

Abstract
Although microbes have been classically viewed as pathogens, it is now well established that the majority of host-bacterial interactions are symbiotic. During development and into adulthood, gut bacteria shape the tissues, cells and molecular profile of our gastrointestinal immune system. This partnership, forged over many millennia of co-evolution, is based on a molecular exchange involving bacterial signals that are recognized by host receptors to mediate beneficial outcomes for both microbe and man. We explore herein how specific aspects of the adaptive immune system are influenced by intestinal commensal bacteria. Understanding the molecular mechanisms which mediate symbiosis between commensal bacteria and humans may redefine how we view the evolution of adaptive immunity, and as a result, the way we approach treating numerous immunologic disorders.
We are (fortunately) not alone- humans provide residence to numerous microbial communities comprised of hundreds of individual bacterial species. Although teleological design may predict that the immune system evolved to eliminate infectious microbes, we now know that almost every environmentally exposed surface of our bodies is teeming with symbiotic microbes (Fig. 1). These polymicrobial communities contribute profoundly to the architecture and function of the tissues they inhabit, and thus play an important role in the balance between health and disease. The notion that commensal microbes critically affect tissue and cell development in humans can be rationalized when this process is viewed from an evolutionary perspective. Bacteria populated the Earth 2 billion years before the first signs of eukaryotic life, and occupy almost every terrestrial and aquatic niche on our planet. Mitochondria and chloroplasts of eukaryotic cells are descended from bacteria, suggesting that bacteria may have had an active role in the evolution of higher organisms. As multicellular metazoans evolved more complex body plans, bacteria acquired the ability to inhabit new anatomical niches. Animals represent a stable, nutrient rich ecosystem for microbes to thrive; hence host health is paramount to the microbiota. In turn, the host benefits from a diverse commensal microbiota which help digest complex carbohydrates and provide essential nutrients to mammals. Symbionts are not the only microbes the host encounters, however. An important challenge faced by the host immune system is distinguishing between beneficial and pathogenic microbes, because both share similar molecular patterns that are recognized by the innate immune system (such as lipopolysaccharide, peptidogycan, lipoproteins, flagellin). Discrimination between specific microbes may be a feature of the adaptive immune system, which can recognize discrete molecular sequences and mount both pro- and anti-inflammatory responses depending on the nature of the antigen. In particular, CD4+ T cells are quite plastic and differentiate into numerous subsets after development in the thymus, and thus are capable of sensing environmental cues from the microbiota. As adaptive immunity evolved in higher vertebrates, the ability of this system to recognize and respond to specific microorganisms may have been driven by evolutionary forces provided by the microbiota itself, resulting in immune functions beyond simply clearing microbial pathogens (which in theory also helps the microbiota by improving host health). Recent evidence shows that the commensal microbiota ‘programs’ many aspects of T cell differentiation, thus augmenting the developmental instructions of the host genome to engender the full function of the adaptive immune system.
Fig. 1.
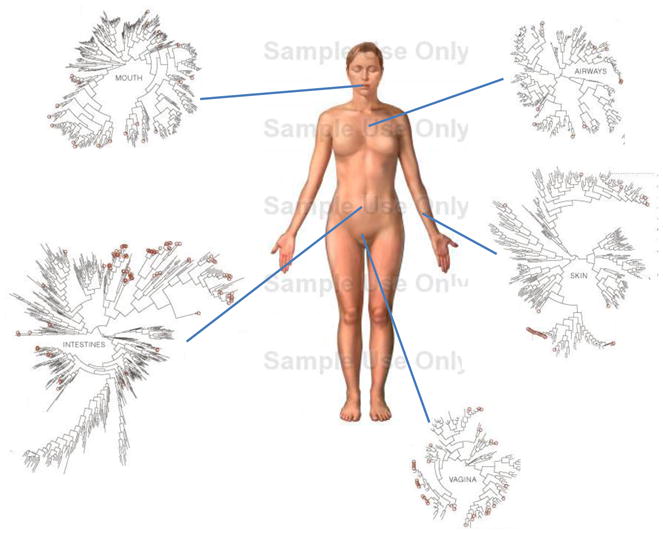
The microbiome of various anatomical location of the human body. Numerous bacterial species colonize the mouth, upper airways, skin, vagina and intestinal tract of humans. The phylogenic trees show the speciation of bacterial clades from common ancestors at each anatomical site. Although the communities in different regions of the body share similarities, they each have a unique site-specific ‘fingerprint’ made of many distinct microbes. Each site has a very high level of diversity, as shown by the individual lines on the dendograms. Data is from the NIH funded Human Microbiome Project, and circles represent bacterial species whose sequences are known. NOTE TO SCIENCE EDITORS: the figure of the body was taken from GOOGLE images and the phylogenic trees were taken from the Human Microbiome Project. Please address any copyright issues and/or redraw figures.
This article will review concepts derived from gnotobiology (Greek for ‘known life’; ‘gnosis’ meaning knowledge and ‘bios’ meaning life) to unravel how commensal bacteria promote the development and function of adaptive immunity. In particular, we will explore how CD4+ T-helper cell subsets within the gastrointestinal and systemic immune system are shaped (perhaps even controlled) by our microbiota, and theorize how and why gut bacteria evolved to so profoundly influence immunologic wellbeing. Understanding human co-evolution with our microbiota may lead to a philosophical and conceptual redefinition of the microbial world, and provide clinical advances toward the treatment of autoimmunity and inflammatory diseases by harnessing the immunomodulatory properties of human commensal bacteria.
How Does the Microbiota Shape Host Immune Development and Function?
Although microbes reside in several anatomical locations including the skin, vagina and mouth, the lower gastrointestinal tract of mammals harbors the greatest density and diversity of commensal microorganisms. These include bacteria, archaea, fungi, viruses, protozoans and in some cases, multicellular helminthes; however, bacteria predominate and reach 100 trillion microbial cells in the colon. Recent efforts to sequence the bacterial genomes of the microbiota (known as the microbiome), have begun to reveal its genetic identity (1) and suggest our microbiome contains over 150 times the number of non-redundant genes than in the human genome (2). For decades, microbiological techniques to culture bacteria in the laboratory have only identified cultivatable organisms, which represent a minority of the microbial species of the gut. The aggregate human microbiota likely contains between 1,000–1,150 bacterial species (spread among all people sampled), with each person harboring approximately 160 bacterial species (2). This suggests an individual’s microbiome is relatively distinct in composition and adaptable to environmental changes and/or host genetics.
Germ-free animals (born and raised in the absence of all microbes) provide important insights into how the microbiota affects the host immune system. The development of gut-associated lymphoid tissues (GALT), the first line of defense for the intestinal mucosa, is defective in germ-free mice. Germ-free mice display fewer and smaller Peyer’s patches, smaller and less cellular mesenteric lymph nodes (MLNs) and less cellular lamina propria of the small intestine compared to animals with a microbiota (3–7). Besides developmental defects in tissue formation, the cellular and molecular profile of the intestinal immune system is also compromised in germ-free mice. In germ-free mice, intestinal epithelial cells (IECs), which line the gut and form a physical barrier between luminal contents and the immune system, exhibit reduced expression of toll-like receptors (TLRs) and major histocompatibility molecule II (MHC II) (8–9), which are involved in pathogen sensing and antigen presentation, respectively. Interspersed between epithelial cells is a specialized population of T cells known as intra-epithelial lymphocytes (IELs). IELs from germ-free mice are reduced in number, and their cytotoxicity is compromised (10–11). Microbial colonization expands specific subsets of intestinal γδ T cells (12). Germ-free mice also have reduced numbers of CD4+ T cells in the lamina propria (13). The development of isolated lymphoid follicles (ILFs), specialized intestinal structures made of mostly dendritic cells (DC) and B cell aggregates, is also dependent on the microbiota (14). Therefore, multiple populations of intestinal immune cells require the microbiota for their development and their function.
The absence of a microbiota also leads to several extra-intestinal defects, including reduced numbers of CD4+ T cells in the spleen, fewer and smaller germinal centers within the spleen and reduced systemic antibody levels, which suggests that the microbiota are capable of shaping systemic immunity (15–17). Beyond development, the microbiota also influences functional aspects of intestinal and systemic immunity, including pathogen clearance. Germ-free mice are more susceptible to infectious agents such as Shigella flexneri, Bacillus anthracis and Leishmania (18). Peptidoglycan from the microbiota enhances neutrophil cytotoxicity following systemic infections by Streptococcus pneumoniae and Staphylococcus aureus (19). During systemic challenge of mice with Listeria monocytogenes, germ-free mice harbored an increased bacterial burden in the liver, spleen and peritoneal cavity (20). Moreover, trafficking of T lymphocytes to the peritoneal cavity in response to Listeria infection is impaired in germ-free mice.
The contributions of the microbiota to the development and function of the immune system appear to be fundamental. A more robust immune system, equipped with a diverse arsenal of cells and molecules, is better able to combat microbial pathogens and ultimately provides a healthier residence for commensal bacteria. This view implies that host mechanisms and the microbiota may have evolved to collaborate against infectious agents. Indeed, several reports now show an antagonistic relationship between overt pathogens and the microbiota. For example, Salmonella trigger intestinal inflammation, which reduces the numbers and diversity of the microbiota, a process which promotes bacterial infection (21). Depletion of the microbiota diminishes intestinal immune responses that help control enteric infections by Citrobacter rodentium and Campylobacter jejuni (22). Given the role of the microbiota in immune system function, harnessing the immunomodulatory capabilities of the microbiota may offer novel avenues for development of anti-microbial therapies for infectious disease.
How does the Microbiota Provide Signals to Instruct Peripheral Regulatory T cell Differentiation?
Although many cell types are influenced by the microbiota, we will focus here on the emerging role of the microbiota on effector CD4+ T cell differentiation. After lineage commitment in the thymus, naïve CD4+ T cells enter the periphery where they sense environmental signals that further instruct their maturation and function. During an infection, microbial and host signals provide cues to naïve CD4+ T cells to induce their differentiation into various pro- and anti-inflammatory subsets. For instance, infection by intracellular pathogens drives the development of T-helper 1 (Th1) cells, whereas extracellular pathogens induce the differentiation of Th2 and Th17 subsets (23). These pro-inflammatory cells coordinate many aspects of the innate and adaptive immune response to clear microbial invaders. CD4+ T cells can also adopt an anti-inflammatory phenotype. Regulatory T cells (Tregs) control unwanted immune system activation and dampen inflammation after microbial infection. Expression of the Treg cell-specific transcription factor Foxp3 (forkhead box P3) induces regulatory phenotypes and functions by CD4+ T cells (24). Foxp3+ T cells develop in the thymus shortly after birth, and deletion or depletion of Foxp3+ T cells leads to severe multi-organ lymphoproliferative disease and autoimmunity (25). Besides the thymus-derived CD4+Foxp3+ T cells (‘natural’ Tregs), various subsets of Tregs can be generated in the gut from naïve T cells (‘inducible’ Tregs), some of which produce the anti-inflammatory cytokine interleukin-10 (26). Moreover, intestinal bacteria may be critically involved in the differentiation of some gut Treg subsets (29–31). Accordingly, several commensal bacteria (e.g., Bifidobacteria infantis, Faecalibacterium prausnitzii) have been shown to induce Foxp3+ Tregs and IL-10 production in the gut (32–33).
Members of the genus Bacteroides are prominent in the mammalian gastrointestinal tract, and are also potent stimulators of the mucosal immune system of mammals (34). The gut microorganism Bacteroides fragilis has emerged as a model system for the study of immune-bacterial symbiosis. During colonization of mice with B. fragilis, the bacterial molecule Polysaccharide A (PSA) directs the cellular and physical development of the immune system (16). Moreover, B. fragilis is able to prevent intestinal pathology in two independent models of experimental colitis in a PSA-dependent manner (35). Furthermore, in mouse models of experimental colitis, oral treatment of mice with purified PSA protects against weight loss, decreases pro-inflammatory cytokine expression in the gut, and inhibits lymphocyte infiltration that is associated with disease. The protective effects of PSA were likely mediated by CD4+ T cell production of IL-10 because CD4+ T lymphocytes from mesenteric lymph nodes of PSA treated mice produced elevated amounts of IL-10. IL-10-deficient CD4+ T cells abolished the protective effects of PSA in both colitis models. These studies identify PSA as a beneficial microbial molecule, which suppresses inflammation-driven host pathology.
No consensus has been reached about whether Foxp3+ Treg cells in the intestinal tissues of germ-free mice are defective (36–40); however, production of IL-10 is reduced within the GALT of germ-free animals (13, 36, 41). Foxp3+ Treg cells in the colon of germ-free mice exhibit reduced IL-10 expression, and colonization of germ-free mice with PSA-producing bacteria (but not PSA-deficient B. fragilis) restores IL-10 expression (42). PSA increases the Foxp3 expression by Treg cells, and colonization of germ-free animals with B. fragilis augments the in vitro suppressive activity of Tregs in a PSA-dependent manner (42). PSA protects and cures animals from experimental colitis by inducing Foxp3+ Treg cells and IL-10 production. These findings imply that optimal Foxp3+ Treg cell differentiation in the colon requires signals from the microbiota and the host genome. They also suggest specific commensal bacteria may have evolved to promote Treg cell differentiation in the gut to actively engender mucosal tolerance to the host. If validated in human disease, these findings may lead to probiotic therapies for colitis based on microbial-driven Treg induction.
How does the Microbiota Instruct T-helper Cell Differentiation?
Although the microbiota has been shown to affect the Th1/Th2 balance in systemic immune compartments (43), studies have not yet observed symbiotic microbial effects on Th1 or Th2 cells at mucosal surfaces. In contrast, Th17 cell development in the gut is specifically impacted by commensal bacteria. Germ-free mice are deficient in the production of IL-17 from CD4+ T cells (the hallmark cytokine of Th17 cells) of the small intestinal lamina propria (39). Only a minor defect was noted for γδ T cells, which suggests that the lack of Th17 cells was not due to an overall deficiency in immune activation, and that specific features of the immune response are sensitive to the microbiota. One mechanism of intestinal Th17 cell differentiation may be production of Adenosine 5′-triphosphate (ATP) in the lamina propria by commensal bacteria, which drives the production of Th17-inducing cytokines by resident lamina propria cells (45). Germ-free animals display a reduction in fecal ATP amounts and treatment of mice with a non-hydrolysable ATP analogue increased the number of gut Th17 cells (45).
Not all bacterial species of the microbiota are similar in their ability to promote non-pathogenic T cell responses during normal colonization of animals. Of the numerous bacterial phylotypes that comprise the normal microbiota of mice, only segmented filamentous bacteria (SFBs) have been shown to direct intestinal T-helper cell development. A role for SFBs was identified by reconstituting germ-free mice with various subsets of bacterial consortia and measuring cytokine production in gut mucosal tissues (41). SFBs, which are known to tightly adhere to the intestinal mucosa (and to Peyer’s patches of the ileum), induced the development of T-helper cells in the lamina propria and in cell aggregates of Peyer’s patches. This activity was not found in even very complex groups of bacteria tested if they were missing SFBs. In a contemporary report, a comparison of the microbiota of mice that contained Th17 cells with mice deficient in these cell types identified SFBs as being sufficient to restore Th17 cells to germ-free mice and conventionally raised mice that lack Th17 cells (46). Gene expression analysis showed that SFBs induce a spectrum of intestinal immune responses including production of cytokines and chemokines, antimicrobial peptides, and serum amyloid A (SAA), which was shown in vitro to support Th17 cell differentiation. SFB colonization protected animals from intestinal infection with Citrobacter rodentium, a bacterial pathogen of animals that causes acute intestinal inflammation similar to enteropathogenic Escherichia coli (EPEC) in humans. Thus, commensal SFBs induce a tonic (or controlled) inflammatory response in the gut through Th17 cell development that does not cause pathology and is protective against infection with pathogenic bacteria. These new studies build on research done several decades ago that SFBs promote germinal center development, mucosal IgA responses and recruitment of intraepithelial lymphocytes (47–49). Collectively, it appears that only a particular subset of bacteria from the gut microbiota directly influences Th17 immune responses.
Are Non-infectious Human Diseases Influenced by the Microbiota?
Numerous autoimmune diseases result from dysregulation of the adaptive immune system. The incidences of autoimmune diseases such as multiple sclerosis (MS), type 1 diabetes (T1D) and rheumatoid arthritis (RA) are rapidly increasing in Western societies, suggesting alterations in environment factors that regulate the adaptive immune system. As appreciation for the immunomodulatory potentials of commensal bacteria have increased, we and others have proposed that lifestyle changes have caused a fundamental alteration in our association with the microbial world (50–51). Altered diets, widespread antibiotic use and other societal factors in developed countries may result in an unnatural shift in the community composition of a ‘healthy’ microbiota, leading to altered microbial colonization known as dysbiosis. Whether dysbiosis causes any human disease is yet unproven (insights may come from microbiome sequencing projects); however, evidence in mice suggests dysbiosis may affect autoimmunity by altering the balance between toleragenic and inflammatory members of the microbiota (Fig. 2). PSA from B. fragilis, previously shown to treat experimental colitis in the gut, is also able to prevent and cure experimental autoimmune encephalomyelitis (EAE; an animal model for multiple sclerosis) (52). Oral treatment of animals with PSA reduced Th17 cell development and increased Treg numbers in the central nervous system (CNS). Furthermore, germ-free animals display reduced Th17 cell numbers in the spleen and spinal cords, and do not develop RA or EAE (inflammation in joints and the CNS, respectively) (53–54). The inflammatory responses in both RA and EAE are promoted by Th17 cells and prevented by Tregs, which suggests that the effects of gut bacteria on the adaptive immune system likely extend beyond the gastrointestinal tract to influence autoimmune diseases that are seemingly unrelated to microbial infections.
Fig. 2.

How the microbiome and the human genome contribute to inflammatory disease. In a simplified model, the community composition of the human microbiome helps to shape the balance between immuneregulatory (Treg) and pro-inflammatory (Th17) T cells. The molecules produced by a given microbiome network work with the molecules produced by the human genome to determine this equilibrium. (A) In a healthy microbiome, there is an optimal proportion of both pro- and anti-inflammatory organisms (represented here by SFBs and B. fragilis), which provide signals to the developing immune system (controlled by the host genome) that leads to a balance of Treg and Th17 cell activities. In this scenario, the host genome can contain ‘autoimmune specific’ mutations (represented by the stars), but disease does not develop. (B, C) The genome of patients with multiple sclerosis, type I diabetes, rheumatoid arthritis and Crohn’s disease contain a spectrum of variants that are linked to disease by genome wide association studies (reviewed in (61)). Environmental influences, however, are risk factors in all of these diseases. Altered community composition of the microbiome due to lifestyle, known as dysbiosis, may represent this disease modifying component. An increase in pro-inflammatory microbes, for example SFBs in animal models, may promote Th17 cell activity to increase and thus predispose genetically susceptible people to Th17-mediated autoimmunity (B). Alternatively, a decrease or absence in anti-inflammatory microbes, for example B. fragilis in animal models, may lead to an under-development of Treg cell subsets (C). The imbalance between Th17 cells and Tregs ultimately leads to autoimmunity.
Why only specific commensal bacteria induce Th17 cell differentiation remains unclear. Th17 responses are critical at mucosal surfaces to control infections by extracellular pathogens. IL-17 production recruits neutrophils to the site of infection and induces antimicrobial peptide expression and other mediators of immunity. If there is an evolutionary rationale for the ability of SFBs to induce Th17 cell differentiation, one interpretation is that they mediate a state of ‘controlled inflammation’, which prepares the gastrointestinal tract for an invading pathogen. SFBs are not overt pathogens and colonize animals as symbionts, and thus Th17 induction may lead to more enhanced immune responses that protect against acute infectious agents (such as C. rodentium). Besides this beneficial outcome, it appears that SFB colonization also leads to adverse host effects. Th1 and Th17 cells of the adaptive immune system promote autoimmunity. As a result, microbes which stimulate T-helper cell development may (inadvertently) also increase the inherent immune reactivity of the host, potentially leading to host-destructive pathologies mediated by the adaptive immune system. This notion is supported by a role for SFBs in promoting RA and EAE during induced animal models, both of which involve Th17 cell inflammation (53–54). The enhancement of RA and EAE by SFBs establishes that the microbiota can adversely influence autoimmune disease outside the gut. Therefore, SFBs can colonize healthy animals without causing illness; but when the host is immunocompromised or under inflammatory conditions, SFBs can be detrimental. We propose that certain microbes, such as SFBs, which can peacefully co-exist with a healthy host but still retain pathogenic potential be termed ‘pathobionts’, to distinguish them from opportunistic pathogens which are acquired from the environment and cause acute infections (55). Pathobionts may represent organisms on the evolutionary continuum between an acute pathogen and commensal, whose sustained relationships with the host induces the development of additional layers of mucosal defense while promoting the unwanted side effect of autoimmune disease. The significance of Th17 cell-inducing organisms such as SFBs to animal models of autoimmunity remains to be further established, as caveats exist such as the fact that animals from colonies devoid of SFBs can develop autoimmune disease. Also, how the microbiota may contribute to human autoimmunity needs to be determined. SFB colonization of animals, however, does provide a model system for testing concepts linking specific gut bacteria to non-intestinal immune disorders. The identification of bacterial molecules required for SFBs to induce Th17 cell responses may reveal why this particular organism is capable of promoting the development of pro-inflammatory T cells. Furthermore, studies that delineate the gene regulatory networks induced by SFB colonization may enhance our understanding of the evolutionary forces that resulted in Th17 lineage development.
Autoimmune diseases such as MS, T1D and RA are associated with a spectrum of genetic polymorphisms as shown by recent genome wide association studies. Given that concordance rates for disease among monozygotic twins averages between 20–40%, environmental factors are crucial for the manifestations of symptoms (56). We predict that autoimmunity can result from the combination of an altered human genome and an altered microbiome (Fig. 2). Patients with autoimmunity likely have a genetic landscape that predisposes them to self-reactivity, and in some cases, certain gut bacteria may promote disease by activating the adaptive immune system. Potential future treatments for autoimmunity may include treatment of dysbiosis, because whereas the human genome is static and intransigent to manipulation, the microbiome is conceivably more amenable to therapeutic alterations. Understanding the molecular mechanisms of how symbiotic microbes affect immune reactions to self antigens may provide insight into the causes, and potential cures, for autoimmune diseases.
Did the Microbiota Influence the Evolution of Adaptive Immunity?
The adaptive immune system distinguishes between self and foreign antigens, and mounts an appropriate response to clear invading pathogens by recognizing non-self molecules. The microbiota presents a challenge to the adaptive immune system, because it contains an enormous foreign antigenic burden, which must be either ignored or tolerated to maintain health. One hypothesis for how this occurs is ‘immunologic ignorance’, whereby spatial separation of bacteria from the immune system or down-modulation of innate immunity prevents overt inflammation (57). This notion is feasible, because the innate immune system cannot distinguish pathogens from symbionts because both share similar molecular patterns (such as TLR ligands). Rather than ignorance, tolerance could also be induced by the microbiota, given the capacity of gut bacteria to induce Treg lineage differentiation. Molecules produced by our microbiome may be considered ‘self’, because inflammatory bowel disease (IBD) is thought to, in part, involve a loss of tolerance to antigens of the microbiota. Therefore it appears that rather than ignore them, we may tolerate the microbiota similar to antigens encoded by our own genome. This then raises the question of whether symbiotic bacteria evolved mechanisms to suppress unwanted inflammation toward the microbiota by actively inducing mucosal tolerance. Several studies now suggest this to be the case (32–33, 42). The necessity for the microbiota to induce tolerance as a requirement for colonization, if true, provides a rationale for why symbiotic bacteria may have influenced critical aspects of the adaptive immune system throughout mammalian evolution.
Although T cells can adopt numerous effector cell fates (such as Th1, Th2, Th3, Th9, etc), there are common mechanistic foundations to Th17 and Treg cell development. Differentiation of both lineages are promoted by transforming growth factor β (TGFβ); Tregs require TGFβ (and retinoic acid), whereas TGFβ and IL-6 promote Th17 development (23). The central transcription factors for Treg cells and Th17 cells (Foxp3 and RORγt, respectively) are co-expressed in naïve CD4+ T cells, physically interact with each other, and differentially respond to cytokine stimulation to help determine lineage commitment between Foxp3+ Treg and Th17 differentiation (58). Furthermore, Th17 cells can develop from Foxp3+ Treg cell precursors (59). As mentioned above, germ-free animals show decreased Th17 cell development in several anatomic locations (39, 45, 53–54) and re-colonization with a microbiota (containing SFBs) promotes Th17 cells (46, 53–54). On the basis of this knowledge, we propose a hypothetical model for how specific commensal bacteria network with an evolving adaptive immune system (Fig. 3). Thymically-derived Foxp3+ Treg cells developed under the control of the evolutionary ancient molecule TGFβ. It is tempting to speculate that commensal microbes (for example B. fragilis) may have ‘learned’ to augment this process by further promoting differentiation of existing Treg cells into expanded subsets, such as those producing IL-10 at mucosal surfaces. This expanded Treg cell repertoire could have provided the host with a mechanism to tolerate foreign antigens of the microbiota. Pathobionts such as SFBs may have further modified the development of adaptive immunity by promoting the differentiation of Th17 cells, in part from Foxp3+ precursors. In support of this notion, IL-17 family members have only been found in vertebrates (60). Other pro-inflammatory cytokines (IL-6, IL-21 and IL-1) along with TGFβ are required to induce IL-17 production, perhaps suggesting Th17 cells might be a more recent invention than Treg cells. Perhaps the evolution of specific immune responses was not mainly driven by pathogens (as is popular assumption), but instead by organisms that developed more sustained relationships with the host such as commensals and pathobionts. As these new symbiotic relationships were forged through host-microbial co-evolution, novel additions to the immune system were introduced over millennia. Although this hypothesis requires validation, Treg and Th17 cells provide a powerful means by which mucosal surfaces can be protected from unwanted inflammatory responses to the microbiota (by Treg cells) while still being capable of potently responding to microbial infections (with Th17 cells). B. fragilis and SFBs represent model organisms that have been experimentally validated to provide these functions; other microbes may possess similar activities. Together, the coordination of Treg and Th17 responses appears ideally suited to benefit both the microbiota and the host, and may represent an important evolutionary partnership for human health.
Fig. 3.

A model for the co-evolution of adaptive immunity with the microbiota. (A) The adaptive immune system develops under the control of the vertebrate genome to produce various cell types. The evolutionarily ancient molecule TGFβ directs the differentiation of Foxp3+ Treg cells. Although the earliest mammals contained a gut microbiota, bacteria may or may not have influenced features of the primordial adaptive immune system. (B) Over millennia of co-evolution, commensal microbes (B. fragilis used as an example here) produced molecules that networked with the primordial immune system to help expand various Treg cell subsets, for example IL-10-producing Foxp3+ Treg cells. This process may have evolved to allow these microorganisms to colonize the gut by inducing antigen-specific tolerance to the microbiota. (C) Pro-inflammatory pathobionts (such as SFBs) may have induced Th17 cell differentiation to increase mucosal defenses against enteric pathogens. (D) The modern adaptive immune system may have arisen from 2 distinct events: Tregs and Th17 cell types evolved independently (A to B and A to C), or through the sequential development of Th17 cells from Treg cell precursors (A to B to C to D). This may have been achieved by a combinatorial signal of TGFβ, augmented by the addition of IL-6 to promote Th17 cell evolution over time (inset). Together, the modulation of Tregs and Th17 cells by commensal and pathobionts, respectively, appears to shape the immune status of the host, and thus represents a possible risk factor for autoimmune diseases which appear to depend on balanced Treg/Th17 proportions.
Co-opting an antigen-specific adaptive immune system by the microbiota may extend beyond simply a host-derived process for controlling microbial infections. During co-habitation with the microbiota, evolution of the vertebrate genome occurred under the influence of signals from symbiotic bacteria. In fact, the evolutionary forces that contributed to immune system development during lifelong microbial associations may be dominant compared to those of transient encounters by microbial pathogens (which are rare and opportunistic). Thus, symbiotic microbes may have influenced features of adaptive immune system evolution and function more profoundly than pathogens, possibly to protect both host and microbiota from invading infections. As an increasing body of knowledge links the microbiota to Treg and Th17 phenotypes that mediate autoimmunity, it is imperative to determine how signals from the microbiota shape gene regulatory networks by CD4+ T cells following their thymic development. If these co-evolutionary interactions are relatively recent inventions, is autoimmunity an unwanted side effect from fine-tuning of the peripheral adaptive immune system by the microbiota? As the influences of the microbiota on autoimmune diseases are unraveled, it can be envisioned that harnessing the ability of the microbiota to induce tolerance through Treg cells may provide novel treatments for autoimmunity by correcting immunologic imbalances found in an evolving adaptive immune system. Finally, as we harbor 10-fold more bacterial cells than human cells, explorations into how the microbiota may have influenced the evolution of adaptive immunity might redefine how we view our ‘microbial selves’.
Acknowledgments
We thank members of the Mazmanian laboratory for their critical review of the manuscript. Work in the laboratory of the authors is supported by funding from the National Institutes of Health (DK078938, DK083633, AI088626), Damon Runyon Cancer Research Foundation and the Crohn’s and Colitis Foundation of America to S.K.M.
References and Notes
- 1.Turnbaugh PJ, et al. Nature. 2007 Oct 18;449:804. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qin J, et al. Nature. Mar 4;464:59. [Google Scholar]
- 3.Falk PG, Hooper LV, Midtvedt T, Gordon JI. Microbiol Mol Biol Rev. 1998 Dec;62:1157. doi: 10.1128/mmbr.62.4.1157-1170.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macpherson AJ, Harris NL. Nat Rev Immunol. 2004 Jun;4:478. doi: 10.1038/nri1373. [DOI] [PubMed] [Google Scholar]
- 5.Pollard M, Sharon N. Infect Immun. 1970 Jul;2:96. doi: 10.1128/iai.2.1.96-100.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoshi H, et al. Tohoku J Exp Med. 1992 Mar;166:297. doi: 10.1620/tjem.166.297. [DOI] [PubMed] [Google Scholar]
- 7.Glaister JR. Int Arch Allergy Appl Immunol. 1973;45:719. doi: 10.1159/000231071. [DOI] [PubMed] [Google Scholar]
- 8.Lundin A, et al. Cell Microbiol. 2008 May;10:1093. doi: 10.1111/j.1462-5822.2007.01108.x. [DOI] [PubMed] [Google Scholar]
- 9.Matsumoto S, Setoyama H, Umesaki Y. Gastroenterology. 1992 Dec;103:1777. doi: 10.1016/0016-5085(92)91434-6. [DOI] [PubMed] [Google Scholar]
- 10.Imaoka A, Matsumoto S, Setoyama H, Okada Y, Umesaki Y. Eur J Immunol. 1996 Apr;26:945. doi: 10.1002/eji.1830260434. [DOI] [PubMed] [Google Scholar]
- 11.Umesaki Y, Setoyama H, Matsumoto S, Okada Y. Immunology. 1993 May;79:32. [PMC free article] [PubMed] [Google Scholar]
- 12.Duan J, Chung H, Troy E, Kasper DL. Cell Host Microbe. 2010 Feb 18;7:140. doi: 10.1016/j.chom.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niess JH, Leithauser F, Adler G, Reimann J. J Immunol. 2008 Jan 1;180:559. doi: 10.4049/jimmunol.180.1.559. [DOI] [PubMed] [Google Scholar]
- 14.Bouskra D, et al. Nature. 2008 Nov 27;456:507. doi: 10.1038/nature07450. [DOI] [PubMed] [Google Scholar]
- 15.Noverr MC, Huffnagle GB. Trends Microbiol. 2004 Dec;12:562. doi: 10.1016/j.tim.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 16.Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. Cell. 2005 Jul 15;122:107. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 17.Bauer H, Horowitz RE, Levenson SM, Popper H. Am J Pathol. 1963 Apr;42:471. [PMC free article] [PubMed] [Google Scholar]
- 18.Smith K, McCoy KD, Macpherson AJ. Semin Immunol. 2007 Apr;19:59. doi: 10.1016/j.smim.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 19.Clarke TB, et al. Nat Med. 2010 Feb;16:228. doi: 10.1038/nm.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inagaki H, Suzuki T, Nomoto K, Yoshikai Y. Infect Immun. 1996 Aug;64:3280. doi: 10.1128/iai.64.8.3280-3287.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stecher B, et al. PLoS Biol. 2007 Aug 28;5:e244. [Google Scholar]
- 22.Lupp C, et al. Cell Host Microbe. 2007 Aug 16;2:119. doi: 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 23.Bettelli E, et al. Nature. 2006 May 11;441:235. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 24.Fontenot JD, et al. Immunity. 2005 Mar;22:329. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 25.Foussat A, et al. J Immunol. 2003 Nov 15;171:5018. doi: 10.4049/jimmunol.171.10.5018. [DOI] [PubMed] [Google Scholar]
- 26.Battaglia M, Gregori S, Bacchetta R, Roncarolo MG. Semin Immunol. 2006 Apr;18:120. doi: 10.1016/j.smim.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 27.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Cell. 2008 May 30;133:775. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 28.Rubtsov YP, et al. Immunity. 2008 Apr;28:546. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 29.Coombes JL, et al. J Exp Med. 2007 Aug 6;204:1757. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boirivant M, Strober W. Curr Opin Gastroenterol. 2007 Nov;23:679. doi: 10.1097/MOG.0b013e3282f0cffc. [DOI] [PubMed] [Google Scholar]
- 31.Coombes JL, Maloy KJ. Semin Immunol. 2007 Apr;19:116. doi: 10.1016/j.smim.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 32.O’Mahony C, et al. PLoS Pathog. 2008;4:e1000112. doi: 10.1371/journal.ppat.1000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sokol H, et al. Proc Natl Acad Sci U S A. 2008 Oct 28;105:16731. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mazmanian SK, Kasper DL. Nat Rev Immunol. 2006 Nov;6:849. doi: 10.1038/nri1956. [DOI] [PubMed] [Google Scholar]
- 35.Mazmanian SK, Round JL, Kasper DL. Nature. 2008 May 29;453:620. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 36.Strauch UG, et al. Gut. 2005 Nov;54:1546. doi: 10.1136/gut.2004.059451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ostman S, Rask C, Wold AE, Hultkrantz S, Telemo E. Eur J Immunol. 2006 Sep;36:2336. doi: 10.1002/eji.200535244. [DOI] [PubMed] [Google Scholar]
- 38.Min B, et al. Eur J Immunol. 2007 Jul;37:1916. doi: 10.1002/eji.200737236. [DOI] [PubMed] [Google Scholar]
- 39.Ivanov, et al. Cell Host Microbe. 2008 Oct 16;4:337. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zaph C, et al. J Exp Med. 2008 Sep 29;205:2191. doi: 10.1084/jem.20080720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaboriau-Routhiau V, et al. Immunity. 2009 Oct 16;31:677. doi: 10.1016/j.immuni.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 42.Round JL, Mazmanian SK. Proc Natl Acad Sci U S A. 2010 Jun 21;107:12204. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dobber R, Hertogh-Huijbregts A, Rozing J, Bottomly K, Nagelkerken L. Dev Immunol. 1992;2:141. doi: 10.1155/1992/57057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ivanov, et al. Cell. 2006 Sep 22;126:1121. [Google Scholar]
- 45.Atarashi K, et al. Nature. 2008 Oct 9;455:808. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- 46.Ivanov, et al. Cell. 2009 Oct 30;139:485. [Google Scholar]
- 47.Snel J, et al. Can J Microbiol. 1998 Dec;44:1177. doi: 10.1139/cjm-44-12-1177. [DOI] [PubMed] [Google Scholar]
- 48.Talham GL, Jiang HQ, Bos NA, Cebra JJ. Infect Immun. 1999 Apr;67:1992. doi: 10.1128/iai.67.4.1992-2000.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Umesaki Y, Okada Y, Matsumoto S, Imaoka A, Setoyama H. Microbiol Immunol. 1995;39:555. doi: 10.1111/j.1348-0421.1995.tb02242.x. [DOI] [PubMed] [Google Scholar]
- 50.Noverr MC, Huffnagle GB. Clin Exp Allergy. 2005 Dec;35:1511. doi: 10.1111/j.1365-2222.2005.02379.x. [DOI] [PubMed] [Google Scholar]
- 51.Round JL, Mazmanian SK. Nat Rev Immunol. 2009 Apr 3;9:313. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ochoa-Reparaz J, et al. Mucosal Immunol. 2010 Jun 9;3:487. doi: 10.1038/mi.2010.29. [DOI] [PubMed] [Google Scholar]
- 53.Wu HJ, et al. Immunity. 2010 Jun 25;32:815. [Google Scholar]
- 54.Lee YK, et al. Proc Natl Acad Sci U S A. 2010 Jul 28; ePub. [Google Scholar]
- 55.Chow J, Mazmanian SK. Cell Host Microbe. 2010 Apr 22;7:265. doi: 10.1016/j.chom.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baranzini SE, et al. Nature. 2010 April 29;464:1351. doi: 10.1038/nature08990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hooper LV. Nat Rev Microbiol. 2009 May;7:367. doi: 10.1038/nrmicro2114. [DOI] [PubMed] [Google Scholar]
- 58.Zhou L, et al. Nature. 2008 May 8;453:236. [Google Scholar]
- 59.Yang XO, et al. Immunity. 2008 Jul 18;29:44. [Google Scholar]
- 60.Weaver CT, Hatton RD, Mangan PR, Harrington LE. Annu Rev Immunol. 2007;25:821. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 61.Baranzini SE. Curr Opin Immunol. 2009 Dec;21:596. doi: 10.1016/j.coi.2009.09.014. [DOI] [PubMed] [Google Scholar]