


Abstract
Feline immunodeficiency virus (FIV) infects many species of cat, and is related to HIV, causing a similar pathology. High-throughput selective 2′ hydroxyl acylation analysed by primer extension (SHAPE), a technique that allows structural interrogation at each nucleotide, was used to map the secondary structure of the FIV packaging signal RNA. Previous studies of this RNA showed four conserved stem–loops, extensive long-range interactions (LRIs) and a small, palindromic stem–loop (SL5) within the gag open reading frame (ORF) that may act as a dimerization initiation site (DIS), enabling the virus to package two copies of its genome. Our analyses of wild-type (wt) and mutant RNAs suggest that although the four conserved stem–loops are static structures, the 5′ and 3′ regions previously shown to form LRI also adopt an alternative, yet similarly conserved conformation, in which the putative DIS is occluded, and which may thus favour translational and splicing functions over encapsidation. SHAPE and in vitro dimerization assays were used to examine SL5 mutants. Dimerization contacts appear to be made between palindromic loop sequences in SL5. As this stem–loop is located within the gag ORF, recognition of a dimeric RNA provides a possible mechanism for the specific packaging of genomic over spliced viral RNAs.
INTRODUCTION
Feline immunodeficiency virus (FIV) is a lentivirus which causes pathology in its host similar to that of HIV in humans (1–5). The virus has a worldwide distribution and infects many feline species, including up to 22% of domestic cats and 80% of African lions (6–8). FIV strains are species specific, and were previously thought to have milder effects upon big cats, but recent evidence refutes this, with similar signs of disease observed across species (9,10). Aside from being a common veterinary pathogen, FIV infection of cats is a small animal model for acquired immune deficiency syndrome (AIDS) (11) and FIV-based vectors are under development for human gene therapy (12). Despite increasing evidence of its profound pathological impact on various feline species, including endangered ones, and its potential therapeutic values, understanding of the molecular biology of the virus is relatively rudimentary. In order to utilize FIV to its full potential, and to develop methods to combat its effect on feline species, a greater understanding of its molecular architecture is essential.
The transcribed unspliced RNA of retroviruses serves as the viral genome and also encodes its core structural and enzymatic proteins. Different lentiviruses may use the same RNA molecule for both functions or separate pools of RNA for translation or packaging (13). In order to package its genome into assembling particles during replication, the virus must specifically recognize and capture its genomic RNA from within the cytoplasmic pool of cellular and spliced viral mRNAs. This is mediated in a highly specific manner by cis-acting packaging signals that are recognized by the viral Gag protein. These sequences are often situated within the 5′-end of the RNA genome, whose structural context contributes to the recognition process (14,15).
Two non-covalently linked copies of the (+) strand RNA genome are encapsidated into the virion. The dimer linkage region is often in close proximity to, or part of, the packaging recognition signal (16–18). Dimer linkage is commonly believed to occur in two or more stages, initiated by the interaction of a palindromic sequence on the two strands to form a ‘kissing loop’ (19–24). This loop is at the tip of a stable helix which subsequently unwinds to form an extended interstrand duplex (25). Although it is clear that packaging and dimerization are intimately linked processes, whether dimerization begins prior to or following an interaction with the Gag polyprotein remains a subject of debate (26–29).
In FIV, the first 511 nt of the genome are necessary and sufficient for optimal packaging, but within this, the virus has an unusual, bipartite, packaging signal comprising the first 150 nt of the 5′ untranslated region (UTR) and the first 100 nt of gag (30–34). The FIV packaging signal region has been examined previously by us (35) and others (36), providing contrasting structural models. Our model shows five conserved stem–loops (SLs), including one of around 150 nt, and one small palindromic SL (SL5) that is a candidate for the genome dimerization initiation site (DIS), as well as extensive, conserved long-range interactions (LRIs) linking the 5′ and 3′ regions. To resolve the disparity between the published structures and to generate more precise structural data on this RNA we used selective 2′ hydroxyl acylation analysed by primer extension (SHAPE), a novel chemo-enzymatic probing strategy which examines the flexibility (a surrogate marker for base-pairing) of the backbone at each nucleotide position (37). SHAPE offers the advantage that all 4 nt can be interrogated with a single reagent, which readily reacts with positions that are single-stranded. In contrast, nucleotides that are base-paired or architecturally constrained are less reactive.
Our results validate the structures of four stable SLs in our original model, but suggest that the 5′ and 3′ interacting regions can adopt alternative structures. In one of these we confirm formation of the conserved LRIs we published previously, and in the alternative structure, a series of small SLs is observed. The two separate monomeric conformers were visualized by native polyacrylamide gel electrophoresis (PAGE), and mutagenesis was used to probe their structures, confirming the two structural models. Previously, we also noted a unique palindromic sequence in the gag region, SL5, which formed a conserved helix loop and seemed a likely candidate for the FIV DIS (35). We therefore performed dimerization assays on a series of in vitro transcribed RNAs containing mutations in this palindrome. Dimerization was reduced following disruption of sequence auto-complementarity in SL5, and SHAPE showed that nucleotides in the palindrome were rendered single-stranded, implicating SL5 as the DIS. Only in the LRI structure is this palindromic DIS accessible to form kissing–loop interactions that can promote the dimerization process, as it is base-paired in the alternative structure. Dimerization assays using LRI-stabilizing and destabilizing mutants support this. This conformational switch between two structures resembles the model proposed in HIV (38,39) in which the DIS can adopt an occluded or accessible configuration, suggesting a mechanism for regulating the monomer/dimer equilibrium in order to facilitate packaging of dimeric RNA over a non-dimerizing monomeric template suited for translation. Our alternative, monomeric structure is less stable than the dimerization-competent structure (−145 kcal/mol versus −181 kcal/mol) which would facilitate translation.
MATERIALS AND METHODS
Preparation of in vitro transcribed RNAs
DNA template corresponding to the first 511 nt of the FIV RNA genome and containing the T7 RNA polymerase promoter was prepared by polymerase chain reaction (PCR) using primers 1 (F) and 511 (R) from template plasmid TR394 (40) as previously described (35), or from mutant plasmids as described in the ‘Results’ section. An extended template for SHAPE was constructed using primer 1 (F) and 711 (R, CAC CGT CAT ATT TAA AAG TCC). Templates of varying length were constructed for dimerization analysis using forward primers 1 and 143 (TAA TAC GAC TCA CTA TAG GCG CCC GAA CAG GGA C) and reverse primers 95 (GAT TAC ACA GAT ACT CGA CAG G), 146 (CGC CAA CTG CGA AGT TCT CGG), 321 (CTG GGC CTT TAA ACA ATG AC), 342 (GAG TCA CCA GAT GTA ATT TAT C), 381 (CTG TCC CTC GGC GAA TCT C), 484 (CCC CTA CTC CTA CAG CAA C), 511 and 564 (CAC CAG GTT CTC GTC CTG TAG). Primer numbers represent the nucleotide position according to the genomic RNA of the 5′ (F primers) or 3′ (R primers) ends of the transcript they produce. PCR products were purified from 1% agarose gels using Qiagen Gel Extraction kits, according to the manufacturer’s protocol. Purification of in vitro transcribed RNA followed one of two protocols. Firstly, 20 μl reactions contained 7.5 mM rNTPs (Ambion), 1× buffer (Megashortscript buffer, Ambion), 2 μg template DNA and 2 μl T7 RNA polymerase (Megashortscript enzyme mix, Ambion) and were incubated for 4 h at 37°C. DNA was degraded with 4 U DNase (turboDNase, Ambion) for 20 min at 37°C, and the size and integrity of RNA transcripts was verified by electrophoresis on 1% agarose gels before purification on filter cartridges (MegaClear, Ambion) and elution in H2O. Large-scale transcription reactions contained 6 mM of each rNTP (Promega), 40 mM DTT, 0.5 U/ml yeast inorganic pyrophosphatase (NEB), 60 U/ml RNase inhibitor (RNAsin Plus, Promega), 4.23 μg/ml T7 RNA polymerase (courtesy of J. Miller, National Cancer Institute, USA), 126 nM DNA template, 80 mM HEPES-KOH pH 7.6, 12 mM MgCl2, 2 mM spermidine, 0.01% Triton X-100 in a volume of 15–30 ml and were incubated at 37°C for 16 h. A further 4.23 μg/ml T7 RNA polymerase, 0.5 U/ml yeast inorganic pyrophosphatase and 10 mM MgCl2 were added and incubated for 3 h at 37°C. DNA was digested with 15 U/ml DNase (RQ1 RNase free DNase, Promega) for 2 h at 37°C. Transcriptions were clarified by centrifugation at 10 000g for 45 min and concentrated to 4 mL using Amicon Ultra Ultracel centrifugal concentrators according to the manufacturer’s instructions. RNA was purified by denaturing PAGE on 10% gels at a constant temperature of 50°C, before visualization by UV-shadowing. Electroeluted RNA was precipitated with 0.3 M sodium acetate and 2.5 vol of ethanol (−20 °C, overnight), recovered by centrifugation, washed with 70% ethanol, dried and resuspended in sterile H2O. Concentrated RNA was stored at −20°C.
Mutagenesis
Mutants AN14, AN20, AN21 and AN26 were prepared as described (41). Mutant AN40 was prepared from plasmid TR394 using primer GAA CCC TGT CGA GTA CCC ATG TAA TCT TTT TTA CCT GTG AGG TC and its reverse complement and the Quikchange II mutagenesis kit (Stratagene), according to the manufacturer’s instructions. The resulting plasmid was verified by sequencing.
Selective 2′hydroxyl acylation analysed by primer extension
Ten picomoles of RNA were resuspended in 20 μl 10 mM Tris pH 8, 100 mM KCl and 0.1 mM ethylenediaminetetraacetic acid (EDTA), heated at 95°C for 3 min and cooled on ice. RNA was refolded by adding 150 μl of 40 mM Tris pH 8, 4 mM MgCl2, 0.2 mM EDTA, 130 mM KCl and 12 U RNase inhibitor (Superasein, Ambion) for 10 min at 37°C. Alternatively, RNA was probed immediately following transcription and column elution, as for previous RNase mapping experiments (35). Reactions were divided into two 72 μl aliquots and 8 μl 40 mM of SHAPE chemical reagent 1M7 or NMIA in dimethyl sulphoxide (DMSO) was added to one. To the other aliquot, 8 μl of DMSO was added. To examine dimerization, 1M7 in DMSO or DMSO only were added to a final concentration of 4 mM after the 4-h incubation (see in vitro dimerization assay method below). Tubes were incubated at 37°C for 2 min and RNA was precipitated at −20°C with 60 ng/μl glycogen, 0.3 M sodium acetate pH 5.2 and 3 vol of cold ethanol. Precipitated RNA was collected by centrifugation, washed once in 70% ethanol and resuspended in 5 μl 5 mM Tris pH 8, 0.1 mM EDTA. Five picomoles of Cy5 or 6FAM-labelled (for SHAPE reagent modified samples) or WellRed D3 or VIC-labelled (for unmodified samples) primer 511 or 384 (35) in 7 μl H2O were annealed to the RNA at 85°C for 1 min, 60°C for 5 min and 35°C for 5 min. RNA was reverse transcribed at 50°C for 20 min with 100 U RT (Invitrogen superscript III), 1× RT buffer (Invitrogen), 5 mM DTT and 500 μM dNTPs (Promega). RNA was hydrolysed with 200 mM NaOH for 5 min at 95°C and reactions were neutralized with an equivalent amount of HCl. Sequencing ladders were prepared using the Epicentre cycle sequencing kit according to the manufacturer’s instructions and primers labelled with WellRed D2 and LicorIR-800 or NED and PET dyes. Modified and control samples were mixed with the sequencing ladders, precipitated as above, dried and resuspended in 40 μl deionized formamide. Primer extension products were analysed on a Beckman CEQ8000 Genetic Analysis System or an Applied Biosystems 3730xl DNA analyser. Electropherograms were processed using the SHAPEfinder programme, following the software developer’s protocol and included the required pre-calibration for matrixing and mobility shift for each set of primers (42). 1M7 or NMIA reactivity at each nucleotide position was normalised as described (43). Briefly, the area under each negative peak was subtracted from that of the corresponding positive peak. The resulting peak area difference at each nucleotide position was then divided by the average of the highest 8% of peak area differences, calculated after discounting any results greater than the 3rd quartile plus 1.5× the interquartile range.
In silico modelling
Minimal free energy modelling was performed using RNAstructure software (44) and illustrated using XRNA. QGRS mapper was used to identify potential G-quadruplexes.
Non-denaturing PAGE
RNAs were mixed with 1/6 volume of native loading buffer (40% (v/v) glycerol, 44 mM tris-borate pH 7, 0.25% orange G dye) and separated by electrophoresis at 100 V for 6 h on gels containing 3.5% polyacrylamide and 1× TBE (89 mM Tris base, 89 mM boric acid, 2 mM EDTA). Gels were subsequently stained for 10 min with 1.3 µM ethidium bromide in 1× TBE
In vitro dimerization assay
Five-hundred nanograms of in vitro transcribed RNA in 10 μl, 10 mM Tris–HCl, pH 7, 200 mM NaCl, 4 mM MgCl2 was incubated at 95°C for 2 min, 4°C for 2 min and 55°C for 4 h, in a thermocycler before the addition of 1/6 volume native loading buffer, as above, and electrophoresis through 1% agarose 1× TBE or TBM (89 mM Tris base, 89 mM boric acid, 0.7 mM MgCl2) gels containing 1.3 µM ethidium bromide in the corresponding buffer, at room temperature.
RESULTS
SHAPE data confirm the structures of SL1-4
SHAPE has been recently developed to study RNA secondary structure (37) by examining the flexibility of the RNA backbone (which depends on base-pairing) at each nucleotide position via reactivity with a specific adduct (1M7 or NMIA). We first applied this technique to the wt FIV packaging signal RNA in order to validate our previous model, examine the conformational stability of individual helices, and identify potential LRIs that could have been overlooked by chemo-enzymatic methods that probe only specific nucleotides and structures. Previous studies had mapped the FIV packaging signal to within a 511-nt element spanning the 5′ UTR and the first 100 nt of gag (31). Although LRIs may exist with parts of the genome further 3′, this region alone was shown to be both necessary and sufficient for optimal packaging, and hence must contain all required structures. Therefore, we chose to examine the structure of the 5′ 511 nt in isolation.
Reactivity to the SHAPE reagent 1M7 at each nucleotide position is shown on Figure 1, in the context of our original model, to assess its validity. Numerical data are given in Supplementary Figure S1. The most reactive, and hence least conformationally constrained nucleotides have a reactivity >0.9 U and are shown in red. Nucleotide positions with reactivity <0.3 U (indicative of fully base paired or otherwise structurally constrained bases) are in black, and nucleotides of intermediate reactivity are colour-coded according to the key. Reactivities of the 3′ nucleotides (G466-A511) were not determined at this stage. Data shown are an average of two or three experiments. Typically the data for any individual nucleotide varied by no more than 0.1 U between experiments, with a Pearson correlation coefficient that was typically 0.8–0.9.
Figure 1.

SHAPE analysis of the FIV packaging signal RNA. In vitro transcribed RNA was modified with 1M7, reverse transcribed using fluorophore-labelled primers and separated by capillary electrophoresis. Nucleotide positions were determined using G and U sequencing ladders. 1M7 reactivity at each nucleotide position was determined by subtraction of the reverse transcription product of unmodified RNA from that of 1M7-modified RNA, using SHAPEfinder software. Nucleotide reactivities are colour-coded as shown in the key. Reactivities of the 3′-end were not determined (shown in grey). The RNA is drawn in our previously published secondary structure, to ascertain its validity. Nts are numbered with a dash every 10 nt. (A) In vitro transcribed packaging signal RNA; 511 nt. (B) In vitro transcribed SL2 RNA. Data shown are an average of at least two independent experiments.
Overall, the SHAPE data support our original model (Figure 1A); this is particularly the case for SLs 1–4. The loops of these four regions contain the most highly reactive, exposed nucleotides. The remainder of the most highly reactive nucleotides are situated in bulges, internal loops, single-stranded regions linking SLs and the closing pairs of helices, with the exception of G 90, U101 and A320. The tip of SL5 contains mainly unreactive nucleotides, possibly indicating kissing–loop interactions involved in dimer initiation. Nucleotides that were predicted to be base-paired in SL1 were unreactive to 1M7, with the exception of A131. As the A residue 3′ of this was predicted to be bulged, and is unreactive, it is likely that the bulge forms consistently at A131 and not at A132 (shown in Figure 2). Predicted base-paired nucleotides in SL2 are relatively unreactive, except for closing pairs and G-U pairs. The exceptions are nucleotides U244–G246, which are highly reactive; this may indicate that the internal loop 3′ of this is larger and includes these nucleotides. Structural predictions using the RNA structure programme (44) and 1M7 reactivities as pseudo free-energy constraints suggest that this is the case in the alternative structure (shown in Figure 2). Minimal free energy predictions suggested that the SL2 structure is maintained independently of adjacent sequences. To validate this hypothesis and search for LRIs that the single-stranded regions of SL2 may form with other areas of the packaging signal, we transcribed SL2 RNA independently, examined its 1M7 reactivity, and compared the data with SL2 in the context of the 511-nt RNA (Figure 1B). Nucleotide reactivities are broadly similar across SL2, whether analysed alone or in the context of the extended 5′ and 3′ structure, suggesting that SL2 forms independently of 5′ and 3′ sub-domains, i.e. independent of LRIs. Subtle differences in nucleotide reactivity were noted between the structures shown in Figure 1A and B, which may reflect minor experimental variation.
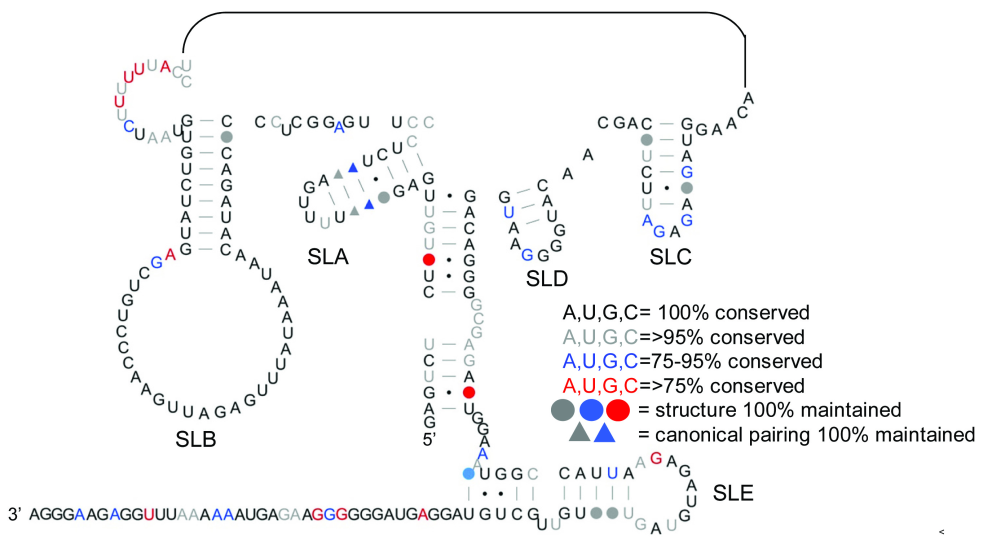
Figure 2.

Alternative (MSL) structure of the FIV packaging signal. 1M7 reactivities determined as before were used as pseudo free-energy constraints in a minimal free energy folding algorithm (RNAstructure). Nucleotide reactivities are colour-coded as shown in the key and numbered with a dash every 10 nt.
SHAPE data provide alternative structures 5′ and 3′ of SLs1–4
The intermediate 1M7 reactivity between regions 5′ of SL1 and 3′ of SL4 was higher than anticipated for paired nucleotides. This was particularly evident between nucleotides 61 and 105. The high G-U content of three of these helices, as well as single and double-strand specific enzymatic cleavage, led us to propose previously that they were metastable, and opened and closed by ‘breathing’, thereby allowing access to both single and double-strand specific nucleases (35). However, disparity between 1M7 reactivity between the 5′ and 3′ side of some of the proposed helices seen in Figure 1A requires an alternative explanation.
The 5′ and 3′ regions can simultaneously adopt two different conformations
Possible alternative RNA conformations which would account for disparity in observed and expected 1M7 reactivities of the LRI regions were examined, using the SHAPE data as pseudo free-energy constraints in the RNAstructure programme (44). Although three structures that correspond more accurately to the SHAPE data were predicted (data not shown), no individual structure was fully confirmatory, and no individual structure convincingly satisfied both SHAPE and our previous biochemical data, which had been observed under similar conditions. The secondary structural model of Figure 1A is supported by strong phylogenetic evidence (69% of nucleotides in the 5′/3′ interacting regions that vary in sequence between FIV isolates maintain the ability to base-pair), and by our probing data, as all enzymatic cleavage sites in this region mapped accurately onto the model (35). We therefore sought an alternative structure of the 5′ and 3′ regions that could be present simultaneously with the LRI structure shown in Figure 1A, and would reconcile both the RNase mapping and the SHAPE data. One of the three predicted structures fulfils these criteria and is shown in Figure 2. SLs 1–4 are maintained, but the LRI region adopts a series of further SLs, designated SL A–E. A mixture of these two structures would account for the disparity in nucleotide reactivity between 5′ and 3′ sides of the LRI region, as in one structure (Figure 1A), most nucleotides are paired, whereas in the other, multiple SLs (MSL) (Figure 2), most 3′ nucleotides remain paired in an alternative conformation, whereas many of the 5′ nucleotides remain single stranded. With a few exceptions, highly reactive nucleotides are single stranded in both structures, whereas those that are single stranded in only one of the two structures exhibit intermediate 1M7 reactivity. Some areas are shown as single stranded in one of the two structures but exhibit little to no 1M7 reactivity, notably the regions connecting SL1–4, and loop nucleotides C402–A409. Although SHAPE data maps more accurately onto the 5′ and 3′ regions of the MSL structure (Figure 2) than the original LRI structure (Figure 1), it does not fully fit the data, and fits our previous enzymatic data less well (7/27 single-stranded cleavage sites map to nucleotides that have Watson–Crick base pairs and in are the middle of helices. 2/20 RNase CV1 cleavage sites are in the middle of a long single-stranded region), suggesting that structure 2 is not present on its own. A mixture of both structures accurately accounts for all the SHAPE and enzymatic data.
Since there were some differences in RNA preparation and purification methods between our enzymatic (35) and SHAPE studies, it was important to determine whether these favoured different RNA conformations, or whether both structures were formed under each condition. NMIA reactivity profiles for RNA prepared by the two methods are very similar (Supplementary Figure S2), eliminating the method of preparation as promoting different structures. Small differences in absolute reactivity do exist, as might be anticipated in such a large data set. The nucleotides that vary most in reactivity are single-stranded, non-canonically paired, or adjacent to a single-stranded or non-canonically paired nucleotide. This variable reactivity profile has been observed by those who developed the SHAPE technique, as the standard deviation is much higher for such nucleotides than for those canonically paired and nested within helices (43,45,46). Crucially, each data set shows the same pattern of reactivity and supports the presence of the two predicted monomeric conformations.
In the event that structural data for the 3′ 50 nt thus far unmapped did not fit our models and suggested alternative structures, we examined the SHAPE reactivity of this region, and the 5′-end of a 711-nt transcript. NMIA sensitivity of the 5′-end of a 511-nt (data not shown) and 711-nt transcript was very similar. Reactivities of the 5′ and 3′-ends of the first 511 nt of the longer transcript are illustrated in Supplementary Figure S3, supporting a mixture of the LRI and MSL structures. Again, most differences in absolute reactivity between the data sets (Figures 1 and 2 versus Supplementary Figure S3) are located at single-stranded or non-canonically paired nucleotides, or those adjacent to them, and are likely to be a reflection of their previously documented variability of reactivity to 1M7 and NMIA (45). Importantly, each data set independently supports the presence of each of the two predicted conformers. The intermediate to high reactivity of the 3′-end is likely to be because this region is single-stranded in the MSL conformation. However, it could be due to the unpredictable reactivity of G-U pairs, or, because of the high number of G-U pairs in this helix, its metastability may contribute to the SHAPE reactivity seen.
The two monomeric conformations can be separated by non-denaturing PAGE
To visualize the two proposed structures we fractionated the RNA on a 3.5% non-denaturing polyacrylamide gel. Two distinct bands were evident around the expected monomeric size of 511 nt, regardless of the RNA preparation method (Figure 3A, lanes wtS and wtT). In order to selectively stabilize the MSL structure, we examined the structure of a mutant, AN14 (41), which disrupts a conserved heptanucleotide helix in the LRI form but should not affect the stability of SLB in the MSL form (Figure 3B). Conversely, to stabilize the LRI form, we mutated 3 nt in the SLB helix, such that it should be selectively destabilized in the MSL conformer (AN40, Figure 3B). Analysis of these mutants by non-denaturing PAGE shows a decrease in the amount of slower-moving relative to faster-moving conformer in the AN14 mutant RNA, and vice versa in the AN40 RNA (Figure 3A). Densitometric analysis of multiple samples shows the changes in ratio to be statistically significant (P < 0.01) (Figure 3C). Dimeric RNA would be expected to migrate at around 1000 nt, but could be detected only as a very faint band in all samples (data not shown).
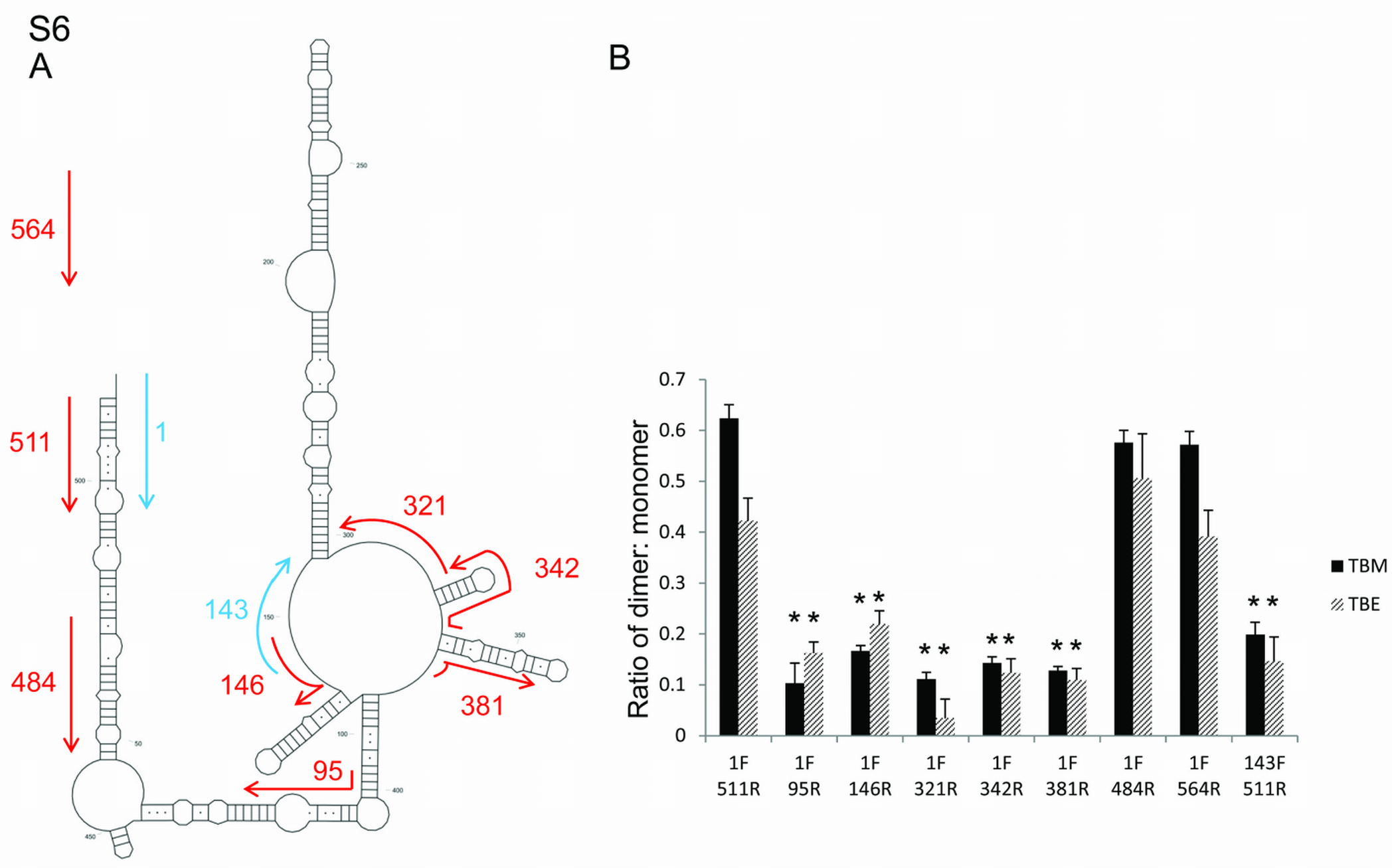
Figure 3.

Analyses of the two conformers by non-denaturing PAGE and SHAPE. (A) FIV packaging signal RNA was electrophoresed on a non-denaturing polyacrylamide gel and visualised by ethidium bromide staining and UV illumination. L; RNA ladder wtS; denatured and renatured wt RNA. AN14; denatured and renatured AN14 RNA. AN40; denatured and renatured AN40 RNA. wtT; wt RNA probed after transcription and column purification. (B) Schematic diagram showing the effects of AN14 and AN40 mutations upon: (Bi) the LRI structure; (Bii) SLB of the MSL structure. (C) Densitometric analysis of the ratio of slower-migrating to faster-migrating conformer. Data represent an average of at least six independent experiments. Stars represent statistical significance relative to wt (P < 0.01) by t-test. Error bars show the S.E.M. (D) NMIA reactivity profile of the 3′ portion of each mutant RNA from nts 380–460. Upper panel, AN14; lower panel, AN40. Grey boxes indicate the location of various structural features of each mutant. Reactivity >4 is shown as 4. Numbers below the graph represent the number of the initial nucleotide of each helix in the LRI structure. Data shown are an average of three independent experiments.
AN14 and AN40 mutants were next analysed by SHAPE. As evident in Figure 3A and C, a small portion of AN14 still adopts the LRI-like conformer, and likewise a small portion of the AN40 RNA assumes the MSL-like conformer. However, as the ratio of each structure differs greatly between the two mutants, we reasoned that this should be reflected in the NMIA reactivity. There was no difference in reactivity of the more static structures SLs 1–4 (data not shown), but as expected the 3′-end shows varying NMIA sensitivity, reflecting the ratios of the two conformers (Figure 3D). Reactivity of the AN40 mutant is low across the expected LRI regions of the LRI structure, with increased reactivity in the connecting loops. Nucleotides 436–438 are within an LRI, but have become more reactive, which is likely to be a reflection of the G-U pairs they form in this conformation. Reactivity of non-canonical base pairs is unpredictable, variable, and can be very high (45). NMIA reactivity of AN14 is low in the stem regions and higher in the loop regions of the MSL structure. The 3′ side of the highly conserved heptanucleotide forming the helix at nts 422–428 is highly reactive when its 5′ complement is mutated to a non-complementary sequence in AN14 (nts 425–428, AN14 Figure 3D). Although these 4 nt are also paired in the MSL conformer, they are non-canonically paired or adjacent to such pairs, rather than paired in a 7-bp helix containing entirely Watson–Crick pairs as they are in the LRI conformation, and hence reactivity to NMIA could be expected to increase. Reactivity of the SL5 region also drops slightly in AN14, where it is predicted to be stably base-paired as part of SLE.
The alternative SLs and helices are structurally conserved between FIV isolates
Previously, we showed that the RNA structure of Figure 1A is highly conserved between 76 FIV isolates. The ability of these domestic cat strains of FIV to form the MSL structure was assessed using our previously published alignment (35), and is illustrated in Figure 4, which shows nucleotides that vary in sequence but maintain the ability to base-pair in all isolates. A more detailed analysis, indicating the degree of sequence variation of each nucleotide, is shown in Supplementary Figure S4. Helices of SLs A and B assume an identical structure in all isolates, while those of sSLs C, D and E form identically in the majority, as do those joining the 5′ and 3′-ends.
Figure 4.

Structural conservation of the 5′- and 3′-ends of the MSL structure. Sequence variation was previously assessed using an alignment of 76 strains of domestic cat FIV. The ability of each strain of FIV to form this MSL structure was determined using this alignment. Grey nucleotides represent those which vary in sequence but maintain the ability to base pair in 100% of isolates. SLs 1–4 are not shown.
SL5 acts as a DIS in vitro
Retroviruses package two copies of their RNA genome, linked as a dimer (16,18). Dimerization begins at the DIS, which is usually found in the 5′UTR and is often a palindromic sequence. Although SL5 is within the gag open reading frame (ORF), it contains the only conserved palindrome in the FIV packaging signal RNA, making it a credible candidate for the DIS. The SL5 helix loop is the only exposed sequence capable of assuming an initial ‘kissing loop’ interaction. This interaction also has the potential to promote an extended dimer, since minimal free energy structural modelling of two copies of the FIV packaging signal produced structures linked at the SL5 palindrome, with the region extending 3′ of this interacting with the 5′ region of the other strand (Supplementary Figure S5). All structures generated by the prediction algorithm were based on the LRI model of Figure 1A, with minor differences in internal loops and bulges (data not shown).
In order to determine whether SL5 might function as the DIS, we performed dimerization assays with a panel of mutants in which SL5 was replaced with either a non-palindromic sequence or an alternative palindrome. Figure 5 shows the results of this assay. Wild-type RNA migrated as two species of ∼500 and 1000 nt, corresponding to the expected size of monomer and dimer species (Figure 5A). Replacing the SL5 sequence with an alternative palindrome also leads to dimer formation (Figure 5A, AN26). However, disrupting the palindromic nature of SL5 by deletion of 5 nt (AN20) or by inserting a non-palindromic sequence of equal length (AN21) abolishes dimer formation. Densitometric analysis of multiple gels is shown in Figure 5B and confirms that there is a statistically significant difference (P < 0.01) between wild type and mutants without a palindromic sequence at SL5, but that there is not a statistically significant difference in ability to dimerize when SL5 is replaced with an alternative palindrome. Similar results in both TBM and TBE gels (Figure 5B) suggest formation of so-called ‘tight dimers’ (47). In order to visualize the structural differences between SL5 mutants, SHAPE was performed by reaction of the RNA with 1M7 immediately after the 4 h incubation at 55°C, and reverse transcription, capillary electrophoresis and analysis as before (see ‘Materials and Methods’ section). In the wild-type RNA, 1M7 reactivity is very low across the central 8 nt of the palindrome, indicating these are most likely base-paired (Figure 5A, WT) which they would be in either the kissing loop model of the LRI structure or in the monomeric MSL structure (Figure 2). 1M7 reactivities remain low across the central 8 nt when an alternative palindrome is present (Figure 5A, AN26). However, nucleotides display increased 1M7 reactivity when the palindrome is disrupted (Figure 5A, AN20 and AN21), indicating loss of base pairing. Reactivity of nucleotides outside these regions is similar (data not shown). Further in vitro dimerization experiments showed that deletions from the 3′-end of the packaging signal RNA abrogated dimerization upon loss of SL5, and that the 5′ side of the LRI region was also necessary (Supplementary Figure S6). Such data argue not only that SL5 acts as the DIS, but also that the LRI structure is a necessary prerequisite for dimerization.
Figure 5.

Dimerization analysis of SL5 mutants. (A) Dimerization assay and 1M7 reactivity of SL5 mutants. In vitro transcribed FIV packaging signal RNAs containing a wt or mutant SL5 sequence were heated to 95°C, snap-cooled, and incubated at 55°C for 4 h in 10 mM Tris–HCl, pH 7, 200 mM NaCl, 4 mM MgCl2 before electrophoresis on an agarose gel in TBM, or SHAPE analysis as described in ‘Materials and Methods’ section. Mutant designations are given above the relevant lanes and their sequences at SL5 are illustrated above or below each lane. L; RNA ladder. Nucleotide colours show 1M7 reactivities as shown on the key. (B) Densitometric analysis of dimerization on TBM (black bars) or TBE gels (striped bars). Stars represent statistical significance relative to wt (P < 0.01) by t-test. Error bars represent the S.E.M.
The palindrome at SL5 must be exposed in order to promote dimerization in vitro
As the SL5 sequence promotes dimerization, and AN40 is more likely to fold into a conformation that exposes SL5, it should dimerize more readily. Conversely, as AN14 is more likely to fold into a structure in which the DIS is occluded, it should dimerize less readily. This was observed in both TBM (Figure 6A and B) and TBE gels (Figure 6B). There is a statistically significant (P < 0.01) increase in the ratio of dimer to monomer in the AN40 RNA relative to wild type and a statistically significant (P < 0.01) decrease in the ratio of dimer to monomer observed in AN14.
Figure 6.

Dimerization of AN40 and AN14 mutants. Wt, AN40 or AN14 RNA underwent dimerization as described in Figure 5, and the percentage of dimers and monomers was visualized by electrophoresis on 1% agarose gels in TBM or TBE. (A) Representative TBM gel. L; RNA ladder. (B) Densitometric analysis of the ratio of dimer to monomer in wt, AN40 or AN14 RNAs in TBM (black bars) or TBE gels (striped bars). Asterisks represent statistical significance relative to wt by t-test (P < 0.01). Error bars represent the SEM.
DISCUSSION
Applying the chemo-enzymatic probing technique SHAPE to the FIV leader has extended our previous work by revealing new insights and resolving areas of uncertainty within this RNA. In particular, an alternative structural model with practical functional implications for this important region of the FIV genome is proposed here. Thus, we have both extended our understanding of FIV RNA cis-acting signals affecting packaging and illustrated additional utilities of this probing technique.
Applying SHAPE to the FIV packaging signal structure revealed only minor differences from our LRI model of SLs 1–4 (compare Figures 1A and 2). SL1 differs in the exact position of the 3′ A bulge; A131 is unreactive, whereas A132 is not. In the LRI model, this bulge was modelled purely by minimal free energy, in silico, predictions, as neither nucleotide was targeted by the enzymes or chemicals previously used (35). Hence, it is likely that this bulge is consistently located at A131. SL2 structure is virtually identical to that of the LRI model, with minor differences in the internal loops around nts A200 and A250. According to SHAPE data, the loop at A250 includes the 2 bp above it. Due to the extremely high 1M7 reactivity of the 3′ side of these pairs, and the absence of nuclease CV1 cleavages and structural conservation of the U-A pair shown previously, the SHAPE-derived structure is likely to be more accurate. The internal loop around nucleotide A200 is suggested by SHAPE to comprise two smaller loops separated by two A-U pairs. These nucleotides vary from unreactive to highly reactive, and some were previously shown to be RNase A-sensitive, indicating single-stranded pyrimidines. Lack of structural conservation of these pairs suggests that they do not consistently form (35). When SL2 is transcribed in isolation 1M7 reactivity of most nucleotides is similar, indicating it is stable outside the context of the entire packaging signal RNA. The largest differences were found in the loop around nucleotide A200 and in the helix above it. When the adjacent 5′ and 3′ regions are absent, there is a slight decrease in 1M7 reactivity of the upper 5′ side of the loop, and an increase in reactivity in the lower 5′ side of the loop as well as the 5′ side of the helix above it, suggesting a stabilizing influence of the 5′ and 3′ regions. It is possible that there are long-range interactions involving a small number of nucleotides in the 5′ side of this loop. SL3 appears to form identically in both models, with metastability of the helix. SL4 is extended at the base in the MSL model.
The area between SL1 and SL2, which contains the primer binding site, was previously shown to be targeted by multiple single strand-specific enzymes (35), but exhibits low 1M7 reactivity. It is possible that in the structural form where the primer binding site (PBS) is single-stranded, backbone flexibility in this region is limited by non-Watson–Crick interactions, which could affect the enzymatic and SHAPE reagents differently. Another region of low 1M7 reactivity is the loop at nts C402–A409. In this case, non-Watson–Crick interactions may also stabilize the backbone, or alternatively, 5′ CUACA 3′ may form a pseudoknot with nucleotides 456–460: 3′ GAUGU 5′. Of the 61 strains of FIV for which there is sequence data for both of these regions, 57 have these sequences, one contains G at position 404 which could pair with U458 and three have single-nucleotide mutations that would not pair (35). Regions of low 1M7 reactivity at the bases of helices may be due to stacking interactions.
Both the intermediate 1M7 reactivity and a comparison of SHAPE results with enzymatic probing data, suggest that the 5′ and 3′ regions assume alternate structures. Minimal free-energy modelling using SHAPE data as pseudo free-energy constraints generated an alternative structure to resolve this. Native PAGE analysis and interrogation by mutagenesis and SHAPE indicate that the RNA adopts alternate monomeric conformations that could be stabilized into the LRI or MSL forms. Phylogenetic evidence for the ability of the RNA to assume alternate structures lies not only in their structural conservation between various FIV isolates, but also the fact that sequence conservation of the nucleotides that pair ‘differently’ in each structure is higher than that of those which pair ‘identically’ (35). Of the paired nucleotides in SLs 1–4, 39% vary in >5% of isolates. Despite this poor degree of sequence conservation, there is an exceptionally high degree of structural similarity between the isolates. Of the nucleotides that are paired differently in both structures, only 9% vary in more than 5% of isolates, indicating selective pressure to maintain these sequences. Although a higher degree of conservation might be expected for the 3′ sequences, as they lie within the gag coding region, the degree of sequence conservation is just as high within the 5′ interacting region, which is untranslated, indicating strong selective pressure to maintain both sequences. This would be expected if the same RNA formed two structures, since in most positions, simultaneous mutations would be necessary to preserve both structures. Many helices in the 5′/3′ interacting regions are G-U rich, which could be functionally important if one molecule of RNA has to switch between structures during the viral lifecycle.
Interestingly, in one of these conformations (LRI), the proposed DIS is exposed, while the AUG start codon is in the middle of a 7-bp helix, and the free energy of the overall structure is lower (ΔG = −181 kcal/mol), possibly favouring dimerization and packaging over translation. The overall analysis suggests that the primer binding site is unpaired and accessible for transfer RNA (tRNA) primer binding which would also be important for subsequent reverse transcription of the genomic RNA after infection.
In the other (MSL) structure, the DIS is occluded, the gag AUG is in a small, 3-bp SL, and the structure on the whole is less stable (ΔG = −145 kcal/mol), possibly facilitating translation over dimerization and packaging. The primer binding site is also occluded. The existence of two structures in the FIV 5′ region provokes comparison with a structural switch mechanism previously proposed for HIV-1 (38,39), and is schematically illustrated in Figure 7. In support of this, the AN14 mutant was recently shown to have impaired packaging (41), and is shown here to have impaired dimerization in vitro, and dimerization is enhanced when the RNA is stabilized in the LRI form.
Figure 7.

Accessibility of the functional elements in each structure. Schematic drawing showing the accessibility of the DIS, poly(A), major splice donor (mSD), PBS and gag AUG in the original LRI structure (left) and MSL structure (right).
The palindromic sequence of SL5 is exposed in only one of these structures. In vitro dimerization assays show that a palindromic sequence at SL5 is necessary for dimer formation, and that the dimers formed are tight dimers. 1M7 reactivity of these nucleotides under conditions favouring dimerization suggest that the region is double-stranded when a palindrome is present, and that nucleotides remain single-stranded when non-palindromic sequences are substituted. Thus, SL5 appears capable of participating in a loop–loop intermolecular interaction consistent with the kissing loop model of dimer initiation. HIV and other retroviruses studied to date contain the DIS in their 5′ UTR, and although retroviruses have been shown to selectively package a dimeric genome, whether dimerization occurs before or after Gag binding is not clear (16,48,49). The FIV DIS is located within the gag ORF, restricting the ability to dimerize efficiently to full-length genomic RNA and thus suggesting a potential packaging mechanism.
This model would also explain the requirement for two separate regions of the RNA for packaging (34), as these stabilize the RNA in the LRI model, exposing the DIS. Not only did successive 3′ truncations of the RNA fail to dimerize efficiently once the DIS sequence was no longer present, but in vitro transcribed packaging signal RNA with SLs 2–4 and the 3′ side of the LRI region intact (and hence the DIS), and the 5′ side deleted, also failed to form dimers as efficiently (Supplementary Figure S6B, 143F 511R).
The alternative FIV packaging signal structure published previously by another group differs from ours except in the structures of SL1, SL3 and SL4 (36). SHAPE data presented here lend further support to the existence of SL2 as a large, stable SL, which in the James and Sargueil model forms four separate SLs (36). However, we observe one of the structural motifs proposed by James and Sargueil (SLA) in our MSL structure which forms the upper helix and loop of SL1 in their model.
SHAPE has proven to be a useful addition to the repertoire of techniques for probing RNA structure. In concert with other methodologies its advantages of high throughput and ability to interrogate each base individually have provided here, as previously (50), a valuable additional insight into structure function relationships in RNA and revealed a potentially critical structural switch in the viral genomic RNA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
The Intramural Research Program of the National Cancer institute, National Institutes of Health (S.F.J.LeG.); the Wellcome Trust (078007/Z/05/Z to A.M.L.L. and T.A.R); and the Biomedical Research Centre (RG52162 to A.M.L.L). Funding for open access charge: Medical Research Council.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Dr Jenny Miller, National Cancer Institute for technical advice and helpful discussion.
REFERENCES
- 1.Olmsted RA, Hirsch VM, Purcell RH, Johnson PR. Nucleotide sequence analysis of feline immunodeficiency virus: genome organization and relationship to other lentiviruses. Proc. Natl Acad. Sci. USA. 1989;86:8088–8092. doi: 10.1073/pnas.86.20.8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Talbott RL, Sparger EE, Lovelace KM, Fitch WM, Pedersen NC, Luciw PA, Elder JH. Nucleotide sequence and genomic organization of feline immunodeficiency virus. Proc. Natl Acad. Sci. USA. 1989;86:5743–5747. doi: 10.1073/pnas.86.15.5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamamoto JK, Sparger E, Ho EW, Andersen PR, O'Connor TP, Mandell CP, Lowenstine L, Munn R, Pedersen NC. Pathogenesis of experimentally induced feline immunodeficiency virus infection in cats. Am. J. Vet. Res. 1988;49:1246–1258. [PubMed] [Google Scholar]
- 4.Pedersen NC, Ho EW, Brown ML, Yamamoto JK. Isolation of a T-lymphotropic virus from domestic cats with an immunodeficiency-like syndrome. Science. 1987;235:790–793. doi: 10.1126/science.3643650. [DOI] [PubMed] [Google Scholar]
- 5.Siebelink KH, Chu IH, Rimmelzwaan GF, Weijer K, van Herwijnen R, Knell P, Egberink HF, Bosch ML, Osterhaus AD. Feline immunodeficiency virus (FIV) infection in the cat as a model for HIV infection in man: FIV-induced impairment of immune function. AIDS Res. Hum. Retroviruses. 1990;6:1373–1378. doi: 10.1089/aid.1990.6.1373. [DOI] [PubMed] [Google Scholar]
- 6.Pecon-Slattery J, McCracken CL, Troyer JL, VandeWoude S, Roelke M, Sondgeroth K, Winterbach C, Winterbach H, O'Brien SJ. Genomic organization, sequence divergence, and recombination of feline immunodeficiency virus from lions in the wild. BMC Genomics. 2008;9:66. doi: 10.1186/1471-2164-9-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olmsted RA, Langley R, Roelke ME, Goeken RM, Adger-Johnson D, Goff JP, Albert JP, Packer C, Laurenson MK, Caro TM, et al. Worldwide prevalence of lentivirus infection in wild feline species: epidemiologic and phylogenetic aspects. J. Virol. 1992;66:6008–6018. doi: 10.1128/jvi.66.10.6008-6018.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakamura Y, Ura A, Hirata M, Sakuma M, Sakata Y, Nishigaki K, Tsujimoto H, Setoguchi A, Endo Y. An updated nation-wide epidemiological survey of feline immunodeficiency virus (FIV) infection in Japan. J. Vet. Med. Sci. 2010;72:1051–1056. doi: 10.1292/jvms.09-0574. [DOI] [PubMed] [Google Scholar]
- 9.Brown MA, Munkhtsog B, Troyer JL, Ross S, Sellers R, Fine AE, Swanson WF, Roelke ME, O'Brien SJ. Feline immunodeficiency virus (FIV) in wild Pallas' cats. Vet. Immunol. Immunopathol. 2010;134:90–95. doi: 10.1016/j.vetimm.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roelke ME, Brown MA, Troyer JL, Winterbach H, Winterbach C, Hemson G, Smith D, Johnson RC, Pecon-Slattery J, Roca AL, et al. Pathological manifestations of feline immunodeficiency virus (FIV) infection in wild African lions. Virology. 2009;390:1–12. doi: 10.1016/j.virol.2009.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elder JH, Lin YC, Fink E, Grant CK. Feline immunodeficiency virus (FIV) as a model for study of lentivirus infections: parallels with HIV. Curr. HIV Res. 2010;8:73–80. doi: 10.2174/157016210790416389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barraza RA, Poeschla EM. Human gene therapy vectors derived from feline lentiviruses. Vet. Immunol. Immunopathol. 2008;123:23–31. doi: 10.1016/j.vetimm.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaye JF, Lever AM. Human immunodeficiency virus types 1 and 2 differ in the predominant mechanism used for selection of genomic RNA for encapsidation. J. Virol. 1999;73:3023–3031. doi: 10.1128/jvi.73.4.3023-3031.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lever AM. HIV-1 RNA packaging. Adv. Pharmacol. 2007;55:1–32. doi: 10.1016/S1054-3589(07)55001-5. [DOI] [PubMed] [Google Scholar]
- 15.D'Souza V, Summers MF. How retroviruses select their genomes. Nat. Rev. Microbiol. 2005;3:643–655. doi: 10.1038/nrmicro1210. [DOI] [PubMed] [Google Scholar]
- 16.Greatorex J. The retroviral RNA dimer linkage: different structures may reflect different roles. Retrovirology. 2004;1:22. doi: 10.1186/1742-4690-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paillart JC, Shehu-Xhilaga M, Marquet R, Mak J. Dimerization of retroviral RNA genomes: an inseparable pair. Nat. Rev. Microbiol. 2004;2:461–472. doi: 10.1038/nrmicro903. [DOI] [PubMed] [Google Scholar]
- 18.Johnson SF, Telesnitsky A. Retroviral RNA dimerization and packaging: the what, how, when, where, and why. PLoS Pathog. 2010;6:e1001007. doi: 10.1371/journal.ppat.1001007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haddrick M, Lear AL, Cann AJ, Heaphy S. Evidence that a kissing loop structure facilitates genomic RNA dimerisation in HIV-1. J. Mol. Biol. 1996;259:58–68. doi: 10.1006/jmbi.1996.0301. [DOI] [PubMed] [Google Scholar]
- 20.Laughrea M, Jette L. Kissing-loop model of HIV-1 genome dimerization: HIV-1 RNAs can assume alternative dimeric forms, and all sequences upstream or downstream of hairpin 248–271 are dispensable for dimer formation. Biochemistry. 1996;35:1589–1598. doi: 10.1021/bi951838f. [DOI] [PubMed] [Google Scholar]
- 21.Laughrea M, Jette L, Mak J, Kleiman L, Liang C, Wainberg MA. Mutations in the kissing-loop hairpin of human immunodeficiency virus type 1 reduce viral infectivity as well as genomic RNA packaging and dimerization. J. Virol. 1997;71:3397–3406. doi: 10.1128/jvi.71.5.3397-3406.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen N, Jette L, Wainberg MA, Laughrea M. Role of stem B, loop B, and nucleotides next to the primer binding site and the kissing-loop domain in human immunodeficiency virus type 1 replication and genomic-RNA dimerization. J. Virol. 2001;75:10543–10549. doi: 10.1128/JVI.75.21.10543-10549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muriaux D, Fosse P, Paoletti J. A kissing complex together with a stable dimer is involved in the HIV-1Lai RNA dimerization process in vitro. Biochemistry. 1996;35:5075–5082. doi: 10.1021/bi952822s. [DOI] [PubMed] [Google Scholar]
- 24.Paillart JC, Skripkin E, Ehresmann B, Ehresmann C, Marquet R. A loop–loop “kissing” complex is the essential part of the dimer linkage of genomic HIV-1 RNA. Proc. Natl Acad. Sci. USA. 1996;93:5572–5577. doi: 10.1073/pnas.93.11.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Windbichler N, Werner M, Schroeder R. Kissing complex-mediated dimerisation of HIV-1 RNA: coupling extended duplex formation to ribozyme cleavage. Nucleic Acids Res. 2003;31:6419–6427. doi: 10.1093/nar/gkg873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner KB, Kohlway AS, Hagan NA, Fabris D. Noncovalent probes for the investigation of structure and dynamics of protein-nucleic acid assemblies: the case of NC-mediated dimerization of genomic RNA in HIV-1. Biopolymers. 2009;91:283–296. doi: 10.1002/bip.21107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hagan NA, Fabris D. Dissecting the protein–RNA and RNA–RNA interactions in the nucleocapsid-mediated dimerization and isomerization of HIV-1 stemloop 1. J. Mol. Biol. 2007;365:396–410. doi: 10.1016/j.jmb.2006.09.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moore MD, Hu WS. HIV-1 RNA dimerization: it takes two to tango. AIDS Rev. 2009;11:91–102. [PMC free article] [PubMed] [Google Scholar]
- 29.Miyazaki Y, Garcia EL, King SR, Iyalla K, Loeliger K, Starck P, Syed S, Telesnitsky A, Summers MF. An RNA structural switch regulates diploid genome packaging by Moloney murine leukemia virus. J. Mol. Biol. 2010;396:141–152. doi: 10.1016/j.jmb.2009.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Browning MT, Mustafa F, Schmidt RD, Lew KA, Rizvi TA. Sequences within the gag gene of feline immunodeficiency virus (FIV) are important for efficient RNA encapsidation. Virus Res. 2003;93:199–209. doi: 10.1016/s0168-1702(03)00098-4. [DOI] [PubMed] [Google Scholar]
- 31.Browning MT, Mustafa F, Schmidt RD, Lew KA, Rizvi TA. Delineation of sequences important for efficient packaging of feline immunodeficiency virus RNA. J. Gen. Virol. 2003;84:621–627. doi: 10.1099/vir.0.18886-0. [DOI] [PubMed] [Google Scholar]
- 32.Kemler I, Azmi I, Poeschla EM. The critical role of proximal gag sequences in feline immunodeficiency virus genome encapsidation. Virology. 2004;327:111–120. doi: 10.1016/j.virol.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 33.Kemler I, Barraza R, Poeschla EM. Mapping the encapsidation determinants of feline immunodeficiency virus. J. Virol. 2002;76:11889–11903. doi: 10.1128/JVI.76.23.11889-11903.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mustafa F, Ghazawi A, Jayanth P, Phillip PS, Ali J, Rizvi TA. Sequences intervening between the core packaging determinants are dispensable for maintaining the packaging potential and propagation of feline immunodeficiency virus transfer vector RNAs. J. Virol. 2005;79:13817–13821. doi: 10.1128/JVI.79.21.13817-13821.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kenyon JC, Ghazawi A, Cheung WK, Phillip PS, Rizvi TA, Lever AM. The secondary structure of the 5′ end of the FIV genome reveals a long-range interaction between R/U5 and gag sequences, and a large, stable stem-loop. RNA. 2008;14:2597–2608. doi: 10.1261/rna.1284908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.James L, Sargueil B. RNA secondary structure of the feline immunodeficiency virus 5′ UTR and Gag coding region. Nucleic Acids Res. 2008;36:4353–4666. doi: 10.1093/nar/gkn447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE) J. Am. Chem. Soc. 2005;127:4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- 38.Abbink TE, Ooms M, Haasnoot PC, Berkhout B. The HIV-1 leader RNA conformational switch regulates RNA dimerization but does not regulate mRNA translation. Biochemistry. 2005;44:9058–9066. doi: 10.1021/bi0502588. [DOI] [PubMed] [Google Scholar]
- 39.Abbink TE, Berkhout B. A novel long distance base-pairing interaction in human immunodeficiency virus type 1 RNA occludes the Gag start codon. J. Biol. Chem. 2003;278:11601–11611. doi: 10.1074/jbc.M210291200. [DOI] [PubMed] [Google Scholar]
- 40.Browning MT, Schmidt RD, Lew KA, Rizvi TA. Primate and feline lentivirus vector RNA packaging and propagation by heterologous lentivirus virions. J. Virol. 2001;75:5129–5140. doi: 10.1128/JVI.75.11.5129-5140.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rizvi TA, Kenyon JC, Ali J, Aktar SJ, Phillip PS, Ghazawi A, Mustafa F, Lever AM. Optimal packaging of FIV genomic RNA depends upon a conserved long-range interaction and a palindromic sequence within gag. J. Mol. Biol. 2010;403:103–119. doi: 10.1016/j.jmb.2010.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vasa SM, Guex N, Wilkinson KA, Weeks KM, Giddings MC. ShapeFinder: a software system for high-throughput quantitative analysis of nucleic acid reactivity information resolved by capillary electrophoresis. RNA. 2008;14:1979–1990. doi: 10.1261/rna.1166808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol. 2008;6:e96. doi: 10.1371/journal.pbio.0060096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reuter JS, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilkinson KA, Vasa SM, Deigan KE, Mortimer SA, Giddings MC, Weeks KM. Influence of nucleotide identity on ribose 2′-hydroxyl reactivity in RNA. RNA. 2009;15:1314–1321. doi: 10.1261/rna.1536209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW, Jr, Swanstrom R, Burch CL, Weeks KM. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009;460:711–716. doi: 10.1038/nature08237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laughrea M, Jette L. HIV-1 genome dimerization: formation kinetics and thermal stability of dimeric HIV-1Lai RNAs are not improved by the 1–232 and 296–790 regions flanking the kissing-loop domain. Biochemistry. 1996;35:9366–9374. doi: 10.1021/bi960395s. [DOI] [PubMed] [Google Scholar]
- 48.Kafaie J, Song R, Abrahamyan L, Mouland AJ, Laughrea M. Mapping of nucleocapsid residues important for HIV-1 genomic RNA dimerization and packaging. Virology. 2008;375:592–610. doi: 10.1016/j.virol.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 49.Sakuragi J, Shioda T, Panganiban AT. Duplication of the primary encapsidation and dimer linkage region of human immunodeficiency virus type 1 RNA results in the appearance of monomeric RNA in virions. J. Virol. 2001;75:2557–2565. doi: 10.1128/JVI.75.6.2557-2565.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deigan KE, Li TW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA structure determination. Proc. Natl Acad. Sci. USA. 2009;106:97–102. doi: 10.1073/pnas.0806929106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}