


Abstract
The conjugative transfer of F-like plasmids such as F, R1, R100 and pED208, between bacterial cells requires TraM, a plasmid-encoded DNA-binding protein. TraM tetramers bridge the origin of transfer (oriT) to a key component of the conjugative pore, the coupling protein TraD. Here we show that TraM recognizes a high-affinity DNA-binding site, sbmA, as a cooperative dimer of tetramers. The crystal structure of the TraM–sbmA complex from the plasmid pED208 shows that binding cooperativity is mediated by DNA kinking and unwinding, without any direct contact between tetramers. Sequence-specific DNA recognition is carried out by TraM’s N-terminal ribbon–helix–helix (RHH) domains, which bind DNA in a staggered arrangement. We demonstrate that both DNA-binding specificity, as well as selective interactions between TraM and the C-terminal tail of its cognate TraD mediate conjugation specificity within the F-like family of plasmids. The ability of TraM to cooperatively bind DNA without interaction between tetramers leaves the C-terminal TraM tetramerization domains free to make multiple interactions with TraD, driving recruitment of the plasmid to the conjugative pore.
INTRODUCTION
Bacterial conjugation diversifies bacterial genomes and enables virulence and antibiotics resistance factors to rapidly spread in medically important human pathogens (1). The F plasmid was the first conjugative plasmid identified (2), and F and its related plasmids are a model system for the elucidation of the fundamental molecular processes that underlie conjugative plasmid transfer (3). F-like plasmids were responsible for some of the earliest outbreaks of antibiotic-resistant Shigella infections in post-Second World War Japan (4), and F-like plasmids continue to confer antibiotic resistance to pathogenic bacteria in recent times (5,6)
During conjugation, a single-stranded DNA is transferred unidirectionally from a donor to a recipient cell in response to an uncharacterized mating signal (7,8). The proteins involved in conjugation are expressed by the plasmid-encoded tra operon. These proteins form the relaxosome, which processes plasmid DNA at the origin of transfer region, oriT and the transferosome, a large transmembrane complex classified as a Type IV secretion system (T4SS) (9). Among the Tra proteins is an auxiliary DNA-processing protein TraM, a cytoplasmic protein that is essential for conjugative transfer of the F and F-like plasmids (7). TraM has been found to enhance the nicking reaction that prepares DNA for transfer (10,11); however, it does not appear to be essential for either nicking or conjugative pore formation (12–15). Instead, TraM has been thought to regulate F conjugation by performing a crucial signaling function to trigger DNA transfer (7).
TraM was first identified as a DNA-binding protein specifically recognizing multiple sites within the origin of transfer (oriT) in the F and F-like plasmids (16–19). TraM binds this region with other factors such as IHF and TraY, which together facilitate the nicking reaction catalyzed by the relaxase/helicase TraI at the transfer initiation site (nic) (20–23). F plasmid TraM cooperatively binds to three sites (sbmA, sbmB, sbmC) within oriT. At high F TraM concentrations, TraM binds to secondary sites that radiate out from these primary DNA-binding sites, suggesting that it aggregates on the DNA in an orderly fashion (24). TraM represses its own gene promoter by binding to sbmA and sbmB (25), whereas sbmC is the most important site of the three for F conjugation (26). Sequence-specific DNA recognition is mediated by the TraM N-terminal domain (residues 2–56 in F TraM), which forms dimers (10,27–29). The C-terminal domain of TraM (residues 58–127 in F TraM) is responsible for tetramerization (27,28,30).
The crystal structure of the C-terminal domain reveals a symmetrical eight-helix bundle composed of four inter-twined protomers that is sensitive to changes in pH and temperature (31). TraM interacts with TraD, an inner membrane component of the conjugative pore, through its C-terminal tetramerization domain (32–35). The interaction is initiated through specific recognition between F TraM58–127 and the very C-terminal eight residues of TraD (33). Extensive genetic analysis indicates that this TraM–TraD interaction is required for F conjugation (32–34).
TraM proteins from F and various F-like plasmids function in a plasmid-specific manner during conjugation that is the result of TraM binding to its cognate sites within oriT (24). However, the mechanism for achieving cooperative DNA binding, and how this relates to ‘signaling’ is not fully understood. Here we show that the N-terminal DNA-binding domain of F plasmid TraM homo-dimerizes to form a ribbon–helix–helix (RHH) DNA-binding module, explaining the mechanism behind the allelic specificity of TraM in cognate DNA binding. We also show that a pair of TraM tetramers is required to cooperatively bind a minimal DNA-binding site with high affinity in both F and F-like pED208 plasmid systems. The crystal structure of pED208 TraM bound to an sbmA DNA reveals that alternating RHH modules from two different TraM tetramers contact staggered target GANTC sequence motifs within sbmA. In this way, each tetramer contacts a pair of GANTC motifs separated by 12 bp. Cooperative DNA recognition is achieved through the coordinated unwinding and kinking of the DNA to align alternating GANTC motifs on the same side of the helix for recognition by TraM. We also show that specific interactions between the TraM tetramerization domain and the C-terminal tail of its cognate TraD are a critical determinant of allelic specificity among F and F-like plasmid systems. This unusual mechanism of cooperative binding allows TraM to dictate plasmid specificity by binding to oriT and mediating relaxosome-coupling protein interactions simultaneously.
MATERIALS AND METHODS
Crystallization
Crystals of pED208 TraM–sbmA were obtained by vapor diffusion in hanging drops. In order to avoid hybrid electron density of bases around a 2-fold crystallographic axis, the sbmA fragment used in crystallization was designed to be palindromic, which required changing only 2 bp and no disruption of the GANTC-binding motifs (Figure 3B). No change in binding affinity resulted from this mutation (data not shown). TraM protein at 15 mg ml−1 in 0.5 M ammonium acetate was mixed with 0.5 mM sbmA DNA in 10 mM Tris pH 7.5, 100 mM NaCl so that the ratio of TraM monomers to DNA was 6:1. 2 µl of the protein–DNA mixture was mixed with 1 or 2 µl of reservoir solution consisting of 100 mM cacodylic acid pH 6.5, 36% MPD and 4.5% PEG2000. Crystals were flash-frozen in liquid nitrogen without any additional cryoprotectant. One microliter of the same TraM–sbmA mixture was mixed with 1 µl of reservoir solution consisting of 50 mM MES pH 6.0, 5% PEG4000 and 5 mM MgSO4. Proteolytic fragments of TraM consisting of the N-terminal domain residues 2–52 crystallized under these conditions (as confirmed by MALDI mass spectrometry, data not shown) in the space group P41212. These crystals were soaked for 10 min in 20% glycerol in reservoir solution prior to flash freezing in liquid nitrogen.
Figure 3.

Crystal structure of pED208 TraM bound to sbmA. (A) Orthogonal views of the overall structure of the TraM–sbmA complex. The α-helices and β-strands are indicated. Disordered linkers are indicated by spheres, one for each Cα that could not be refined. (B) sbmA sequence used for crystallization. Mutated residues are highlighted. GANTC TraM-binding motifs are underlined and numbered. (C) Interactions between the N-terminal domain of TraM and DNA. Hydrogen bonds are indicated by purple-dashed lines. The GAATC-binding motif consisting of bases G7 to T11 of sbmA is indicated with purple letters. (D) Schematic diagram of TraM–sbmA interactions for one TraM tetramer. Hydrogen bonds are indicated with dashed lines. (E) Sequence alignment of pED208 TraM and F TraM. Secondary-structure elements are indicated. Conserved hydrophobic core residues are highlighted in orange, DNA-contacting residues in yellow, TraD C-terminal tail-contacting residues in cyan and position 88, responsible for protonation-mediated destabilization of the F TraM tetramer, in green.
Structure solution
Native datasets for both crystal forms were collected at ALS 12.3.1 and 8.3.1 at Berkeley, CA and CLS at Saskatoon. Data from the TraM–sbmA complex was collected to 2.90 Å, and from the TraM N-terminal domain to 1.30 Å. Diffraction images were processed with HKL2000 (36).
Selenomethionine-substituted pED208 TraM did not bind DNA or crystallize, ruling out the possibility of determining the structure of the TraM–sbmA complex by Se-met MAD phasing. Initial phases were instead determined by molecular replacement utilizing the F TraM C-terminal domain (31) as a search model in MOLREP (37). Following refinement with NCS restraints on the four chains of the C-terminal tetramer, electron density for helices of the N-terminal domain was visible. The N-terminal domain was solved by manually building the N-terminal helices into visible electron density, followed by refinement with NCS restraints between the four copies of α1 and α2. The helices were then replaced with a model based on the high-resolution structure of the MetJ RHH domain (PDB ID: 1CMB) (38) and refined. The resulting electron density map revealed the DNA phosphate backbone and base pairs, into which a B-DNA model of the sbmA duplex was placed using Coot (39). The TraM–sbmA model was further refined by cycles of manual building in Coot, followed by TLS and NCS-restrained maximum likelihood refinement and simulated annealing refinement, carried out with REFMAC (40) and CNS (41), respectively.
The high-resolution structure of the N-terminal domain was phased by molecular replacement with MOLREP (37) using the structure of the N-terminal domain from the low-resolution structure as a search model. flex–wARP (42) was used to build the residues of the N-terminal domain into electron density, and the model was refined with REFMAC (40). The high-resolution pED208 N-term domain structure was placed instead of the MetJ model in the TraM–sbmA complex, followed by further refinement of the TraM–sbmA complex.
The crystallographic asymmetric unit contains one TraM tetramer and one strand of sbmA DNA. The biologically relevant complex containing duplex sbmA and a pair of TraM tetramers is obtained by a crystallographic 2-fold rotation. Figures were made in Pymol (http://www.pymol.org). DNA conformation was analyzed with 3DNA (43).
Electrophoretic mobility shift assay of pED208 TraM and sbmA
sbmA oligos were 32P-labeled with T4 polynucleotide kinase (Invitrogen) and unincorporated nucleotides were removed by P-30 Micro Bio-Spin columns buffered in 10 mM Tris, pH 7.4 (Bio-Rad). TraM–sbmA-binding buffer was 50 mM Tris, pH 7.5, 10% glycerol, 30 ng µl−1 bovine serum albumin (Pierce), 20 ng µl−1 polydI·dC (Roche). To each binding reaction containing the indicated amount of TraM and incubated, 0.1 nM of sbmA oligo was added for 10 min at room temperature. TraM–sbmA mixtures were run on 1× TBE-buffered 12% 29:1 acrylamide gels at 200 V for 45 min at 4°C. Bands were visualized by phosphor screen and band intensities were determined with Imagequant. Kd values were calculated by fitting binding curves to the equation y = a × x/[1 + (a × x)], where a is the Ka, y is the ratio of bound to unbound DNA, and x is the protein concentration, using SigmaPlot (http://www.sigmaplot.com).
Donor ability assays
Escherichea coli XK1200 and ED24 were used as donor and recipient strains, respectively. The mating experiments were performed as previously described (27,44). Donor ability was calculated as the number of transconjugants divided by the number of donors. Each assay was repeated three times and the averaged values are reported. Standard deviations of all mating assays were within one log unit. Additional Materials and Methods are provided in Supplemental Data available online.
Accession numbers
Coordinates and structure factors have been deposited in the Protein Data Bank (http://www.rcsb.org/pdb) under ID codes 3ON0 (TraM–sbmA complex) and 3OMY (TraM N-terminal domain).
RESULTS
The N-terminal DNA-binding domain of TraM is an RHH fold
NMR analysis of the N-terminal domain of the R1 TraM protein suggested that this domain adopts a monomeric, helix-turn-helix structure, with a disordered N-terminal region at pH 4.0 (45). However, sedimentation analyses of the DNA-binding domains of F and R1 TraM clearly revealed that this domain adopts a dimeric structure at neutral pH (27,30). Furthermore, analyses of the DNA-binding properties of an extensive series of F TraM point mutants clearly indicated a critical role for the 10 N-terminal residues in specific DNA recognition (28). To reconcile these results, we hypothesized that the TraM DNA-binding domain adopts a dimeric RHH fold (46) (Figure 1A), which is commonly found in bacterial transcriptional repressors, as well as the R388 plasmid auxiliary DNA-processing protein TrwA (47).
Figure 1.

The N-terminal domain of pED208 TraM binds to sbmA as a ribbon-helix-helix fold. (A) Primary structure alignment of TraM homologues and RHH fold domains. DNA-contacting β-strand residues are boxed in purple. Conserved residues are highlighted. Hydrophobic, orange; Aromatic, magenta; acidic, red; basic, cyan; Gly/Pro, green; polar aliphatic, yellow. Secondary structure elements of TraM are indicated. F TraM mutations shown in previous studies to disrupt F plasmid conjugation are indicated. R24 and R29, F TraM residues protected from trypsin digestion upon binding to sbmA, are boxed in blue. (B) Complementation of TraM proteins in a TraM-deficient F-derived plasmid system. (C) EMSA of F, R100 and R100 R3K:I5N TraM binding to 30-bp F sbmA. Concentrations in each lane are 0, 2, 5, 10, 20, 50, 100, 250, 600, 1000, 2500, 6000, 10 000 and 25 000 nM.
The β-ribbon of the RHH fold provides critical residues for DNA recognition that directly contact DNA base pairs. While members of the TraM family are quite similar throughout their N-terminal domains, many bind different DNA elements, providing a basis for allelic specificity (10,29,44). For example, R100 TraM functions poorly in complementation assays for conjugative transfer of a TraM-deficient F plasmid derivative, pOX38-MK3 (Figure 1B). To understand the basis for this specificity, we generated a double mutant (R3K:I5N) of R100 TraM such that its predicted β-ribbon region is the same as that of the F plasmid TraM (Figure 1A). The side chains of residues 3 and 5 are predicted to be exposed on the surface of the RHH β-ribbon and in contact with DNA. When complementing pOX38-MK3, this R100 TraM mutant resulted in a 60 000-fold increase in conjugation efficiency compared to the wild-type R100 TraM. As a control, the F plasmid TraM N5D mutant, which is defective in DNA binding, was cloned in the same vector, resulting in undetectable levels of conjugation (Figure 1B). EMSA analysis indicated that the R100 double mutant has a dramatically increased binding affinity for F plasmid cognate DNA, sbmA, compared to the wild type (Figure 1C). Both F conjugation efficiency and sbmA-binding ability of the R100 TraM double mutant are slightly lower than those of F plasmid TraM, suggesting that some residues outside the N-terminal β-ribbon sequence might also contribute to cognate DNA binding.
Pairs of TraM tetramers cooperatively bind DNA
F TraM binds to three related DNA sequences, termed sbmA, sbmB and sbmC, all located within an ∼200-bp region of DNA (oriT) located between the traM promoter and the plasmid nic site (24). sbmA is bound with highest affinity and is distinguished by the highest level of sequence symmetry. Each sbmA contains four-sequence repeats, A(G/C)CG(G/C)T, arranged around a centre of palindromic symmetry (Figure 2A). TraM itself is a tetramer (27,30,31). To characterize the stoichiometry of binding of F TraM to its cognate sbmA, we used multi-angle laser light scattering (MALLS) to determine the molecular mass of TraM–DNA complexes purified by size exclusion chromatography (Figure 2B). This analysis showed that the TraM–sbmA complex forms a ∼132-kDa complex. Given the mass of the sbmA DNA is ∼19.8 kDa and the mass of a single TraM protomer is ∼14.4 kDa, this result suggests that the stable complex formed contains two TraM tetramers bound to a single DNA. This result was confirmed using an electrophoretic mobility shift assay (EMSA) carried out at protein and DNA concentrations well in excess of the Kd of the complex. These results clearly demonstrate that 8 molar equivalents of TraM are required to bind the sbmA duplex (Figure 2C). No evidence of higher mobility species corresponding to a single tetramer bound to sbmA was observed even at sub-stoichiometric amounts of TraM, indicating that pairs of TraM tetramers cooperatively recognize a single sbmA. In a similar experiment, 8 molar equivalents of pED208 TraM bound to its cognate DNA with no intermediate species, demonstrating that the requirement for two TraM tetramers to cooperatively bind sbmA is conserved between the two F-like plasmids (Supplementary Figure S3A). Interactions between F TraM and a larger 75-bp DNA containing both the sbmA and sbmB sites was also characterized by MALLS (Figure 2B). This DNA formed a stable complex with TraM with molecular weight of ∼260 kDa, consistent with a binding stoichiometry of four tetramers per DNA (predicted M.W. = 274 kDa), indicating that a pair of F TraM tetramers also bind sbmB.
Figure 2.
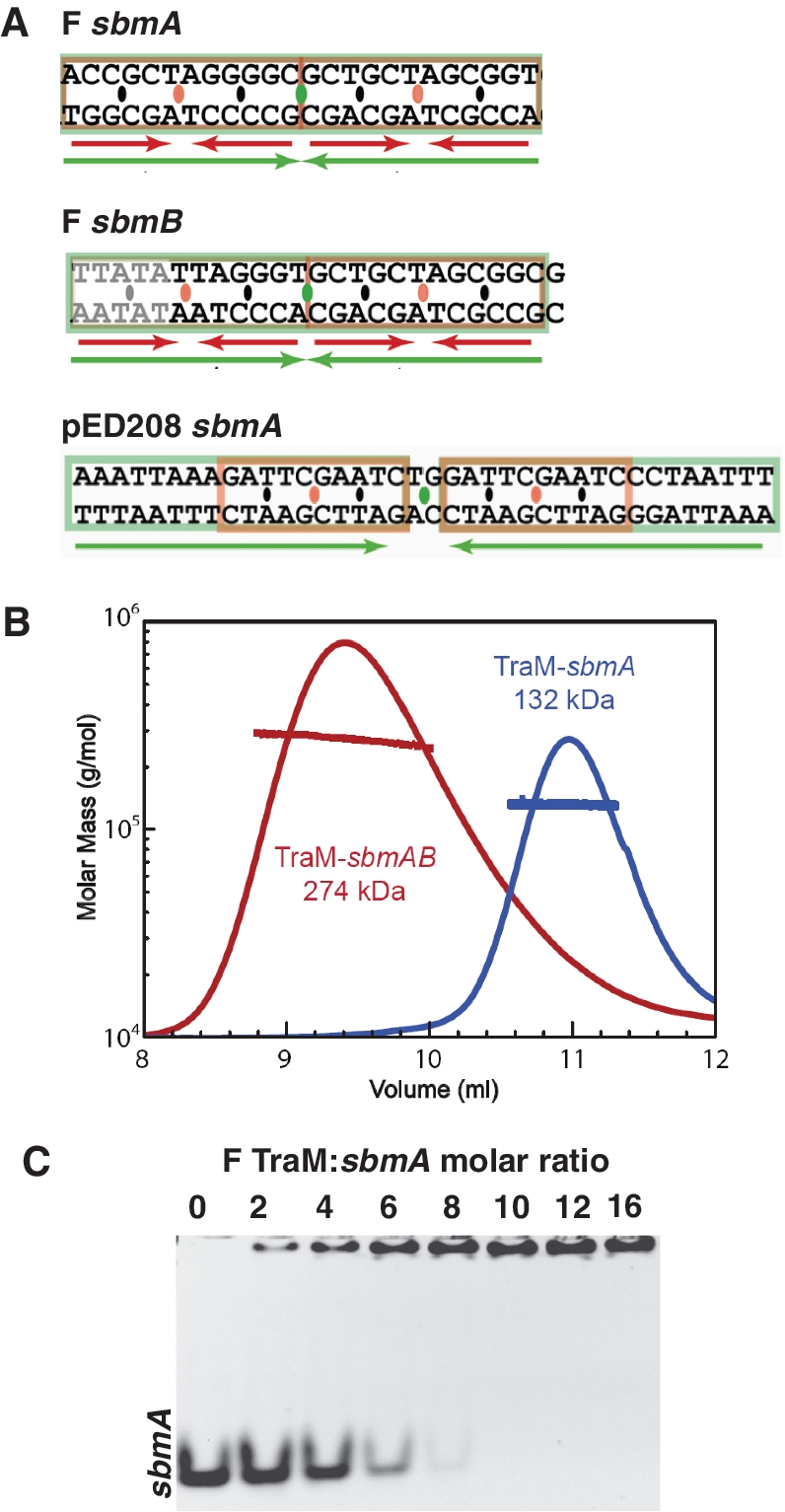
Binding of F and pED208 TraM to sbm DNA. (A) The sbm sequences from F and pED208 plasmids. Inverted repeats are indicated with green and red arrows. TraM-binding motifs are separated by orange ovals, and their centers of symmetry are indicated by black ovals. The center of symmetry of the sbm site is indicated by a green oval. (B) MALLS analysis of molecular weight of F TraM 30-bp sbmA complex and TraM 75-bp sbmAB complex. The traced peaks are the refractive index of the eluted material and the superimposed lines indicate the molecular weight of the protein or protein–DNA complex over the corresponding portion of the elution peaks. (C) Binding stoichiometry analyzed by titration of F TraM with 30-bp sbmA.
Overall structure of the TraM–sbmA complex
In order to gain insights into the mechanism of TraM cooperative binding to sbmA, we set out to crystallize and determine the structure of TraM bound to an sbmA-containing DNA. While we were unable to obtain diffraction quality crystals of F TraM bound to its cognate sbmA DNA, we were able to crystallize and determine the structure of the related pED208 TraM bound to its sbmA site at 2.90 Å resolution (Table 1, Supplementary Figure S1A, Materials and Methods).The structure of the isolated N-terminal domain of pED208 TraM was also determined to 1.30 Å resolution (Table 1, Supplementary Figure S2A, Materials and Methods). pED208 belongs to the IncFV incompatibility group and is a transfer-derepressed derivative of the Folac plasmid originally isolated from Salmonella typhi (48,49). F TraM and pED208 TraM are 38% identical at the amino acid sequence level (Figure 3E and Supplementary Figure S3D) and they bind distinct sbmA DNAs. While both proteins bind sbmA sites containing sequence motifs spaced 12-bp apart with an inverted repeat symmetry, the core sequence motif bound by pED208 TraM, GANTC, which is also palindromic, is distinct from that bound by F TraM (Figure 2A).
Table 1.
Data collection and refinement
| TraM–sbmA complex | TraM N-terminal domain | |
|---|---|---|
| Data collection | ||
| Space group | C2221 | P41212 |
| Cell dimensions | ||
| a, b, c (Å) | 93.0, 154.7, 167.6 | 54.1, 54.1, 67.3 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Wavelength (Å) | 1.1158 | 1.1158 |
| Resolution (Å) | 2.90 | 1.30 |
| Rsymb | 0.070 (0.433)a | 0.044 (0.33) |
| I/σI | 23.0 (3.0) | 39.2 (6.6) |
| Redundancy | 6.3 (3.5) | 7.5 (7.4) |
| Completeness (%) | 99.3 (94.2) | 99.85 (100.0) |
| Refinement | ||
| Resolution (Å) | 50.0–2.90 | 50.00–1.30 |
| Number of unique reflections | 27 583 (2568) | 25 217 (2459) |
| Rworkc/Rfreed (%) | 25.0/27.8 | 15.1/17.3 |
| Number of protein atoms in asymmetric unit | 4226 | 1284 |
| B-factor (overall) | 32.4 | 10.0 |
| Bond angle rmsd (°) | 1.35 | 1.38 |
| Bond length rmsd (Å) | 0.011 | 0.011 |
aData of the highest resolution shell (2.90–3.00 Å for TraM–sbmA complex, 1.30–1.35 Å for TraM N-terminal domain) are shown in parentheses.
bRsym = ΣhklΣi |Ii(hkl) − <I(hkl)>|/ΣhklΣi Ii(hkl), where Ii(hkl) is the intensity for an observation of a reflection and <I(hkl)> is the average intensity of all symmetry-related observations of a reflection.
cRwork = Σhkl| |Fobs| − |Fcalc| |/Σhkl|Fobs|.
dRfree = Rwork calculated for 5% of reflections excluded from refinement.
The structure of the TraM–sbmA complex reveals that two TraM tetramers bind sbmA (Figure 3A), validating the MALLS and EMSA results (Figure 2B and C, and Supplementary Figure S3A). As predicted by the specificity swap experiments, the N-terminal regions adopt dimeric RHH folds that each contact one of the GANTC sites within sbmA. The two tetramers contact sbmA in a staggered arrangement in which the two N-terminal domains of one tetramer bind the fiirst and third GANTC motifs, while those of the second tetramer bind the second and fourth GANTC repeats (Figure 3B). In this way, the two tetramers are arranged on nearly opposite sides of the DNA. Each RHH domain is connected to the C-terminal tetramerization domain via flexible peptide linkers corresponding to residues 56–60. The end of α2 preceding the linker is unwound to different degrees in different protomers (Supplementary Figure S1B), providing additional conformational flexibility to the tetramer which may facilitate TraM–DNA binding. The RHH and tetramerization domains do not contact each other, suggesting that these domains are flexibly tethered to one another and otherwise do not interact.
N-terminal domain structure and TraM–sbmA interactions
The N-terminal strands of each RHH domain form a two-stranded anti-parallel β-sheet that provides amino acid side chains, Lys3, Gln5 and Tyr7, that enter the DNA major groove and contact the pseudo-palindromic GANTC repeat in a sequence-specific and symmetric manner (Figure 3C and D). The conserved A–T base pair is recognized by Gln5, which makes a pair of hydrogen bonds with the face of the adenine base, an interaction commonly observed in protein–nucleic acid interactions (50). The orientation of the Gln5 side chain is stabilized through an additional hydrogen bond to Lys3 in the other β-strand. The conserved G–C pair is recognized by Tyr7. Tyr7 also forms a hydrogen bond with Lys3, which likely helps to position this side chain and ensure that the terminal hydroxyl group is oriented to donate a hydrogen bond to the guanine N7 atom. Lys3 may also directly contribute to recognition of the guanine through a long hydrogen bond between Lys3 Nε and guanine O6. The RHH domain also makes strong, symmetric interactions with the DNA backbone on either side of the GANTC motif. The interaction involves the N-terminus of α2, which is aligned so that its helix dipole, the main chain NH of Leu33, and the hydroxyl groups of Ser32 and Ser34, interact with the DNA phosphate backbone of the base preceding the G of the GANTC motif (Figure 3C and D). A comparison of the structure of the DNA-bound form of TraM and the free TraM RHH domains reveals subtle conformational change induced by DNA binding (Supplementary Figure S2). The α1–α2 loop comes into closer contact with the DNA backbone (Supplementary Figure S2B), and reorientation and stabilization of the β1 DNA-contacting side chains (Supplementary Figures S2C and D) occurs upon DNA binding.
pED208 TraM binds its cognate sbmA with high affinity (Kd = 4.8± 0.7 nM, Supplementary Figure S3B), similar to the affinity of F TraM for its cognate sbmA site (24). Tetramerization is essential for high-affinity binding, as the isolated N-terminal domain binds with ∼500 times lower affinity (Supplementary Figure S3C). We used a competitive EMSA assay to test the relative importance of the conserved GC and AT base pairs within the GANTC motifs for TraM binding (Figure 4A and B). Mutation of GtoC or AtoT is deleterious to TraM binding, and AtoT mutations have a greater binding defect. These results demonstrate that TraM recognizes the four GANTC motifs in sbmA in a highly specific and cooperative manner.
Figure 4.

Role of residue-specific interactions in TraM–sbmA interaction. (A) sbmA oligos used in competition assays. (B) Effect of mutation of G or A in the GANTC motifs of sbmA on competition of wild-type pED208 TraM from TraM–sbmA complexes. Oligos with mutations in similar numbers of GANTC motifs are aligned vertically for comparison. Amounts of competitor in each lane, as number of times labeled DNA are 0, 100×, 1000×, 2500×, 7500×, 15 000× and 25 000×.
TraM–DNA interactions are stabilized by cooperative DNA unwinding and distortion
The crystal structure of the TraM–sbmA complex reveals that binding of sbmA is achieved without direct contact between the two TraM tetramers, suggesting that the mechanism of cooperative DNA binding must act through the DNA itself (Figure 5). Analysis of the DNA structure reveals that it is significantly distorted compared to standard B-DNA. Strikingly, the DNA is significantly underwound with an average helical twist of 32° between staggered GANTC sites, compared to 36° per base pair in standard B-DNA. This unwinding results in a duplex structure with ∼12 bp per turn, which aligns alternating GANTC sites on the same side of the DNA and facilitates recognition of the alternating GANTC sites by RHH domains from a single tetramer. The binding of one tetramer to one face of the sbmA DNA would thus unwind and align the unbound GANTC sites on the opposite side of the DNA such that they would be positioned to interact with the second TraM tetramer, thereby facilitating cooperative recognition of sbmA.
Figure 5.

Cooperative recognition of sbmA DNA by TraM is mediated by DNA unwinding and kinking. (A) Alignment of N-terminal domains of one TraM tetramer, as observed in the crystal structure. Binding of TraM to sbmA results in DNA unwinding and alignment of the N-terminal DNA-binding domains of one tetramer on the same side of the DNA. (B) Model of TraM N-terminal domains bound to sbmA in an ideal B-DNA conformation. (C) Electrostatic surface potential map of TraM bound to sbmA. The DNA helix axis (indicated by pink spheres) appears to be bent by attraction to the basic β-sheet surface and repulsion by the acidic loops between α1 and α2. (D) DNA kinking induced in sbmA is driven by repulsion of the DNA backbone by the α1–α2 loop. The negatively charged side chains of Glu29 and Glu30, indicated by red spheres, repel the phosphate backbone. The DNA helix axis is indicated by a grey line. GANTC-binding motifs are indicated in blue and green letters. (E) Effect of varying the number of base pairs between two GANTC motifs in sbmA on the binding of pED208 TraM measured by EMSA. Each DNA contains only two GANTC motifs (highlighted in red) separated by either 12 (left panel), 11 (centre panel) or 10 bp (right panel). pED208 TraM concentrations in each lane are 0, 2, 5, 10, 20, 50, 100, 250, 600, 1000, 3000, 10 000 and 30 000 nM. (F) F TraM and pED208 TraM can cooperatively bind a hybrid sbmA. Binding of pED208 TraM (left), F TraM (center) and a 1:1 mixture of F and pED208 TraM (right) to a hybrid sbmA containing the GANTC motifs of pED208 in positions 1 and 3 (highlighted in red), and the A(G/C)CG(G/C)T motifs of F in positions 2 and 4 (highlighted in blue) of sbmA were assessed by EMSA. TraM concentrations in each lane are 0, 2, 5, 10, 20, 50, 100, 250, 600, 1000, 3000, 10 000 and 25 000 nM.
We reasoned that if DNA unwinding is important for cooperativity, then binding of a single TraM tetramer to DNA containing a pair of GANTC repeats could be achieved by reducing the spacing between the repeats to better match the helical pitch of B-DNA (Figure 5A and B). To test this idea, we compared the ability of pED208 TraM to bind a double-stranded DNA containing only a single pair of GANTC sites separated by 12 bp (as in sbmA), with DNAs in which the pair of GANTC sites are separated by either 11 or 10 bp using EMSA (Figure 5E). Interaction between TraM and DNA with the 12-bp spacing was weak. However, reduction of the spacing to 11 bp significantly enhanced TraM binding. Further reduction of the spacing to 10 bp essentially abrogated binding. Modeling suggests that docking of two RHH modules on GANTC sites separated by 10 bp on a B-form double helix would lead to significant clashes between the α1 helices and α1–α2 loops, explaining the loss of binding with this DNA. While these experiments support the importance of the positioning of the GANTC sites on the same side of the DNA double helix for TraM binding, the fact that the binding of the 11-bp spacer DNA is still much weaker than the intact sbmA (Supplementary Figure S3B) indicates that other mechanisms must also facilitate cooperative TraM–DNA interactions.
Further comparison of the sbmA structure with that of ideal B-DNA reveals significant kinking of the DNA helix axis, mainly localized to the junctions between the GANTC repeats (Figure 5C and D). This kinking appears to be largely due to interactions between TraM and the DNA backbone. As noted above, the N-termini of the α2 helices bind the phosphate backbone, anchoring the RHH domain to both sides of the DNA major groove, facilitating a conformational change that results in the unwinding of the DNA within the GANTC site, as well as widening and deepening of the major groove. At the same time, a pair of acidic residues, Glu29 and Glu30, is positioned in the loop between α1 and α2 to make unfavorable electrostatic interactions with the DNA helical backbone. In response, the DNA bends into the major groove to minimize these interactions (Figure 5C and D). This push of the backbone away from one RHH helps to wrap the next GANTC site around its RHH domain. The distortions induced in sbmA upon TraM binding are illustrated by animations in which the DNA is morphed from idealized B-form DNA to that observed in the crystal structure (Supplementary Movie S1 and 2).
We next asked whether similar deformations in DNA structure are involved in the cooperative DNA interactions by other TraM proteins. Although the binding motifs are not conserved between the F and pED208 plasmids, the 12-bp spacing between sites bound by the same TraM tetramer is (Figure 2A). To ascertain whether the DNA deformations induced by pED208 TraM are similar to those induced by F TraM, we tested the ability of pED208 and F TraM to bind a hybrid sbmA DNA in which the first- and third-binding motifs correspond to those found in pED208 sbmA, while the second and fourth motifs correspond to F sbmA (Figure 5F). While either F TraM or pED208 TraM bound this DNA with significantly reduced affinity in EMSA, an equimolar mixture of F and pED208 TraM tetramers bound this DNA at high affinity, similar to the affinity of either F TraM or pED208 TraM for their cognate sbmA. Moreover, the mobility of the shifted species derived from the pED208/F TraM mixture was distinct from that of either the F TraM complex or the pED208 complex, demonstrating that the complex derived using the protein mixture contains both F and pED208 TraM tetramers (Figure 5F). Thus, this result indicates that the structural changes induced in the DNA by these two tetramers are similar enough to facilitate cooperative binding on the hybrid sbmA, and suggest that similar distortions will facilitate DNA recognition within the family of TraM proteins.
Selective TraM–TraD interactions also govern allelic specificity
In addition to DNA binding, TraM also must contact the conjugative pore protein, TraD, to effect conjugation (34). TraM–TraD interactions critically depend on the recognition of the C-terminal tail of TraD by a groove on the surface of the TraM tetramerization domain (33). To test the relative importance of TraM–DNA and TraM–TraD interactions for plasmid specificity, we assessed the ability of F or pED208 TraM and TraD, as well as chimeric molecules derived from these proteins, to rescue conjugative transfer of a TraM- and TraD-deficient F plasmid derivative (Figure 6). A critical role for plasmid specific TraM–TraD interactions was demonstrated by the finding that a chimeric TraM with an F N-terminal domain and a pED208 C-terminal domain, TraM[F1–55:pED20856–127], does not complement a TraM-deficient F plasmid (Figure 6A), despite the fact that the chimeric protein binds to F sbmA with wild type affinity (Supplementary Figure S4). Likewise, substitution of the eight C-terminal residues of F TraD with those of pED208 TraD in TraD[F1–709:pED208729–736] also disrupts conjugation when co-expressed with F TraM. Significantly, mating is rescued when both chimeric proteins, TraM[F1–55pED20856–127] and TraD[F1–709:pED208729–736], are co-expressed, demonstrating plasmid specific TraM–TraD interactions rely on specific interactions between the C-terminal TraD tail and the TraM tetramerization domain (Figure 6A).
Figure 6.

Conserved mechanisms of allelic specificity of TraM–sbmA and TraM–TraD interaction. (A) Double complementation of a TraM and TraD-deficient F-derived plasmid system by F and pED208 TraM and TraD mutants. F plasmid components are in cyan, pED208 components are in magenta. (B) Sequence alignment of C-terminal tails of TraD. Conserved acidic residues are highlighted in red, basic residues in cyan, glycines in green and aromatic residues in magenta. The residues that show charge exchange between pED208 and F are boxed. (C) View of the TraD-binding pocket in the F TraM C-terminal tetramer (33). (D) Model of pED208 TraM C-terminal domain—TraD C-terminal tail interaction based on the F structure.
A comparison of the sequences of the TraD tails and the structures of TraD-binding pockets on TraM in the two plasmid systems suggests an explanation for this specificity. pED208 TraD contains a single positively charged residue (Arg734) in its otherwise highly negatively charged tail at a position that is negatively charged in F TraD (Figure 6B). Modeling of the pED208 TraM–TraD interaction based on the structure of F TraM–TraD complex suggests that pED208 TraD Arg734 will be juxtaposed with a negatively charged residue (Glu81) in the pED208 TraM pocket (Figure 6D). In F TraM, this residue is positively charged (Lys83) (Figure 6C). To test the hypothesis that complementary charge interactions between TraM and TraD may help define specificity, we mutated Arg734 to Asp in the chimeric TraD protein TraD[F1–709:pED208729–736:R734D]. This protein significantly rescued conjugation compared to TraD[F1–709:pED208729–736] providing further support that side chain-specific interactions analogous to those observed in the F plasmid system (33) are necessary in vivo for conjugation in pED208 (Figure 6A). These interactions define the binding specificity of the F and pED208 TraD C-terminal tail for their cognate TraM.
DISCUSSION
TraM is a member of the RHH family of plasmid regulatory proteins
Our work reveals that TraM is a member of the large class of bacterial repressor proteins that bind DNA through a RHH DNA recognition module (46). RHH modules appear to be a common DNA-recognition module among plasmid regulatory proteins. For example, F-like plasmids encode at least one other RHH DNA-binding protein, TraY, which is critical for the activation of plasmid nicking through interactions with the nickase/relaxase, TraI (14,15,51). The R388 plasmid encodes a single RHH protein, TrwA, which likely fulfills both TraY and TraM functions in that it appears to interact with and activate both the TraD-like coupling protein, TrwB (52,53), as well as the nickase/relaxase, TrwC (47). The smallest of the plasmid regulatory RHH proteins is CopG, which regulates copy number of the streptococcal pMV158 plasmid (54). Particularly intriguing is the Agrobacterium tumefaciens T DNA-regulatory protein, VirC2, which binds DNA through a novel, tandem repeat RHH module (55). In this case, the RHH dimer is formed from a single polypeptide chain, where the loop connecting the two RHH motifs wraps around the structure to form a topological knot (56).
Cooperative DNA binding is mediated through DNA distortion
The regulation of bacterial transcription often involves the formation of hierarchical assemblies of oligomeric DNA-binding proteins on large, complex DNA elements. In general, these higher order interactions are mediated by protein–protein interactions, such as the interactions between dimers of λ repressor bound to adjacent DNA elements (57), or the interactions of tetrameric TgtV repressor with DNA containing two adjacent recognition elements (58).Thus, it was highly surprising that pairs of TraM tetramers cooperatively bind the high affinity sbmA DNA without direct contact between the tetramers. Instead, cooperativity is mediated by a protein-induced distortion of the DNA that facilitates simultaneous binding of both tetramers. TraM binding induces an unwinding of the DNA to an ∼12 bp per turn form that aligns alternating GANTC motifs on the same side of the DNA double helix for recognition by two RHH domains presented from a single tetramer (Figure 3A). In addition, the protein also kinks the DNA and deforms the groove widths, through a combination of attractive electrostatic and hydrogen-bonding interactions, as well as repulsive interactions between negatively charged residues in the α1–α2 loop and the DNA phosphodiester backbone (Figure 5C and D). The fact that the 12-bp spacing of sequence elements is conserved in the sbm sites of other plasmids suggests that similar mechanisms will mediate DNA-binding cooperativity in other TraM proteins (Figure 2A). Our demonstration that a hybrid pED208-F sbmA site in which DNA-binding motifs alternate between pED208 and F, is only bound with high affinity by a mixture of pED208 TraM and F TraM, and not by the individual proteins, strongly suggests that the DNA deformations that facilitate DNA-binding cooperativity are conserved throughout the TraM family (Figure 5F). We suggest that cooperative DNA recognition by TraM tetramers proceeds through the model outlined in Figure 7A. Initially, a single tetramer binds DNA, inducing the underwound and kinked DNA conformation in an unstable, high-energy-intermediate state. The DNA is thus primed for the binding of the second tetramer to the opposite face of the DNA, thereby stabilizing the cooperative TraM–DNA complex.
Figure 7.

Models for cooperative recognition of DNA and TraD by TraM. (A) Model for cooperative binding of sbmA by TraM. sbmA DNA exists in a B-like conformation in the absence of TraM (left). A single TraM tetramer (blue) binds a pair of GANTC elements via its two RHH domains, thereby unwinding and kinking the DNA to form an unstable intermediate complex (center panel). Binding of the first tetramer induces a DNA conformation that aligns the remaining free pair of GANTC elements on the opposite side of the DNA helix, which facilitates binding of the second tetramer and stabilization of the complex (right). (B) Stabilization of TraM–TraD interactions through an avidity effect. TraD (gold) exists as a hexamer ring complex and forms the cytoplasmic face of the conjugative pore, imbedded within the cytoplasmic membrane (purple). While isolated interactions between a single TraD tails and a TraM tetramerization domain are weak, cooperative TraM–DNA complexes provide multiple contact points for the C-terminal tails of TraD (yellow).
Interestingly, similar mechanisms of protein-induced DNA distortions have been uncovered in other bacterial transcriptional repressors. For example, three repressor proteins in the TetR family, QacR, IcaR and CgmR, bind DNA sites in a staggered arrangement in which the DNA is underwound and kinked (59–61). Similarly, members of the iron-dependent regulator family (IdeR and DtxR), which are activated by the binding of divalent metal ions, also bind DNA in a similar manner, utilizing DNA deformation and not protein–protein interactions to effect cooperativity (62–65).
Role of TraM in the definition of allelic specificity
Core components of conjugative type IV secretion pores can transfer proteins with the appropriate translocation signals (66), whereas the conjugative pore requires both a T4SS and a coupling protein that recognizes the relaxosome in preparation for DNA transfer. In F-like plasmids, this recognition event prepares the relaxase, which contains complex internal translocation signals and is covalently bound to the nic site, for translocation to the recipient cell (67). Thus, the relaxosome accessory protein, TraM, and the coupling protein, TraD, confer a high level of selectivity for the cognate relaxosome (68), with the DNA-binding specificity of TraM being key for this selectivity. Both R1 and pED208 TraM RHH modules bind DNA elements containing GANTC motifs, which is explained by the critical DNA contacts made by residues at positions 3, 5 and 7 in the β ribbon that are conserved in these two proteins (Figure 1A). In contrast, the F TraM RHH binds an unrelated, 6-bp DNA motif, A(G/C)CG(C/G)T. Interestingly, this difference is likely largely dictated by a single, conservative Gln–Asn substitution in the RHH β ribbon. Gln5 is critical for recognition of the AT base pairs in the GANTC in the pED208 system. In F, the shorter Asn5 side chain is not expected to recognize an AT pair in the same manner, and may also interact differently with Lys3, leading to a rearrangement at the protein–DNA interface. In addition, subtle changes to the way in which the RHH contacts the DNA backbone may also impact the DNA geometry, which may in turn modulate sequence specificity. For example, Glu29 and Glu30 in the α1–α2 loop repel the phosphodiester backbone and contribute to DNA kinking in the pED208 TraM (Figure 5D). In F, R1 and R100, this loop is two residues longer and Glu29 is substituted with either an Arg or Lys residue (Figure 1A). Thus, a combination of direct readout, mediated through interactions involving residues of the RHH β-ribbon, and indirect, structural effects, mediated by interactions between the RHH and the DNA backbone, together govern DNA-binding specificity.
In addition to specificity at the level of TraM–DNA interactions, our work also reveals that TraM–TraD contacts are also plasmid-specific. Central to TraM–TraD interaction is the recognition of the C-terminal Phe and main chain carboxylate of the TraD tail by a hydrophobic pocket in TraM (33). The carboxylate group is recognized by conserved positively charged residues that form one side of this otherwise hydrophobic pocket (Figure 6C). Negatively charged residues in the tail make a number of long-range electrostatic interactions with the overall positively charged surface of the TraM interaction site. The putative TraD-binding pocket in pED208 TraM is well conserved with F TraM, and includes a pocket that could bind the C-terminal Tyr of the pED208 TraD tail, positively charged residues to recognize the TraD C-terminal carboxylate, as well as a number of charged residues surrounding the binding site (Figures 3E, 6C and D and Supplementary Figure 3D). The reversal of a potential charge–charge interaction in the pED208 TraM–TraD complex (TraM Glu81 to TraD Arg734) compared to the F complex (TraM Lys83 to TraD Asp715) plays a role in helping to define binding specificity between these two plasmid systems (Figures 3E and 6B).
Implications of cooperative TraM–DNA interactions for coupling of the plasmid to the transferosome
While recruitment of the plasmid to the conjugative pore depends upon interactions between the TraM tetramerization domain and the TraD C-terminal tail, interactions between the isolated tail peptide and the tetramerization domain are extremely weak in solution (33). Our finding that TraM binds DNA as a cooperative dimer of tetramers could provide multiple TraD contact points to enhance the affinity of the TraM-bound plasmid for the conjugative pore through an avidity effect (Figure 7B). The flexibility imparted by the TraM linker, as well as the predicted flexibility of the C-terminal tail of TraD, may be important to facilitate these interactions. In this regard, the fact that DNA-binding cooperativity is mediated through induced DNA conformational change, and not through extensive protein–protein interactions between tetramers, leaves the C-terminal TraM domain unencumbered and available for interaction with TraD.
Intriguingly, the mechanism of TraM–TraD interactions may be analogous to systems that underlie plasmid segregation. In both R1 and pSK41 plasmids, an RHH protein, ParR, binds cooperatively to multiple DNA repeats within the centrosome, forming a helical protein–DNA filament (69,70). The C-terminal domain of ParR specifically contacts a filamentous actin-like ATPase, ParM, most likely capping the filament in a cooperative interaction that may be analogous to the interaction between DNA-bound TraM and the hexameric TraD ATPase (69,70).
While this work has begun to reveal the mechanism of cooperative TraM interactions with DNA, higher order TraM complexes, perhaps facilitated by other partner proteins, likely form in and around the plasmid nic site. TraM binds multiple sbm sites in all F-like plasmids characterized to date and also appears to bind together with the plasmid-encoded TraY, as well as the host factor, IHF. TraY appears to be particularly critical to facilitate plasmid nicking via TraI (14,15,51). IHF is an architectural protein, creating nearly 180° ‘U-turns’ in the bound DNA (71). Interestingly, the pED208 sbmA site contains a central TraM site, flanked by symmetric IHF-binding sites (72). Binding of both TraM and IHF to this DNA would create a looped DNA structure, which may facilitate interactions between additional TraM tetramers and the TraD hexamer, as well as TraY and IHF. In addition, such DNA wrapping could potentially introduce torsional strain that could facilitate DNA unwinding that in turn, facilitates TraI binding and nicking (20,73–76). A better understanding of these processes will provide a basis to ultimately regulate important modes of bacterial horizontal gene transfer, which underlie the rapid acquisition of antibiotic resistance and virulence genes in human pathogens.
ACCESSION NUMBERS
PDB ID 3ON0, 3OMY.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Canadian Institutes of Health Research (grant FRN-42447 to J.N.M.G.); Howard Hughes International Scholarship (to J.N.M.G.). Funding for open access charge: Canadian Institutes of Health Research.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Pallen MJ, Wren BW. Bacterial pathogenomics. Nature. 2007;449:835–842. doi: 10.1038/nature06248. [DOI] [PubMed] [Google Scholar]
- 2.Lederberg J, Tatum EL. Sex in bacteria; genetic studies, 1945–1952. Science. 1953;118:169–175. doi: 10.1126/science.118.3059.169. [DOI] [PubMed] [Google Scholar]
- 3.de la Cruz F, Frost LS, Meyer RJ, Zechner EL. Conjugative DNA metabolism in gram-negative bacteria. FEMS Microbiol. Rev. 2010;34:18–40. doi: 10.1111/j.1574-6976.2009.00195.x. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe T. Infective heredity of multiple drug resistance in bacteria. Bacteriol. Rev. 1963;27:87–115. doi: 10.1128/br.27.1.87-115.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coque TM, Novais A, Carattoli A, Poirel L, Pitout J, Peixe L, Baquero F, Canton R, Nordmann P. Dissemination of clonally related escherichia coli strains expressing extended-spectrum beta-lactamase CTX-M-15. Emerg. Infect. Dis. 2008;14:195–200. doi: 10.3201/eid1402.070350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strahilevitz J, Jacoby GA, Hooper DC, Robicsek A. Plasmid-mediated quinolone resistance: a multifaceted threat. Clin. Microbiol. Rev. 2009;22:664–689. doi: 10.1128/CMR.00016-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Willetts N, Wilkins B. Processing of plasmid DNA during bacterial conjugation. Microbiol. Rev. 1984;48:24–41. doi: 10.1128/mr.48.1.24-41.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lanka E, Wilkins BM. DNA processing reactions in bacterial conjugation. Annu. Rev. Biochem. 1995;64:141–169. doi: 10.1146/annurev.bi.64.070195.001041. [DOI] [PubMed] [Google Scholar]
- 9.Lawley TD, Klimke WA, Gubbins MJ, Frost LS. F factor conjugation is a true type IV secretion system. FEMS Microbiol. Lett. 2003;224:1–15. doi: 10.1016/S0378-1097(03)00430-0. [DOI] [PubMed] [Google Scholar]
- 10.Kupelwieser G, Schwab M, Hogenauer G, Koraimann G, Zechner EL. Transfer protein TraM stimulates TraI-catalyzed cleavage of the transfer origin of plasmid R1 in vivo. J. Mol. Biol. 1998;275:81–94. doi: 10.1006/jmbi.1997.1436. [DOI] [PubMed] [Google Scholar]
- 11.Ragonese H, Haisch D, Villareal E, Choi JH, Matson SW. The F plasmid-encoded TraM protein stimulates relaxosome-mediated cleavage at oriT through an interaction with TraI. Mol. Microbiol. 2007;63:1173–1184. doi: 10.1111/j.1365-2958.2006.05576.x. [DOI] [PubMed] [Google Scholar]
- 12.Fekete RA, Frost LS. Mobilization of chimeric oriT plasmids by F and R100-1: Role of relaxosome formation in defining plasmid specificity. J. Bacteriol. 2000;182:4022–4027. doi: 10.1128/jb.182.14.4022-4027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howard MT, Nelson WC, Matson SW. Stepwise assembly of a relaxosome at the F plasmid origin of transfer. J. Biol. Chem. 1995;270:28381–28386. [PubMed] [Google Scholar]
- 14.Karl W, Bamberger M, Zechner EL. Transfer protein TraY of plasmid R1 stimulates TraI-catalyzed oriT cleavage in vivo. J. Bacteriol. 2001;183:909–914. doi: 10.1128/JB.183.3.909-914.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson WC, Howard MT, Sherman JA, Matson SW. The traY gene product and integration host factor stimulate escherichia coli DNA helicase I-catalyzed nicking at the F plasmid oriT. J. Biol. Chem. 1995;270:28374–28380. [PubMed] [Google Scholar]
- 16.Abo T, Inamoto S, Ohtsubo E. Specific DNA binding of the TraM protein to the oriT region of plasmid R100. J. Bacteriol. 1991;173:6347–6354. doi: 10.1128/jb.173.20.6347-6354.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Laurenzio L, Frost LS, Finlay BB, Paranchych W. Characterization of the oriT region of the IncFV plasmid pED208. Mol. Microbiol. 1991;5:1779–1790. doi: 10.1111/j.1365-2958.1991.tb01927.x. [DOI] [PubMed] [Google Scholar]
- 18.Di Laurenzio L, Frost LS, Paranchych W. The TraM protein of the conjugative plasmid F binds to the origin of transfer of the F and ColE1 plasmids. Mol. Microbiol. 1992;6:2951–2959. doi: 10.1111/j.1365-2958.1992.tb01754.x. [DOI] [PubMed] [Google Scholar]
- 19.Schwab M, Gruber H, Hogenauer G. The TraM protein of plasmid R1 is a DNA-binding protein. Mol. Microbiol. 1991;5:439–446. doi: 10.1111/j.1365-2958.1991.tb02127.x. [DOI] [PubMed] [Google Scholar]
- 20.Byrd DR, Matson SW. Nicking by transesterification: The reaction catalysed by a relaxase. Mol. Microbiol. 1997;25:1011–1022. doi: 10.1046/j.1365-2958.1997.5241885.x. [DOI] [PubMed] [Google Scholar]
- 21.Byrd DR, Sampson JK, Ragonese HM, Matson SW. Structure-function analysis of escherichia coli DNA helicase I reveals non-overlapping transesterase and helicase domains. J. Biol. Chem. 2002;277:42645–42653. doi: 10.1074/jbc.M205984200. [DOI] [PubMed] [Google Scholar]
- 22.Frost LS, Ippen-Ihler K, Skurray RA. Analysis of the sequence and gene products of the transfer region of the F sex factor. Microbiol. Rev. 1994;58:162–210. doi: 10.1128/mr.58.2.162-210.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matson SW, Sampson JK, Byrd DR. F plasmid conjugative DNA transfer: the TraI helicase activity is essential for DNA strand transfer. J. Biol. Chem. 2001;276:2372–2379. doi: 10.1074/jbc.M008728200. [DOI] [PubMed] [Google Scholar]
- 24.Fekete RA, Frost LS. Characterizing the DNA contacts and cooperative binding of F plasmid TraM to its cognate sites at oriT. J. Biol. Chem. 2002;277:16705–16711. doi: 10.1074/jbc.M111682200. [DOI] [PubMed] [Google Scholar]
- 25.Penfold SS, Simon J, Frost LS. Regulation of the expression of the traM gene of the F sex factor of escherichia coli. Mol. Microbiol. 1996;20:549–558. doi: 10.1046/j.1365-2958.1996.5361059.x. [DOI] [PubMed] [Google Scholar]
- 26.Fu YH, Tsai MM, Luo YN, Deonier RC. Deletion analysis of the F plasmid oriT locus. J. Bacteriol. 1991;173:1012–1020. doi: 10.1128/jb.173.3.1012-1020.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller DL, Schildbach JF. Evidence for a monomeric intermediate in the reversible unfolding of F factor TraM. J. Biol. Chem. 2003;278:10400–10407. doi: 10.1074/jbc.M212502200. [DOI] [PubMed] [Google Scholar]
- 28.Lu J, Zhao W, Frost LS. Mutational analysis of TraM correlates oligomerization and DNA binding with autoregulation and conjugative DNA transfer. J. Biol. Chem. 2004;279:55324–55333. doi: 10.1074/jbc.M409352200. [DOI] [PubMed] [Google Scholar]
- 29.Schwab M, Reisenzein H, Hogenauer G. TraM of plasmid R1 regulates its own expression. Mol. Microbiol. 1993;7:795–803. doi: 10.1111/j.1365-2958.1993.tb01170.x. [DOI] [PubMed] [Google Scholar]
- 30.Verdino P, Keller W, Strohmaier H, Bischof K, Lindner H, Koraimann G. The essential transfer protein TraM binds to DNA as a tetramer. J. Biol. Chem. 1999;274:37421–37428. doi: 10.1074/jbc.274.52.37421. [DOI] [PubMed] [Google Scholar]
- 31.Lu J, Edwards RA, Wong JJ, Manchak J, Scott PG, Frost LS, Glover JN. Protonation-mediated structural flexibility in the F conjugation regulatory protein, TraM. EMBO J. 2006;25:2930–2939. doi: 10.1038/sj.emboj.7601151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu J, Frost LS. Mutations in the C-terminal region of TraM provide evidence for in vivo TraM-TraD interactions during F-plasmid conjugation. J. Bacteriol. 2005;187:4767–4773. doi: 10.1128/JB.187.14.4767-4773.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu J, Wong JJ, Edwards RA, Manchak J, Frost LS, Glover JN. Structural basis of specific TraD-TraM recognition during F plasmid-mediated bacterial conjugation. Mol. Microbiol. 2008;70:89–99. doi: 10.1111/j.1365-2958.2008.06391.x. [DOI] [PubMed] [Google Scholar]
- 34.Beranek A, Zettl M, Lorenzoni K, Schauer A, Manhart M, Koraimann G. Thirty-eight C-terminal amino acids of the coupling protein TraD of the F-like conjugative resistance plasmid R1 are required and sufficient to confer binding to the substrate selector protein TraM. J. Bacteriol. 2004;186:6999–7006. doi: 10.1128/JB.186.20.6999-7006.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Disque-Kochem C, Dreiseikelmann B. The cytoplasmic DNA-binding protein TraM binds to the inner membrane protein TraD in vitro. J. Bacteriol. 1997;179:6133–6137. doi: 10.1128/jb.179.19.6133-6137.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 37.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 38.Rafferty JB, Somers WS, Saint-Girons I, Phillips SE. Three-dimensional crystal structures of escherichia coli met repressor with and without corepressor. Nature. 1989;341:705–710. doi: 10.1038/341705a0. [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 40.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 41.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 42.Cohen SX, Ben Jelloul M, Long F, Vagin A, Knipscheer P, Lebbink J, Sixma TK, Lamzin VS, Murshudov GN, Perrakis A. ARP/wARP and molecular replacement: The next generation. Acta Crystallogr. D Biol. Crystallogr. 2008;64:49–60. doi: 10.1107/S0907444907047580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu XJ, Olson WK. 3DNA: A versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat. Protoc. 2008;3:1213–1227. doi: 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu J, Manchak J, Klimke W, Davidson C, Firth N, Skurray RA, Frost LS. Analysis and characterization of the IncFV plasmid pED208 transfer region. Plasmid. 2002;48:24–37. doi: 10.1016/s0147-619x(02)00007-0. [DOI] [PubMed] [Google Scholar]
- 45.Stockner T, Plugariu C, Koraimann G, Hogenauer G, Bermel W, Prytulla S, Sterk H. Solution structure of the DNA-binding domain of TraM. Biochemistry. 2001;40:3370–3377. doi: 10.1021/bi002031c. [DOI] [PubMed] [Google Scholar]
- 46.Schreiter ER, Drennan CL. Ribbon-helix-helix transcription factors: Variations on a theme. Nat. Rev. Microbiol. 2007;5:710–720. doi: 10.1038/nrmicro1717. [DOI] [PubMed] [Google Scholar]
- 47.Moncalian G, de la Cruz F. DNA binding properties of protein TrwA, a possible structural variant of the arc repressor superfamily. Biochim. Biophys. Acta. 2004;1701:15–23. doi: 10.1016/j.bbapap.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 48.Falkow S, Baron LS. Episomic element in a strain of salmonella typhosa. J. Bacteriol. 1962;84:581–589. doi: 10.1128/jb.84.3.581-589.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Finlay BB, Paranchych W, Falkow S. Characterization of conjugative plasmid EDP208. J. Bacteriol. 1983;156:230–235. doi: 10.1128/jb.156.1.230-235.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luscombe NM, Laskowski RA, Thornton JM. Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Inamoto S, Fukuda H, Abo T, Ohtsubo E. Site- and strand-specific nicking at oriT of plasmid R100 in a purified system: Enhancement of the nicking activity of TraI (helicase I) with TraY and IHF. J. Biochem. 1994;116:838–844. doi: 10.1093/oxfordjournals.jbchem.a124604. [DOI] [PubMed] [Google Scholar]
- 52.Llosa M, Zunzunegui S, de la Cruz F. Conjugative coupling proteins interact with cognate and heterologous VirB10-like proteins while exhibiting specificity for cognate relaxosomes. Proc. Natl Acad. Sci. USA. 2003;100:10465–10470. doi: 10.1073/pnas.1830264100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tato I, Matilla I, Arechaga I, Zunzunegui S, de la Cruz F, Cabezon E. The ATPase activity of the DNA transporter TrwB is modulated by protein TrwA: Implications for a common assembly mechanism of DNA translocating motors. J. Biol. Chem. 2007;282:25569–25576. doi: 10.1074/jbc.M703464200. [DOI] [PubMed] [Google Scholar]
- 54.Gomis-Ruth FX, Sola M, Acebo P, Parraga A, Guasch A, Eritja R, Gonzalez A, Espinosa M, del Solar G, Coll M. The structure of plasmid-encoded transcriptional repressor CopG unliganded and bound to its operator. EMBO J. 1998;17:7404–7415. doi: 10.1093/emboj/17.24.7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu J, den Dulk-Ras A, Hooykaas PJ, Glover JN. Agrobacterium tumefaciens VirC2 enhances T-DNA transfer and virulence through its C-terminal ribbon-helix-helix DNA-binding fold. Proc. Natl Acad. Sci. USA. 2009;106:9643–9648. doi: 10.1073/pnas.0812199106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bolinger D, Sulkowska JI, Hsu HP, Mirny LA, Kardar M, Onuchic JN, Virnau P. A stevedore’s protein knot. PLoS Comput. Biol. 2010;6:e1000731. doi: 10.1371/journal.pcbi.1000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stayrook S, Jaru-Ampornpan P, Ni J, Hochschild A, Lewis M. Crystal structure of the lambda repressor and a model for pairwise cooperative operator binding. Nature. 2008;452:1022–1025. doi: 10.1038/nature06831. [DOI] [PubMed] [Google Scholar]
- 58.Lu D, Fillet S, Meng C, Alguel Y, Kloppsteck P, Bergeron J, Krell T, Gallegos MT, Ramos J, Zhang X. Crystal structure of TtgV in complex with its DNA operator reveals a general model for cooperative DNA binding of tetrameric gene regulators. Genes Dev. 2010;24:2556–2565. doi: 10.1101/gad.603510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schumacher MA, Miller MC, Grkovic S, Brown MH, Skurray RA, Brennan RG. Structural basis for cooperative DNA binding by two dimers of the multidrug-binding protein QacR. EMBO J. 2002;21:1210–1218. doi: 10.1093/emboj/21.5.1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jeng WY, Ko TP, Liu CI, Guo RT, Liu CL, Shr HL, Wang AH. Crystal structure of IcaR, a repressor of the TetR family implicated in biofilm formation in staphylococcus epidermidis. Nucleic Acids Res. 2008;36:1567–1577. doi: 10.1093/nar/gkm1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Itou H, Watanabe N, Yao M, Shirakihara Y, Tanaka I. Crystal structures of the multidrug binding repressor corynebacteriumglutamicum CgmR in complex with inducers and with an operator. J. Mol. Biol. 2010;403:174–184. doi: 10.1016/j.jmb.2010.07.042. [DOI] [PubMed] [Google Scholar]
- 62.Wisedchaisri G, Holmes RK, Hol WG. Crystal structure of an IdeR-DNA complex reveals a conformational change in activated IdeR for base-specific interactions. J. Mol. Biol. 2004;342:1155–1169. doi: 10.1016/j.jmb.2004.07.083. [DOI] [PubMed] [Google Scholar]
- 63.Pohl E, Holmes RK, Hol WG. Crystal structure of a cobalt-activated diphtheria toxin repressor-DNA complex reveals a metal-binding SH3-like domain. J. Mol. Biol. 1999;292:653–667. doi: 10.1006/jmbi.1999.3073. [DOI] [PubMed] [Google Scholar]
- 64.Pohl E, Holmes RK, Hol WG. Crystal structure of the iron-dependent regulator (IdeR) from mycobacterium tuberculosis shows both metal binding sites fully occupied. J. Mol. Biol. 1999;285:1145–1156. doi: 10.1006/jmbi.1998.2339. [DOI] [PubMed] [Google Scholar]
- 65.White A, Ding X, vanderSpek JC, Murphy JR, Ringe D. Structure of the metal-ion-activated diphtheria toxin repressor/tox operator complex. Nature. 1998;394:502–506. doi: 10.1038/28893. [DOI] [PubMed] [Google Scholar]
- 66.Alvarez-Martinez CE, Christie PJ. Biological diversity of prokaryotic type IV secretion systems. Microbiol. Mol. Biol. Rev. 2009;73:775–808. doi: 10.1128/MMBR.00023-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lang S, Gruber K, Mihajlovic S, Arnold R, Gruber CJ, Steinlechner S, Jehl MA, Rattei T, Frohlich KU, Zechner EL. Molecular recognition determinants for type IV secretion of diverse families of conjugative relaxases. Mol. Microbiol. 2010;78:1539–1555. doi: 10.1111/j.1365-2958.2010.07423.x. [DOI] [PubMed] [Google Scholar]
- 68.Sastre JI, Cabezon E, de la Cruz F. The carboxyl terminus of protein TraD adds specificity and efficiency to F-plasmid conjugative transfer. J. Bacteriol. 1998;180:6039–6042. doi: 10.1128/jb.180.22.6039-6042.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Salje J, Lowe J. Bacterial actin: Architecture of the ParMRC plasmid DNA partitioning complex. EMBO J. 2008;27:2230–2238. doi: 10.1038/emboj.2008.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schumacher MA, Glover TC, Brzoska AJ, Jensen SO, Dunham TD, Skurray RA, Firth N. Segrosome structure revealed by a complex of ParR with centromere DNA. Nature. 2007;450:1268–1271. doi: 10.1038/nature06392. [DOI] [PubMed] [Google Scholar]
- 71.Rice PA, Yang S, Mizuuchi K, Nash HA. Crystal structure of an IHF-DNA complex: A protein-induced DNA U-turn. Cell. 1996;87:1295–1306. doi: 10.1016/s0092-8674(00)81824-3. [DOI] [PubMed] [Google Scholar]
- 72.Di Laurenzio L, Scraba DG, Paranchych W, Frost LS. Studies on the binding of integration host factor (IHF) and TraM to the origin of transfer of the IncFV plasmid pED208. Mol. Gen. Genet. 1995;247:726–734. doi: 10.1007/BF00290404. [DOI] [PubMed] [Google Scholar]
- 73.Csitkovits VC, Zechner EL. Extent of single-stranded DNA required for efficient TraI helicase activity in vitro. J. Biol. Chem. 2003;278:48696–48703. doi: 10.1074/jbc.M310025200. [DOI] [PubMed] [Google Scholar]
- 74.Dostal L, Schildbach JF. Single-stranded DNA binding by F TraI relaxase and helicase domains are coordinately regulated. J. Bacteriol. 2010;192:3620–3628. doi: 10.1128/JB.00154-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Datta S, Larkin C, Schildbach JF. Structural insights into single-stranded DNA binding and cleavage by F factor TraI. Structure. 2003;11:1369–1379. doi: 10.1016/j.str.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 76.Hekman K, Guja K, Larkin C, Schildbach JF. An intrastrand three-DNA-base interaction is a key specificity determinant of F transfer initiation and of F TraI relaxase DNA recognition and cleavage. Nucleic Acids Res. 2008;36:4565–4572. doi: 10.1093/nar/gkn422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.