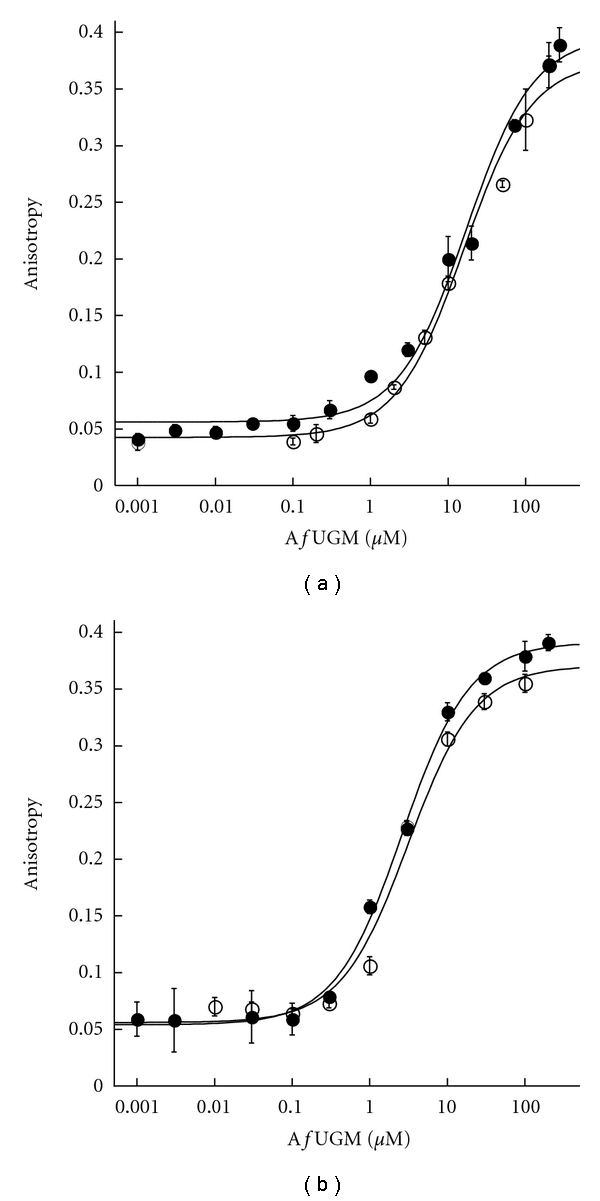
FP binding assay to determine K d of the chromophores. (a) Chromophores 1 (○) and 2 (⬤) (excitation at 492 nm and emission at 524 nm). (b) Chromophores 3 (⬤) and 4 (○) (excitation at 544 nm and emission at 584 nm).
Table 1.
K d values of UDP-fluorescent probes.
| Chromophore | K d for AfUGM (μM) | K d for MtUGM (μM) |
|---|---|---|
| 1 | 17 ± 3 | >30 |
| 2 | 16 ± 3 | 0.10 ± 0.01 |
| 3 | 2.6 ± 0.2 | 0.73 ± 0.07 |
| 4 | 3.0 ± 0.7 | >30 |
With the UDP-TAMRA analogs (chromophores 3 and 4), the binding to AfUGM was ~6 times better than with the UDP-fluorescein analogs, and significant anisotropy change was measured (Figure 5(b)). Interestingly, the length of the linker had little or no effect on the binding affinities (Table 1), suggesting that with AfUGM the interaction between the chromophore and some components of the active site or perhaps directly with the flavin cofactor play a major role in binding. Compound 3 and 4 bound to MtUGM with similar affinities as chromophores 1 and 2, respectively. In contrast to AfUGM, in the bacterial enzymes, the length of the linker plays a major role in binding with longer linkers increasing the affinity, further demonstrating that the active-site architecture varied among the UGM enzymes. We selected 3 as the FP probe for further characterization of the binding assay.
3.3. Determination of Competitive Binding Using FP Assay
FP competitive inhibition binding assay was conducted to confirm that the FP probes bind to the active site on AfUGM. First, the Z′ factor as a function of AfUGM was determined to establish the proper enzyme concentration to be used in the assay. The Z′ factor is a statistical parameter that reports on the quality of the assay [18]. As shown in Figure 6, the FP assay exhibits excellent quality at an AfUGM concentration higher than 2 μM with a Z′ factor above 0.8. The minimum value (2 μM) in this range was selected as the optimal assay concentration.
Figure 6.
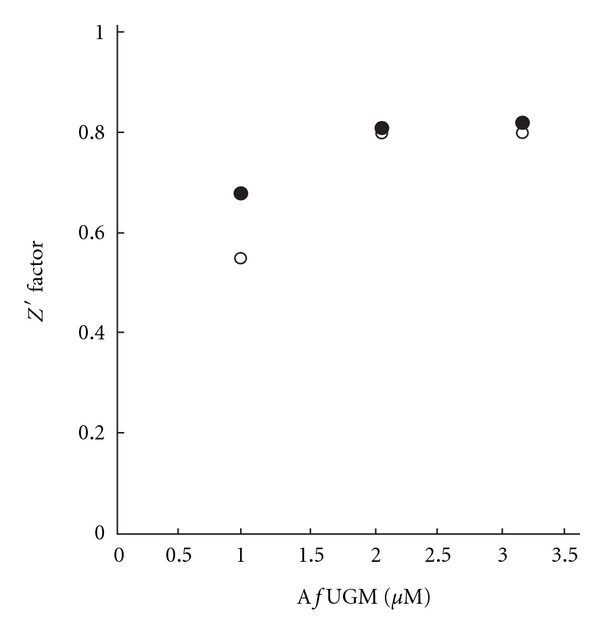
Determination of optimal AfUGM concentration to use in the FP assay with chromophore 3 (⬤) and chromophore 4 (○).
The K d for UDP was determined using the FP assay by titrating AfUGM with serial dilutions of UDP. A value of 9.0 ± 1.7 μM was obtained, which is in good agreement with the K d (33 ± 9 μM) previously determined by directly monitoring the flavin fluorescence (Figure 7(a)) [7].
Figure 7.
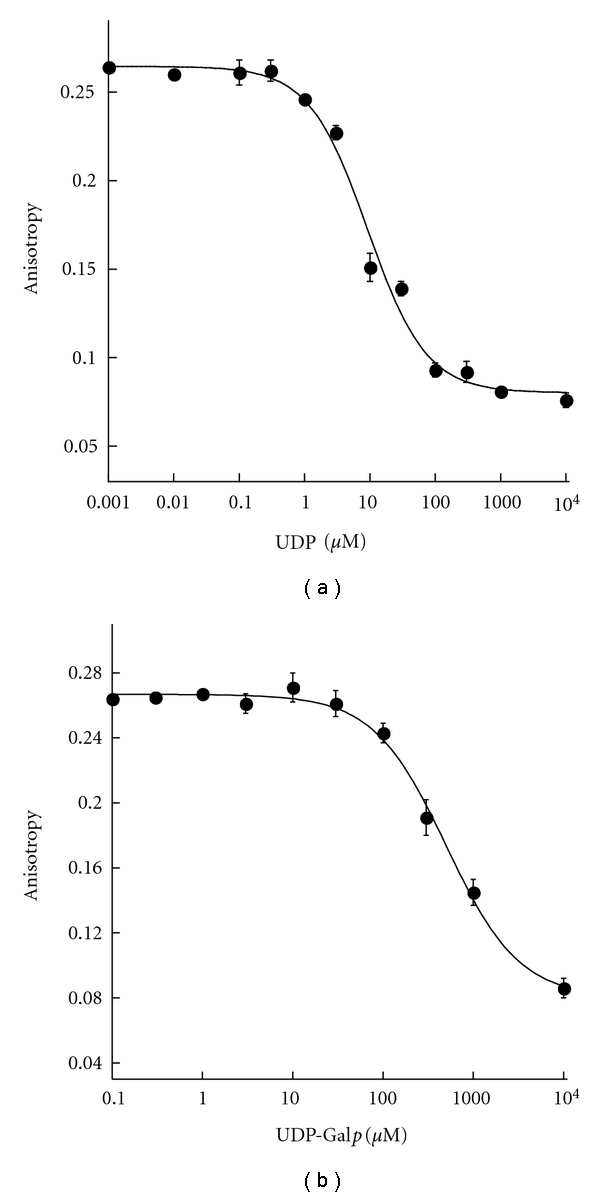
FP competitive binding assay with UDP (a) and UDP-Galp (b).
UDP-Galp was the second ligand tested in the FP inhibition assay, and a K d value of 495 ± 66 μM was calculated (Figure 7(b)), indicating that UDP-Galp is a poor ligand for AfUGM, which agrees well with the observation previously reported by Oppenheimer et al. [7].
Recently, a series of prokaryotic UGM inhibitors were identified from chemical libraries by high-throughput screening (HTS) [14]. In our FP assay, we tested two of the best prokaryotic UGM inhibitors, compound 7 and compound 8 (Figure 8). Interestingly, they behaved differently on AfUGM. Compound 7 turned out to be a poor ligand for AfUGM with a K d of 140 ± 9 μM (Figure 9(a)). In contrast, compound 8 exhibits much better binding to AfUGM (Figure 9(b)), and its K d was found to be 11 ± 0.4 μM (Table 2). We also tested these two compounds in a secondary assay, directly monitoring the activity of AfUGM to see if these molecules function as inhibitors. The HPLC chromatograms (Figure 10) indicated that both of the compounds inhibit the activity of AfUGM. These results confirm that the FP assay identifies ligands that bind to the active site of AfUGM and that these molecules inhibit the activity of the enzyme in a secondary assay that directly measures product formation.
Figure 9.
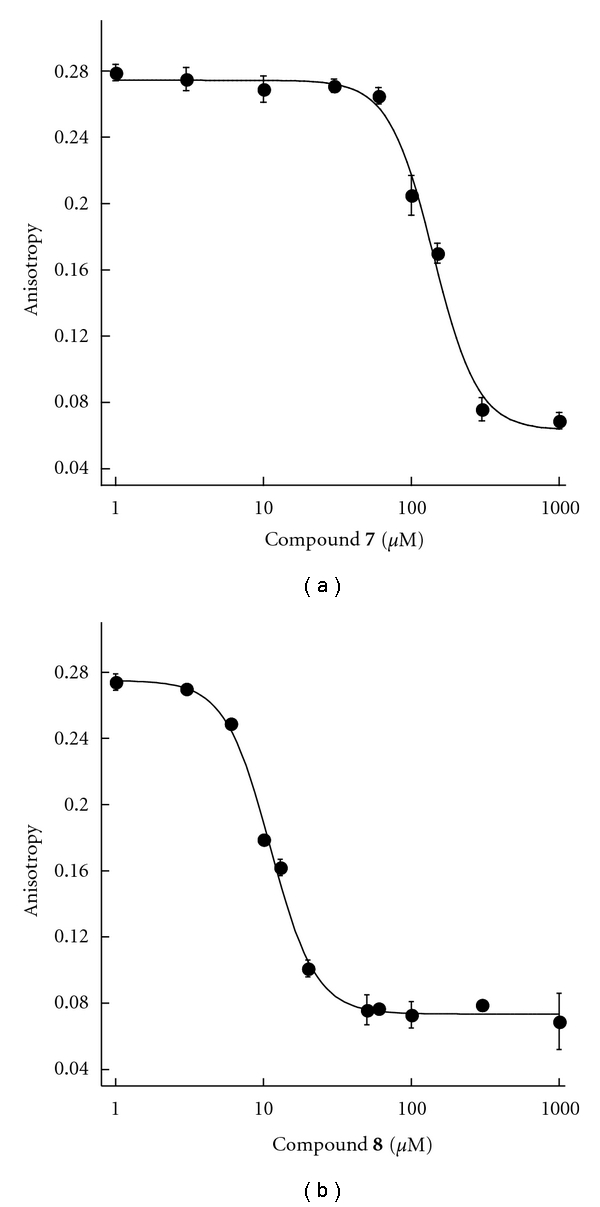
FP inhibition assay with compounds 7 (a) and 8 (b).
Table 2.
K d values of UGM ligands.
| Ligand | K d for AfUGM (μM) | K d for MtUGM (μM) |
|---|---|---|
| UDP | 9.0 ± 1.7 | 15 ± 2 |
| UDP-Galp | 495 ± 66 | 563 ± 75 |
| 7 | 140 ± 9 | 21 ± 1 |
| 8 | 11 ± 0.4 | 25 ± 2 |
Figure 10.
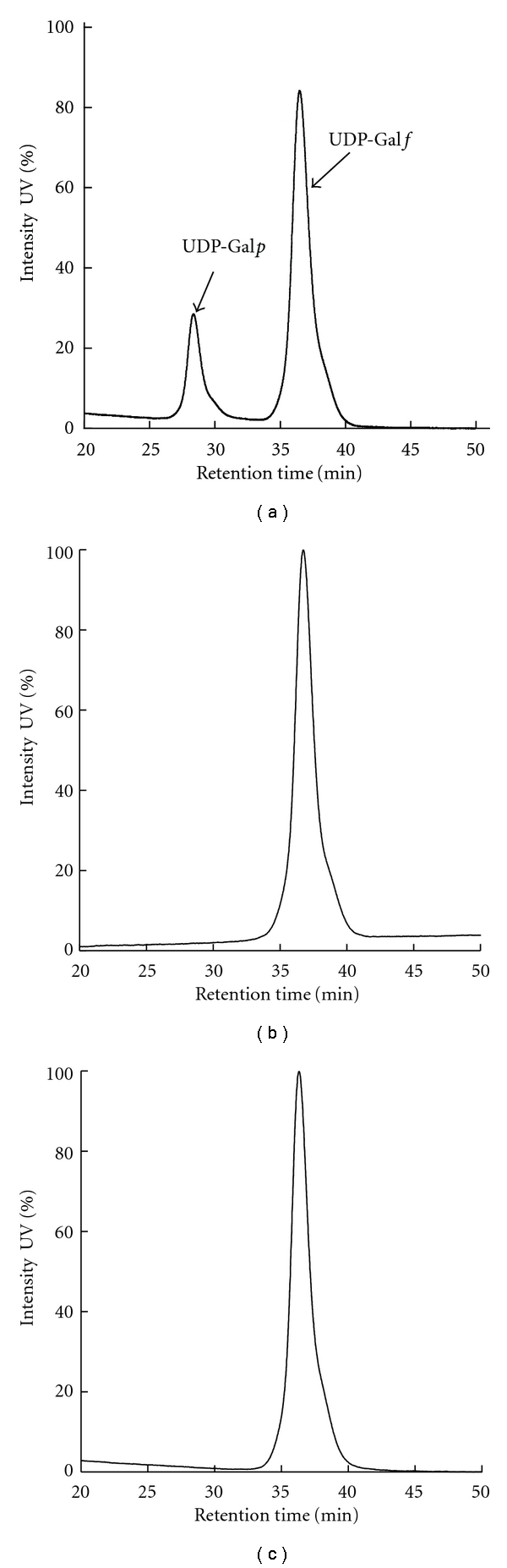
AfUGM activity assay. The HPLC chromatograms at 262 nm are shown. (a) AfUGM activity in the absence of inhibitor. (b) In the presence of 7 (500 μM). (c) In the presence of 8 (50 μM).
3.4. FP Assay Quality
The Z′ factor value using chromophore 3 was calculated to be 0.79 ± 0.02. An assay with a Z′ factor greater than 0.5 is considered a good assay; therefore, our FP assay is suitable for HTS (Figure 6). We also estimated the tolerance of the FP assay to DMSO by calculating the Z′ factors at various DMSO concentrations, because a majority of compounds in HTS libraries are dissolved in DMSO. The Z′ factors were plotted against DMSO concentrations to generate a DMSO calibration curve (Figure 11), and our assay maintains excellent quality with DMSO concentration up to 5% (v/v).
Figure 11.
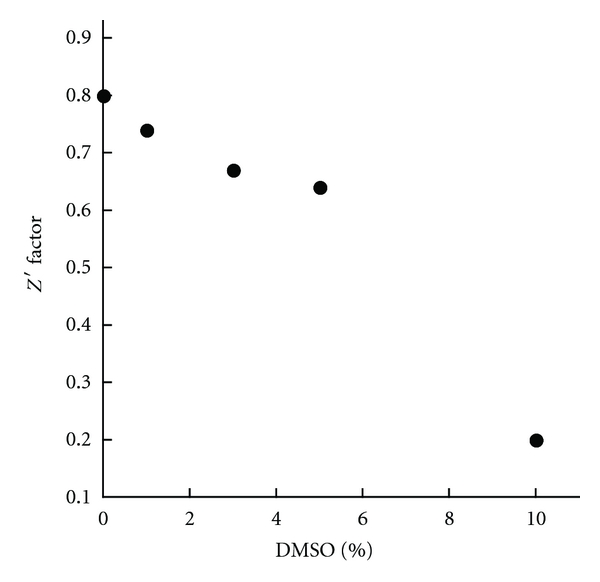
Tolerance to DMSO.
4. Conclusion
In conclusion, four fluorescently labeled UDP derivatives (1–4) were synthesized and tested for binding to AfUGM. Different from the bacterial UGM, the nature of the chromophore enhanced binding to AfUGM while the length of the linkers did not. UDP-TAMRA analogs (3 and 4) bind to AfUGM with high affinities. Binding of chromophore 3 to the active site of AfUGM was demonstrated by a competition experiment using UDP and UDP-Galp. Furthermore, binding of known inhibitors of bacterial UGM was tested against AfUGM, and it was found that these compounds bound AfUGM, however, with lower affinities. Inhibition of AfUGM, measuring product formation by HPLC, was demonstrated with compounds 7 and 8. A Z′ factor of 0.79 was calculated, and the assay was shown to exhibit good tolerance to DMSO. We expect that the FP assay described here will allow fast identification of AfUGM inhibitors from chemical libraries. We believe that inhibitors of AfUGM that block the biosynthesis of Galf could lead to novel therapeutics against A. fumigatus-related diseases.
Supplementary Material
The Supporting Material includes the UV-visible spectrum and SDS-PAGE of AfUGM, and 1H NMR and High-resolution MS spectra of the four UDP chromophores. UV-visible spectrum was obtained on Agilent photospectrometer. NMR spectral data were obtained using a JEOL Eclipse spectrometer at 500 MHz, or a Varian Inova spectrometer at 400 MHz. Chemical shifts were reported as σ-values relative to known solvent residue peaks. High-resolution mass spectra (HRMS) were obtained in the Mass Spec Incubator of Department of Biochemistry at Virginia Tech.
Acknowledgments
This work was supported in part by NIH Grants RO1 GM094469 (P. Sobrado, PI) and RO1 AI082542 (R. Tarleton, PI). M. Oppenheimer was supported by a fellowship from the American Heart Association.
References
- 1.Trof RJ, Beishuizen A, Debets-Ossenkopp YJ, Girbes ARJ, Groeneveld ABJ. Management of invasive pulmonary aspergillosis in non-neutropenic critically ill patients. Intensive Care Medicine. 2007;33(10):1694–1703. doi: 10.1007/s00134-007-0791-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kradin RL, Mark EJ. The pathology of pulmonary disorders due to Aspergillus spp. Archives of Pathology and Laboratory Medicine. 2008;132(4):606–614. doi: 10.5858/2008-132-606-TPOPDD. [DOI] [PubMed] [Google Scholar]
- 3.Chong S, Kim TS, Koh WJ, Cho EY, Kim K. Invasive pulmonary aspergillosis complicated by pulmonary artery occlusion in an immunocompetent patient. Clinical Radiology. 2006;61(3):287–290. doi: 10.1016/j.crad.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 4.Virnig C, Bush RK. Allergic bronchopulmonary aspergillosis: a US perspective. Current Opinion in Pulmonary Medicine. 2007;13(1):67–71. doi: 10.1097/MCP.0b013e328010c812. [DOI] [PubMed] [Google Scholar]
- 5.Chamilos G, Luna M, Lewis RE, et al. Invasive fungal infections in patients with hematologic malignancies in a tertiary care cancer center: an autopsy study over a 15-year period (1989–2003) Haematologica. 2006;91(7):986–989. [PubMed] [Google Scholar]
- 6.Latge JP. Galactofuranose containing molecules in Aspergillus fumigatus . Medical Mycology. 2009;47(1):S104–S109. doi: 10.1080/13693780802258832. [DOI] [PubMed] [Google Scholar]
- 7.Oppenheimer M, Poulin MB, Lowary TL, Helm RF, Sobrado P. Characterization of recombinant UDP-galactopyranose mutase from Aspergillus fumigatus . Archives of Biochemistry and Biophysics. 2010;502(1):31–38. doi: 10.1016/j.abb.2010.06.035. [DOI] [PubMed] [Google Scholar]
- 8.Schmalhorst PS, Krappmann S, Vervecken W, et al. Contribution of galactofuranose to the virulence of the opportunistic pathogen Aspergillus fumigatus . Eukaryotic Cell. 2008;7(8):1268–1277. doi: 10.1128/EC.00109-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stynen D, Goris A, Sarfati J, Latge JP. A new sensitive sandwich enzyme-linked immunosorbent assay to detect galactofuran in patients with invasive Aspergillosis. Journal of Clinical Microbiology. 1995;33(2):497–500. doi: 10.1128/jcm.33.2.497-500.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costachel C, Coddeville B, Latgé JP, Fontaine T. Glycosylphosphatidylinositol-anchored fungal polysaccharide in Aspergillus fumigatus . Journal of Biological Chemistry. 2005;280(48):39835–39842. doi: 10.1074/jbc.M510163200. [DOI] [PubMed] [Google Scholar]
- 11.Simenel C, Coddeville B, Delepierre M, Latgé JP, Fontaine T. Glycosylinositolphosphoceramides in Aspergillus fumigatus . Glycobiology. 2008;18(1):84–96. doi: 10.1093/glycob/cwm122. [DOI] [PubMed] [Google Scholar]
- 12.Nassau PM, Martin SL, Brown RE, et al. Galactofuranose biosynthesis in Escherichia coli K-12: identification and cloning of UDP-galactopyranose mutase. Journal of Bacteriology. 1996;178(4):1047–1052. doi: 10.1128/jb.178.4.1047-1052.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engel J, Schmalhorst PS, Dörk-Bousset T, Ferrières V, Routier FH. A single UDP-galactofuranose transporter is required for galactofuranosylation in Aspergillus fumigatus . Journal of Biological Chemistry. 2009;284(49):33859–33868. doi: 10.1074/jbc.M109.070219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carlson EE, May JF, Kiessling LL. Chemical Probes of UDP-Galactopyranose Mutase. Chemistry and Biology. 2006;13(8):825–837. doi: 10.1016/j.chembiol.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Dykhuizen EC, Kiessling LL. Potent ligands for prokaryotic UDP-galactopyranose mutase that exploit an enzyme subsite. Organic Letters. 2009;11(1):193–196. doi: 10.1021/ol802094p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haugland RP. Handbook of Fluorescent Probes and Research Chemicals. Eugene, Ore, USA: Molecular Probes; 1996. [Google Scholar]
- 17.Yao X, Bleile DW, Yuan Y, et al. Substrate directs enzyme dynamics by bridging distal sites: UDP-galactopyranose mutase. Proteins. 2009;74(4):972–979. doi: 10.1002/prot.22206. [DOI] [PubMed] [Google Scholar]
- 18.Zhang JH, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of Biomolecular Screening. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supporting Material includes the UV-visible spectrum and SDS-PAGE of AfUGM, and 1H NMR and High-resolution MS spectra of the four UDP chromophores. UV-visible spectrum was obtained on Agilent photospectrometer. NMR spectral data were obtained using a JEOL Eclipse spectrometer at 500 MHz, or a Varian Inova spectrometer at 400 MHz. Chemical shifts were reported as σ-values relative to known solvent residue peaks. High-resolution mass spectra (HRMS) were obtained in the Mass Spec Incubator of Department of Biochemistry at Virginia Tech.