

Abstract
AMPA receptor-mediated responses to the agonist kainate differ from those of glutamate in two important respects. Glutamate is a full agonist that elicits strongly desensitizing responses, whereas kainate is a partial agonist with responses that are often described as weakly desensitizing or non-desensitizing. The efficacy of kainate relative to glutamate has previously been shown to be increased by mutations in the AMPA receptor ligand-binding cleft (Mano et al., 1996) and by coexpression with the auxiliary subunit stargazin (Tomita et al., 2005; Turetsky et al., 2005), but much less is known about factors that affect kainate desensitization. We therefore designed experiments to compare kainate and glutamate desensitization and efficacy in wild-type and mutant AMPA receptors expressed with and without stargazin in HEK293 cells. Desensitization to the two agonists was differentially affected by mutations in the helices participating in bonds between two subunits in the active state of the receptor (Sun et al., 2002), indicating that the protein interactions maintaining the stability of the dimer interface differ depending on which agonist is bound. Kainate efficacy was affected by factors distinct from ligand-binding cleft closure, including mutations in the dimer interface and channel vestibule as well as receptor composition. The increase in kainate responses for AMPA receptors coexpressed with stargazin was the result of both reduced kainate desensitization and increased kainate efficacy. These results provide critical new insights into the agonist dependence of both AMPA receptor activation and desensitization and the mechanism of the effects of stargazin on responses of partial agonists.
Introduction
The ionotropic glutamate receptors that mediate fast excitatory synaptic transmission are critical for the normal development and function of the nervous system. The glutamate receptor subunits GluA1–4 (Hollmann et al., 1989; Boulter et al., 1990; Keinänen et al., 1990) form AMPA-preferring receptors comprising four subunits arrayed around a central pore (Rosenmund et al., 1998). Crystallization of the ligand-binding domain of GluA2 (Armstrong et al., 1998) provided insights important for focused electrophysiological investigation of receptor activation, gating, desensitization, and the mechanism of partial agonist action (Jin et al., 2003; Horning and Mayer, 2004).
AMPA receptor-mediated responses to various agonists can differ in two respects, the degree of desensitization and relative efficacy, with glutamate and AMPA considered as full agonists. The AMPA receptor partial agonist kainate is often described as a non-desensitizing ligand despite several previous reports demonstrating that kainate produces rapidly desensitizing responses (Raman and Trussell, 1992; Patneau et al., 1993; Stern-Bach et al., 1998; Plested and Mayer, 2009). The relative magnitude of macroscopic desensitization observed (>50%), however, is much less than for strongly desensitizing agonists such as glutamate (98–99%).
Crystallization of the GluA2flop receptor with various agonists revealed differences in the ligand-binding core with glutamate versus kainate and led to the hypothesis that the degree of ligand-binding cleft closure determines relative agonist efficacy (Armstrong and Gouaux, 2000; Armstrong et al., 2003). Additional studies with the willardiine family of partial agonists provided further support for this hypothesis (Jin et al., 2003). However, subsequent experiments have identified additional conformational changes that may contribute to efficacy (Holm et al., 2005; Bjerrum and Biggin, 2008; Birdsey-Benson et al., 2010). For example, mutations in the agonist-binding sites L650T and L651V increase kainate efficacy, but only the former increases cleft closure (Armstrong et al., 2003; Birdsey-Benson et al., 2010). Interestingly, both of these mutations also alter kainate desensitization (Mano et al., 1996; Madden et al., 2004), but the relationship between agonist-induced desensitization and efficacy has not been examined previously.
Kainate is more efficacious relative to glutamate in neurons as compared with recombinant receptors. Discovery that AMPA receptor auxiliary subunits [transmembrane AMPA receptor regulating proteins (TARPs)], which are present in native receptors, increase kainate efficacy may explain this difference (Tomita et al., 2005; Turetsky et al., 2005). However, the mechanism underlying this effect of TARPs on kainate efficacy is unknown. The prototypical TARP stargazin (γ2) also reduces glutamate-evoked desensitization (Priel et al., 2005; Tomita et al., 2005; Turetsky et al., 2005), and it is possible that some of its effects on kainate efficacy reflect changes in kainate-induced desensitization.
We therefore compared kainate and glutamate desensitization in human embryonic kidney 293 (HEK293) cells expressing recombinant AMPA receptors with and without stargazin. Our experiments indicate that kainate and glutamate binding differentially affect the conformation of the dimer interface. We further show that kainate efficacy can be increased by manipulations that would not be expected to affect the ligand-binding cleft. Our experiments with stargazin reveal a previously overlooked effect on kainate desensitization that provides new insights into the mechanism of action of stargazin.
Materials and Methods
Cell culture and transfection.
Human embryonic kidney 293 cells were obtained from the American Type Culture Collection. Cells were grown in MEM supplemented with 10% FBS, and were maintained in a humidified 37°C, 5% CO2 incubator. Transfections were performed using Lipofectamine (Invitrogen). For each 35 mm dish to be transfected, 2 μg of Qiagen-purified DNA and 8 μl of Lipofectamine were diluted in OPTI-MEM (Invitrogen) and incubated for 20 min at room temperature. Culture medium was replaced with this transfection solution, and cells were returned to the 37°C incubator for 3 h. Afterward, the DNA/lipid complexes were washed off the cells, and cultures were fed with MEM + 10% FBS containing 50 μm 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX) to block activation of expressed AMPA receptors by traces of glutamate in the medium. For all experiments, pEGFP (Clontech) was included to allow identification of transfected cells during subsequent electrophysiology experiments. For homomeric receptors, the GluA1:γ2:GFP ratio was 1:1:1.64 (0.55 μg + 0.55 μg + 0.9 μg), with conditions not containing a γ2 subunit having additional GFP added to compensate. For heteromeric receptors without γ2, the GluA1:GluA2:GFP ratios were 1:1:0.1 (0.9 μg + 0.9 μg + 0.2 μg). For heteromeric receptors with γ2, the GluA1:GluA2:γ2:GFP ratios were 1:1:2:0.44 (0.45 μg + 0.45 μg + 0.9 μg + 0.2 μg). The AMPA receptor expression constructs were generously provided by Dr. P. H. Seeburg. Cacng2 (stargazin; γ2) was cloned from adult rat cerebellum total RNA using the ProSTAR Ultra HF RT-PCR system (Stratagene) and subcloned into the expression vector pCMV-Script (Stratagene).
Site-directed mutagenesis.
Point mutations were introduced into GluA1flip using the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene, Agilent Technologies), which employs thermal cycling to generate a mutant plasmid, followed by DpnI digestion of the parental DNA template. All mutant constructs were sequenced at the Oklahoma State University Recombinant DNA/Protein Resource Facility to confirm that the expected mutation was present and that no collateral mutations had been introduced.
Electrophysiology.
Electrophysiology experiments were conducted 36–42 h after transfection. Whole-cell patch-clamp recordings were obtained at room temperature (22−25°C) with cells voltage-clamped at −60 mV. Recording electrodes were pulled from borosilicate glass and fire-polished; typical electrode resistance in series with the cell was 2–7 MΩ and was compensated by 60–80% using an Axopatch 200A patch-clamp amplifier (Molecular Devices). Recorded data were filtered at 1–3 kHz and sampled at 2–10 kHz using pClamp (Molecular Devices).
Extracellular saline contained (in mm) 145 NaCl, 5.4 KCl, 1 MgCl2, 1.8 CaCl2, 5 HEPES, 5.5 glucose, and 0.01 mg/ml phenol red. The osmolarity was adjusted to 300 mOsm and the pH to 7.3 with NaOH. Kynurenate (500 μm) was added to the bath to protect cells from pre-exposure to agonists. Patch pipettes were filled with intracellular solution containing (in mm) 135 CsCl, 10 CsF, 2 MgCl2, 0.5 CaCl2, 5 Cs4BAPTA, 10 HEPES, and 2 Na2ATP. Osmolarity was adjusted to 295 mOsm and pH to 7.2 with CsOH. Rapid agonist application was achieved using a glass flowpipe array of 12 parallel barrels, each 400 μm in diameter, placed directly in front of a voltage-clamped cell. Perfusion was gravity-fed and solution application was gated with mini solenoid valves (Lee Company). Valve opening and closing were coordinated with flowpipe movement using a Warner Instrument Corporation SF-77B Perfusion Fast-Step system.
To verify incorporation of GluA2 in heteromeric GluA1/A2 recombinant receptors, rectification ratios for glutamate peak at +40/-60 mV were calculated and only cells with rectification ratios >0.6 were included in the dataset.
Analysis.
Clampfit 10 (Molecular Devices) was used for peak, steady-state amplitude, and kinetic measurements of electrophysiological data. Cells that had peak glutamate currents >10 nA or cells that did not recover to 70% of the original glutamate peak were excluded from the dataset. To normalize for transfection efficiency, each experiment was compared with time-matched controls. Thus, there is slight variation between the control data included in each figure. Because peak agonist responses in whole-cell recordings may be affected by desensitization, maximal responses to both glutamate and kainate were determined in the presence of trichlormethiazide (TCM), a benzothiadiazine-positive allosteric modulator with efficacy similar to cyclothiazide but faster association and dissociation kinetics (500 μm TCM reduces glutamate-evoked desensitization in hippocampal neurons to a similar degree as 100 μm cyclothiazide; 96 ± 4% of peak response to cyclothiazide, n = 14). Percentage desensitization to glutamate and kainate was determined relative to maximal agonist responses [% desensitization = (1 − agonist-evoked steady-state/agonist + TCM peak) × 100]. One exception was desensitization of mutants on the dimer interface (see legend for Fig. 5), where some mutations almost fully blocked macroscopic desensitization and TCM inhibited the maximal current, similar to what is observed with cyclothiazide (Patneau et al., 1993; Stern-Bach et al., 1998). In this case, percentage desensitization of glutamate was calculated relative to glutamate peak [% desensitization = (1 − glutamate-evoked steady-state/glutamate peak) × 100]. Efficacy of the partial agonist kainate was determined relative to maximal glutamate responses with desensitization for both agonists blocked by TCM (kainate + TCM steady-state/glutamate + TCM peak). Kainate efficacy was also calculated relative to glutamate peak (kainate steady-state/glutamate peak) to illustrate the effect of desensitization on this calculation (see Fig. 2B).
Figure 5.

Mutations in the dimer interface that reduce or block glutamate desensitization differentially affect kainate desensitization. A, Because the positive modulator TCM decreases responses to glutamate in L479Y and to a lesser degree in L479F, percentage desensitization for glutamate was calculated relative to the peak of the response to glutamate alone [(1 − glutamate steady-state/glutamate peak) × 100]. Percentage desensitization for kainate was determined relative to the kainate response with desensitization blocked by TCM [(1− KainSS/Kain + TCMSS) × 100]. *Significant difference between kainate and glutamate desensitization within each construct, p < 0.05. #Significant difference in glutamate desensitization compared with GluA1flip, p < 0.01. †Significant difference in kainate desensitization compared with GluA1flip, p < 0.01. B, Kainate efficacy was calculated relative to the maximal response to glutamate in the presence of TCM. †Significant difference in kainate efficacy, compared withGluA1flip, p < 0.05, n = 29 for GluA1 and n = 6–11 for mutated receptors. C, Representative responses to glutamate and kainate in the absence (solid black line) and presence (dashed blue line) of TCM are overlaid. The blue bar indicates application of TCM and black bar agonist application. Note that the response to glutamate with TCM for the L479Y mutant is smaller than glutamate alone (indicated by arrow), consistent with previous reports that the related positive modulator cyclothiazide has a nonspecific inhibitory effect (Patneau et al., 1993; Stern-Bach et al., 1998).
Figure 2.

Effects of stargazin on AMPA receptor-mediated responses to kainate reflect both decreased desensitization and increased efficacy. A, Mean data for HEK293 cells transfected with GluA1flip with and without stargazin (γ2). Percentage steady-state desensitization was determined relative to maximal current in response to glutamate or kainate in the presence of TCM. *Significant difference between kainate and glutamate desensitization, p < 0.05. #Significant difference in percentage desensitization for glutamate with and without stargazin, p < 0.01. †Significant difference in percentage desensitization for kainate with and without stargazin, p < 0.01. B, Two methods of determining kainate efficacy for HEK293 cells expressing GluA1 with and without γ2 are compared. Efficacy was first calculated by dividing kainate steady-state by the peak glutamate response (KainSS/GlutPk, violet bars). Absolute efficacy with desensitization blocked was then calculated by dividing kainate steady-state in the presence of TCM by glutamate peak in the presence of TCM (Kain + TCMSS/Glut + TCMPk, blue bars). †Significant effect of stargazin on kainate efficacy, p < 0.01, n > 20 cells. C, Representative responses of HEK293 cells transfected with GluA1 or GluA1 plus γ2 to glutamate and kainate in the absence (solid black line) and presence (dashed blue line) of TCM are overlaid. The blue bar indicates application of TCM and black bar agonist application.
Statistical analyses were performed using Sigma Stat 3.1 (Systat). Significance was determined using one-way or two-way ANOVA followed by Student–Newman–Keuls (SNK) post hoc comparisons. Power for all experiments was >0.95. Main effects were considered significant for p < 0.05. For two-way ANOVA with significant main effects and a significant interaction, subsequent one-way ANOVA followed by SNK post hoc comparisons was used to determine the nature of the interaction; to control for experiment-wise error, only results with p < 0.01 were considered significantly different for these post hoc comparisons. Correlations for kainate desensitization versus glutamate desensitization (see Fig. 7A) or kainate efficacy (see Fig. 7B) were calculated using Pearson product–moment correlation.
Figure 7.

Kainate desensitization is not correlated with either glutamate desensitization or kainate efficacy. A, Scatter plot of glutamate desensitization [(1 − (glutamate steady-state/glutamate + TCM peak) × 100] versus kainate desensitization [1 − (kainate steady-state/kainate + TCM steady-state) × 100] for all receptors presented in this paper except L479Y because we could not determine percentage glutamate desensitization relative to maximal responses with TCM (see Materials and Methods and Fig. 5). The correlation coefficient for these data was 0.18 (p = 0.53). B, Scatter plot of kainate efficacy (kainate + TCM steady-state/glutamate + TCM peak) versus kainate desensitization for all receptors presented in this paper. Statistical analysis found no relationship, with a correlation coefficient of 0.17 (p = 0.56). Black indicates wild-type homomeric receptors. Violet indicates heteromeric AMPA receptors. Red indicates constructs with mutations in the dimer interface. Blue indicates mutations in the channel vestibule. Green indicates mutation in the binding cleft.
Materials.
Cs4BAPTA was purchased from Invitrogen. Kainate, NBQX, and kynurenate were purchased from Tocris Bioscience. All other reagents were obtained from Sigma unless noted otherwise.
Results
AMPA receptor subunit composition affects kainate efficacy and kainate desensitization
Although kainate-evoked responses recorded from AMPA receptors are often referred to as non-desensitizing, there is clear desensitization produced by kainate whether observed macroscopically in outside-out patches (Patneau et al., 1993; Plested and Mayer, 2009) or as evidenced by the increase in response to kainate produced by drugs that block desensitization such as cyclothiazide (Patneau et al., 1993; Partin et al., 1994). Figure 1 compares glutamate and kainate steady-state desensitization calculated relative to maximal responses with desensitization blocked by TCM. Kainate was only slightly less desensitizing than glutamate (96% for kainate vs 99% for glutamate) for GluA1flip. Note that kainate responses were potentiated almost 30-fold by TCM (Fig. 1C). Because it is well established that flip-flop alternative splicing affects desensitization (Mosbacher et al., 1994), we first explored the effect of alternative splicing on kainate desensitization. Whereas glutamate desensitization was comparable between GluA1flip and GluA1flop as well as heteromeric AMPA receptors made up of GluA1 and GluA2flip (Fig. 1A), kainate desensitization was reduced relative to GluA1flip for GluA1flop and for all heteromeric receptors comprised of GluA1 and GluA2 (Fig. 1A).
Figure 1.

Kainate efficacy and desensitization are affected by receptor composition. A, Percentage steady-state desensitization for saturating concentrations of glutamate (3 mm) and kainate (600 μm) was calculated relative to the peak response with desensitization blocked by 500 μm TCM for homomeric GluA1flip (1i), GluA1flop (1o), GluA2flip(Q), or GluA2flip(R) and heteromeric GluA1flip/2flip, GluA1flip/2flop, or GluA1flop/2flop. Error bars in this and subsequent figures indicate SEM. Significance for this and all subsequent figures was determined using one-way or two-way ANOVA followed by SNK post hoc comparisons. For two-way ANOVA with significant main effects and a significant interaction, subsequent one-way ANOVA followed by SNK post hoc comparisons were used to determine the nature of the interaction (to control for experiment-wise error, only results with p < 0.01 were considered significantly different for these post hoc comparisons). *Significant difference between kainate and glutamate desensitization within receptor type, p < 0.05. #Significance between different receptor composition for glutamate desensitization relative to GluA1flip, p < 0.01. †Significance between different receptor composition for kainate desensitization relative to GluA1flip, p < 0.01. B, Kainate efficacy was calculated relative to peak glutamate responses with desensitization for both agonists blocked by TCM. †Significant difference relative to GluA1flip, p < 0.05. $Significant difference from GluA1flip/2flip, p < 0.05. Each bar in A and B is the mean of data from six to nine cells. C, Representative responses to glutamate and kainate in the absence (solid black line) and presence (dashed blue line) of TCM are overlaid. The blue bar indicates application of TCM and black bar agonist application.
The reduced kainate desensitization observed in heteromers could have been due to flip-flop alternative splicing or to the inclusion of the GluA2 subunit. Because GluA2 is edited from Q to R at position 582 (Sommer et al., 1991), we tested both variants of GluA2flip. GluA2(R), but not GluA2(Q), exhibited significantly less desensitization for both glutamate and kainate than GluA1flip, suggesting that the inclusion of GluA2(R) contributed to the reduced desensitization in heteromeric GluA1/GluA2(R) receptors. We observed a similar pattern of apparent dominance of GluR2(R) for control of kainate desensitization in experiments comparing homomeric GluR4 with heteromeric receptors made up of GluR4 and GluR2R (data not shown).
Kainate efficacy relative to glutamate, which is considered a full agonist, was calculated using maximal responses for both agonists with desensitization blocked by TCM. By this measure, the efficacy of kainate in GluA1flip was 0.10 (Fig. 1B,C). Relative kainate efficacy was significantly increased relative to GluA1 for GluA2(R), but not GluA2(Q). Heteromeric GluA1/2 receptors also exhibited increased kainate efficacy compared with GluA1flip (Fig. 1B,C), with the most pronounced effect for receptors containing a flop subunit. The efficacy of kainate for flop receptors, however, may be overestimated because glutamate responses for receptors containing only flop subunits are in general less sensitive to the effects of the benzothiadiazine-positive modulators (Partin et al., 1993). Thus, the calculation of kainate efficacy for GluA1flop/2flop receptors could be inflated by incomplete block of desensitization in glutamate responses. Comparison of the shape of the glutamate and kainate responses in the presence of TCM in Figure 1C emphasizes the prominent decay of the glutamate peak for GluA1flop/2flop receptors and the fact that kainate responses are not affected in the same way. This further suggests that the conformational changes underlying desensitization are distinct for the two agonists.
Stargazin affects both kainate efficacy and desensitization
Previous research has shown two striking effects of stargazin (γ2) on agonist-evoked responses. As illustrated in Figure 2, stargazin reduces glutamate desensitization and increases the relative size of the response to the partial agonist kainate, which has been interpreted as an increase in kainate efficacy (Tomita et al., 2005; Turetsky et al., 2005). The effect of stargazin on kainate desensitization, however, has not been previously examined. As shown in Figure 2A, stargazin decreased kainate desensitization (from 96% for GluA1 alone to 23% with γ2) to a greater extent than glutamate desensitization (from 99% to 95%).
Figure 2B shows the effect of stargazin on kainate efficacy relative to the full agonist glutamate. Previous papers have calculated kainate efficacy by comparing steady-state responses of kainate relative to the peak of the glutamate response. By this calculation, stargazin increased apparent kainate efficacy 55-fold. However, this measure does not take into consideration relative levels of desensitization in response to either glutamate or kainate. Thus, a more valid measure of kainate efficacy is to compare maximal kainate and glutamate responses in the presence of TCM. With desensitization blocked, stargazin increased kainate efficacy eightfold, indicating that a substantial component of the effect of stargazin on apparent kainate efficacy is a result of reduced kainate desensitization.
Because both receptor composition and coexpression with stargazin increase kainate efficacy and decrease kainate desensitization, we examined the effects of stargazin on heteromeric AMPA receptors comprising GluA1flip/2flip, GluA1flip/2flop, and GluA1flop/2flop. As shown in Figure 3, kainate and glutamate desensitization in heteromeric receptors were differentially affected by stargazin. Glutamate desensitization was significantly less for GluA1flip/2flip coexpressed with stargazin compared with either homomeric GluA1flip or other GluA1/A2 combinations (Fig. 3A,C). Stargazin reduced kainate desensitization to <20% and, unlike glutamate desensitization, there was no significant difference between receptors of different composition.
Figure 3.

Effects of stargazin on glutamate and kainate desensitization in heteromeric AMPA receptors are distinct. To facilitate comparison between conditions with and without stargazin, data from Figure 1 (shown in light bars) are reproduced here. [All conditions with stargazin were significantly different from the same condition without stargazin (γ2), p < 0.05.] A, Percentage steady-state desensitization for glutamate and kainate was calculated relative to the peak response with desensitization blocked by TCM. HEK293 cells were cotransfected with stargazin and GluA1flip (1i), GluA1flip/2flip (1i/2i), GluA1flip/2flop (1i/2o), or GluA1flop/2flop (1o/2o). *Significant difference between kainate and glutamate desensitization within each receptor type, p < 0.05. #Significant difference relative to GluA1flip for glutamate desensitization, p < 0.01. B, Kainate efficacy was calculated relative to peak glutamate responses with desensitization for both agonists blocked by TCM. *Significant effect of stargazin, p < 0.05. †Significant difference from all other conditions for kainate efficacy, p < 0.01, n = 8–11 cells. C, Representative responses to glutamate and kainate in the absence (solid black line) and presence (dashed blue line) of TCM are overlaid. The blue bar indicates application of TCM and black bar agonist application.
Kainate efficacy was comparable in both homomeric and heteromeric receptors coexpressed with stargazin (∼0.80) (Fig. 3B), with the exception of GluA1flop/2flop, which appeared to be more efficacious than glutamate. However, because kainate efficacy is calculated compared with the peak glutamate response, which still exhibits prominent decay in the presence of TCM for GluA1flop/2flop, relative kainate efficacy is probably overestimated for this construct. Thus, it appears that even with stargazin, kainate efficacy in heteromeric receptors is limited to ∼80% of the maximal response to glutamate.
A binding cleft mutation that increases kainate efficacy also affects macroscopic desensitization of kainate responses
Mutations in the agonist-binding cleft of AMPA receptors can increase kainate affinity and efficacy (Mano et al., 1996; Madden et al., 2004). One of these, L646T (L650T in GluA2), increases the degree of agonist-binding cleft closure (Armstrong et al., 2003). Mano et al. (1996) concluded that the L646T mutation on a GluA1flop background also affected desensitization because they observed greater potentiation of kainate currents by cyclothiazide but were not able to observe macroscopic desensitization in oocyte recordings. We made this mutation on a GluA1flip background and expressed it in HEK293 cells. Percentage steady-state desensitization for both glutamate and kainate was not significantly different between the L646T mutant receptor and wild-type GluA1 (Fig. 4A). However, responses to kainate for GluA1 L646T exhibited rapid macroscopic desensitization (Fig. 4C), with a mean steady-state to peak ratio of 0.4. Consistent with previous reports, kainate efficacy for the L646T mutant was increased (0.30 vs 0.08 for GluA1 control) (Fig. 4B). Thus, with this mutation in the agonist-binding cleft, kainate activates the receptor in a manner more similar to glutamate, both in terms of efficacy and macroscopic desensitization.
Figure 4.

Mutation in the AMPA receptor agonist-binding cleft affects both kainate efficacy and desensitization. HEK293 cells were transfected with GluA1flip or GluA1 L646T with and without stargazin. A, Percentage desensitization for glutamate and kainate steady-state was calculated relative to the peak response with desensitization blocked by TCM. *Significant difference between glutamate and kainate desensitization, p < 0.05. #Significant effect of stargazin coexpression on glutamate desensitization, p < 0.01. †Significant effect of stargazin coexpression on kainate desensitization, p < 0.01. B, Relative kainate efficacy was calculated by dividing kainate steady-state in the presence of TCM by glutamate peak in the presence of TCM (Kain + TCMSS/Glut + TCMPk). *Significant effect of stargazin on kainate efficacy, p < 0.05. †Significant effect of the L646T mutation on kainate efficacy, p < 0.01, n = 9–13 cells. C, Representative responses to glutamate and kainate in the absence (solid black line) and presence (dashed blue line) of TCM are overlaid. The blue bar indicates application of TCM and black bar agonist application. The boxes show magnified responses to kainate that illustrate the macroscopic desensitization of control kainate responses for L646T (stimulation artifacts were masked for clarity).
We were also interested in whether the effects of stargazin would be additive with the effects of the L646T mutation on kainate desensitization and efficacy. As shown in Figure 4A, stargazin reduced kainate desensitization of the L646T mutant receptors to an extent comparable to wild-type receptors. In contrast, coexpression of L646T with stargazin significantly increased kainate efficacy relative to GluA1 with stargazin (0.88 vs 0.75) (Fig. 4B), suggesting that stargazin and the mutation do not share the same mechanism of action.
Kainate and glutamate desensitization are differentially affected by mutations in the dimer interface
AMPA receptor desensitization is believed to reflect dissociation of contacts between helices J and D on the interface between two subunits in a dimer (Sun et al., 2002), and mutations in this location have been demonstrated to affect the kinetics of glutamate desensitization (Horning and Mayer, 2004). To examine the role of the dimer interface in kainate desensitization, we made a series of mutations in positions 479 and 482 of helix D and 748 of helix J. The L479Y mutation on a GluA3 background (L507Y; Stern-Bach et al., 1998) was previously reported to almost fully block glutamate desensitization. Consistent with previously published data (Stern-Bach et al., 1998; Horning and Mayer, 2004), glutamate desensitization was decreased by three mutations at the L479 positions Y, F, A, but was unaffected in L479E (Fig. 5A).
Although kainate desensitization for L479Y and L479F was significantly decreased relative to GluA1, kainate responses were still strongly desensitizing (72% and 85%, respectively). This provides an explanation for the previous observation of Armstrong et al. (2003) that kainate efficacy is greater for GluA2Qflip when determined in the presence of cyclothiazide in comparison to the L483Y background. Surprisingly, kainate was more strongly desensitizing than control for L479A and L479E (98% and 99%, respectively, compared with 96% for GluA1). The E482A mutation (Horning and Mayer, 2004) significantly reduced desensitization of kainate, but not glutamate, responses in our experiments, whereas the K748A mutation in helix J affected glutamate, but not kainate, desensitization relative to GluA1 (Fig. 5A,C). These results provide strong evidence that the conformational rearrangements underlying desensitization are different for kainate and glutamate.
The variation in kainate desensitization of these dimer interface mutant receptors offers the opportunity to explore the relationship between kainate desensitization and kainate efficacy (Fig. 5A,B). Interestingly, none of the mutations with significantly less kainate desensitization were more efficacious than control, whereas the two mutations with the most strongly desensitizing kainate responses (L479A and L479E) exhibited the highest kainate efficacy (0.18 and 0.16, respectively, compared with 0.10 for GluA1). K748A also had significantly greater kainate efficacy, but without any effect on kainate desensitization relative to GluA1. Thus, these data clearly indicate the lack of relationship between the effects of dimer interface mutations on kainate desensitization and kainate efficacy (see also Fig. 7B). Despite the significant differences in kainate efficacy between different mutations in the dimer interface, coexpression with stargazin increased kainate efficacy to an equivalent value of ∼0.75 for all mutations (data not shown).
Mutations in the external channel vestibule of the AMPA receptor increase kainate efficacy
In kainate receptors GluK1 and GluK2, kainate is a high-affinity, efficacious agonist that exhibits desensitization similar to glutamate. One approach to understanding the differences in pharmacology between AMPA and kainate receptors is to mutate residues in AMPA receptors to their corresponding kainate receptor residues. We focused on regions that couple ligand-binding domain closure to channel gating and mutated residues on a GluA1 background to their GluK2 equivalents. Two mutations in the external channel vestibule A518S/Y519P in the region linking the ligand-binding domain and first transmembrane domain of the channel and V788I at the beginning of transmembrane domain 4 (previously made on a GluA3 background by Balannik et al., 2005, in their paper examining noncompetitive antagonism) altered kainate responses and increased the effects of stargazin on glutamate responses. As shown in Figure 6C, A518S/Y519P and V788I increased kainate efficacy more than twofold, but did not significantly affect either glutamate (Fig. 6A) or kainate (Fig. 6B) steady-state desensitization. Coexpression of A518S/Y519P and V788I with stargazin significantly decreased glutamate, but not kainate, desensitization relative to GluA1 with stargazin (compare Fig. 6A,B). The efficacy of these channel vestibule mutants coexpressed with stargazin was comparable to GluA1 with stargazin (Fig. 6C), indicating that the effects of these mutations and stargazin on kainate efficacy were not additive.
Figure 6.
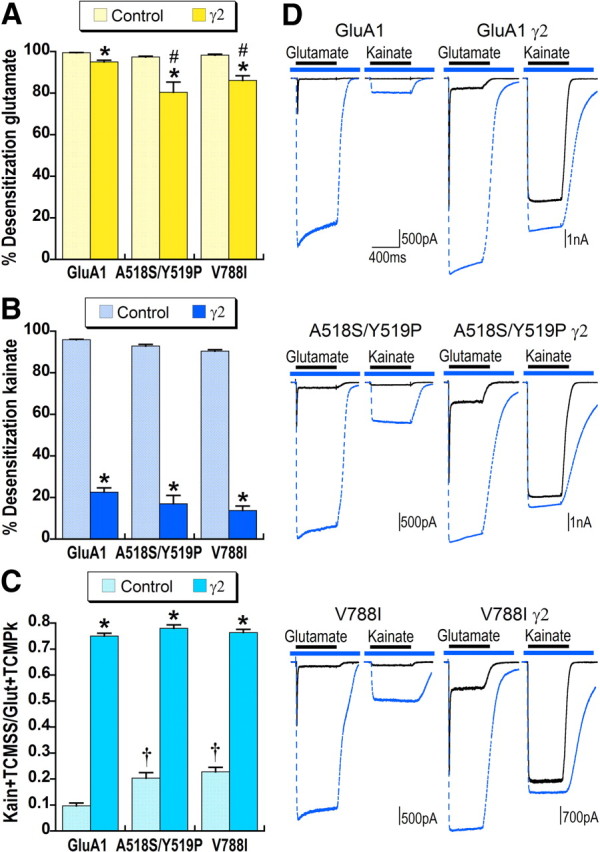
Mutations in the external channel vestibule increase relative kainate efficacy. A, B, Percentage desensitization for glutamate (A) and kainate (B) was calculated relative to maximal responses with desensitization blocked by TCM for GluA1flip or GluA1flip carrying mutations A518S/Y519P or V788I expressed alone or with stargazin (γ2). *Significant effect of stargazin on desensitization, p < 0.05. #Significant difference in glutamate desensitization compared with control GluA1γ2, p < 0.01. C, Kainate efficacy for GluA1flip or GluA1flip carrying mutations A518S/Y519P or V788I expressed alone or with stargazin was calculated relative to maximal glutamate responses with desensitization blocked by TCM. †Significant effect of the mutation on kainate efficacy compared with GluA1flip, p < 0.01, n > 20 cells for GluA1 and GluA1γ2, and n = 7–19 for cells expressing mutated AMPA receptors. *Significant effect of stargazin on kainate efficacy, p < 0.05. D, Representative responses to glutamate and kainate in the absence (solid black line) and presence (dashed blue line) of TCM are overlaid. The blue bar indicates application of TCM and the black bar agonist application.
Kainate desensitization is not correlated with either kainate efficacy or glutamate desensitization
The design of our experiments allowed exploration of the relationship between kainate and glutamate steady-state desensitization in a way that has not been examined previously. To ensure that estimates of glutamate and kainate desensitization were comparable, we determined percentage steady-state desensitization relative to maximal responses with desensitization blocked by trichlormethiazide for both agonists. In Figure 7A, kainate desensitization for both the wild-type and mutated AMPA receptors in Figures 1–6 is plotted against glutamate desensitization. As is evident from examination of this plot, and confirmed by statistical analysis, there is no correlation between kainate and glutamate desensitization. This lack of correlation indicates that the stability of the dimer interface is differentially affected by glutamate and kainate.
A previous study examining the relationship between domain closure and efficacy of several partial agonists, the 5-substituted willardiines, found that both efficacy and desensitization were correlated with degree of agonist-binding cleft closure (Jin et al., 2003), but it did not explore the relationship between desensitization and efficacy. Our experiments offered the opportunity for a direct comparison between efficacy and desensitization of a single partial agonist across receptors that vary in both kainate efficacy and desensitization. A scatter plot of kainate efficacy versus kainate desensitization of the data from Figures 1–6 is shown in Figure 7B. The lack of relationship between desensitization and efficacy for kainate is evident from examination of this graph and suggests that independent conformational rearrangements control agonist efficacy and desensitization.
Discussion
The experiments presented here provide critical new insights into the agonist dependence of AMPA receptor desensitization and activation. Direct comparison of responses to glutamate and kainate in the same cells revealed a striking lack of correspondence between desensitization to the two agonists in both recombinant receptors of varying composition and in receptors bearing mutations in the dimer interface or channel vestibule. Kainate efficacy was affected by receptor composition as well as mutations in the dimer interface and channel vestibule, indicating that ligand-binding cleft closure is only one of multiple factors controlling agonist efficacy. Finally, these data provide a framework for understanding the effects of stargazin on kainate responses as reflecting both increased efficacy and reduced desensitization.
Glutamate and kainate desensitization involve distinct conformational changes
Macroscopic desensitization of AMPA receptor-mediated kainate responses in whole-cell recordings is typically not observed. In outside-out patch recordings, kainate responses exhibit 30–50% desensitization as determined by steady-state to peak ratio (Patneau et al., 1993; Plested and Mayer, 2009). Our experiments reveal kainate as a much more desensitizing agonist in recombinant AMPA receptors than previously believed (96% for GluA1flip). The kainate desensitization previously observed in hippocampal neurons (Patneau et al., 1993) was ∼30%, which is substantially less than we observed in recombinant receptors. This is likely due to differences in receptor composition, most notably that most neuronal receptors are heteromeric and contain auxiliary TARP subunits. The extent of macroscopic kainate desensitization observed in outside-out patch recordings from recombinant receptors with very rapid perfusion techniques (Plested and Mayer, 2009) was also substantially less (50%). More significantly, the kainate peak to glutamate peak ratio in Plested and Mayer (2009) was 0.009, compared with the 0.10 relative kainate efficacy we observed in the presence of TCM (Fig. 1). Thus, even with fast perfusion on outside-out patches, it appears that >90% of the kainate-bound receptors never activate. This indicates that the lack of macroscopic desensitization in whole-cell recordings is only partially explained by very rapid onset of desensitization for kainate (Patneau et al., 1993) and that a major difference between kainate and glutamate desensitization is the large number of receptors that desensitize without opening. Whether this is because kainate binding fails to establish the dimer interface (Gonzalez et al., 2010) or is insufficient to maintain its stability long enough for the channel to open is unknown.
Our data taken as a whole (Fig. 7A) provide strong physiological evidence that the conformation of the AMPA receptor dimer interface and the strength of interactions across the interface differ for kainate and glutamate. The L479Y and L479F mutations, which almost fully block macroscopic glutamate-evoked desensitization, had only modest effects on kainate responses, which were still >70% desensitizing. Thus, introducing tyrosine or phenylalanine at the 479 position does not stabilize the dimer interface with kainate bound, suggesting the position of helices J and D relative to each other is agonist-dependent. Further support for differences in the dimer interface was provided by the L479A mutation, which decreased glutamate desensitization while significantly increasing kainate desensitization, and the E482A mutation, which reduced kainate desensitization while leaving glutamate desensitization unaffected. Finally, flop alternative splicing substantially reduced kainate desensitization, with minimal affects on glutamate desensitization.
However, because the crystal structure of a dimeric or tetrameric AMPA receptor in complex with kainate has not been determined, identification of the specific conformational differences in the dimer interface awaits further experiments. Both X-ray crystallography and NMR spectroscopy have, however, provided substantial support for agonist-dependent rearrangements of the ligand-binding domain as well as differences in protein dynamics between glutamate and kainate (Bjerrum and Biggin, 2008; Fenwick and Oswald, 2010; Maltsev and Oswald, 2010). While this paper was under review, an article was published (Nayeem et al., 2011) in which comparison of the crystal structure of a dimeric kainate receptor (GluK2) ligand-binding domain revealed rotation of the dimer interface by 2.5° with kainate bound relative to the glutamate-bound structure. A similar motion in the dimer interface of AMPA receptors with kainate bound would alter relationships between binding partners across the dimer interface, thus explaining the disparate effects of dimer interface mutations on kainate and glutamate desensitization.
Kainate efficacy is controlled by multiple mechanisms
A long-standing question for AMPA receptors is what controls agonist efficacy. The agonist-binding cleft closure model (Armstrong and Gouaux, 2000) is intuitively appealing and has received substantial experimental support (Sun et al., 2002; Jin et al., 2003). In agreement with Mano et al. (1996), we observed that the L646T mutation, which increases the degree of cleft closure in response to kainate (Armstrong et al., 2003), does significantly increase kainate efficacy. But other aspects of our data are in agreement with reports suggesting that this model is incomplete and that additional mechanisms contribute to agonist efficacy. Two of these are sideways or twisting motions of domain 2 relative to domain 1 (Bjerrum and Biggin, 2008; Birdsey-Benson et al., 2010) and differential rotation of adjacent subunits in a dimer, as reported for GluK2 (Nayeem et al., 2011). We suggest that alternative splicing and the dimer interface mutations that increase kainate efficacy may induce conformational changes in domain 1 of the intact receptor that could affect such twisting motions or, alternatively, the stability of the ligand-binding domain (Fenwick and Oswald, 2010).
Two aspects of our data indicate that the channel environment also contributes to agonist efficacy: the increased kainate efficacy observed with mutations in the channel vestibule, and the greater efficacy of GluA2(R) relative to GluA2(Q). Because the movements initiated by ligand binding are ultimately coupled via the linkers to rearrangement of the channel helixes, these last two are perhaps not so surprising. Finally, it is clear that the large effect of stargazin coexpression on kainate efficacy could involve any or all of the above mechanisms, as discussed in more detail below. In summary, our data indicate that multiple factors that impact the coupling between ligand binding and channel gating contribute to agonist efficacy.
In our experiments, the L646T mutation on a GluA1flip background, in addition to increasing kainate efficacy (Fig. 4) (Mano et al., 1996), changed the properties of kainate desensitization such that we could observe substantial macroscopic desensitization of kainate currents even in whole-cell recording. However, the extent of steady-state desensitization was comparable to control GluA1, in keeping with the lack of relationship between desensitization and efficacy shown in Figure 7B. This finding suggests that the degree of binding cleft closure may affect the kinetics, if not the extent, of desensitization. Thus, increased cleft closure with kainate bound stabilizes or promotes formation of the dimer interface such that receptors gate before desensitizing. In contrast, macroscopically weakly or non-desensitizing kainate responses in control GluA1 that nonetheless exhibit 96% steady-state desensitization must reflect a high proportion of receptors that desensitize without gating.
Effects of stargazin on kainate desensitization and efficacy
Stargazin reduced kainate steady-state desensitization (Fig. 2A) substantially more than glutamate desensitization, providing further evidence that kainate and glutamate desensitization are distinct. Our data suggest that the majority of receptors with kainate bound desensitize without opening, and that stargazin must therefore reduce kainate desensitization by favoring the activated conformation of the receptor. This, in concert with the lack of strain on the dimer interface because the agonist-binding cleft remains relatively open with kainate bound, results in a large, macroscopically non-desensitizing current. In contrast, the much smaller effect of stargazin on glutamate steady-state desensitization indicates that stargazin cannot reduce the strain on the dimer interface caused by the greater degree of binding cleft closure and therefore has a smaller effect on glutamate steady-state desensitization.
The consistency of the effect of stargazin on maximal kainate efficacy was striking. For most of the homomeric GluA1 receptor constructs coexpressed with stargazin, kainate efficacy was ∼0.75. It appears that stargazin cannot convert kainate into a full agonist, but in all cases, it maximally affects coupling between agonist binding and channel gating. One possibility is that the residual difference between kainate and glutamate efficacy (∼25%) reflects the contribution of incomplete cleft closure when kainate is bound. In support of this conclusion, kainate efficacy for the L646T construct coexpressed with stargazin was significantly increased relative to GluA1 from 0.75 to 0.88. The effect of the increased cleft closure in this mutation was therefore additive with the effect of stargazin on kainate efficacy, suggesting that stargazin does not increase the degree of cleft closure. In contrast, there was no additivity between the effects of stargazin and mutations in the vestibule of the channel that significantly increased kainate efficacy (Fig. 6), suggesting that these mutations and stargazin affect efficacy by a similar mechanism. The transmembrane domains of stargazin are conveniently positioned to physically interact with the channel, and this may be a major contributor to the increase in kainate efficacy (Soto et al., 2007). The extracellular portions of stargazin, in contrast, are positioned to interact with the linker regions and the lower portions of domain 2, affecting inter-subunit rotation and linker separation. These interactions might also contribute to the effects of stargazin on kainate desensitization.
Footnotes
This work was supported by National Institutes of Health Grant NS051496.
References
- Armstrong N, Gouaux E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron. 2000;28:165–181. doi: 10.1016/s0896-6273(00)00094-5. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Sun Y, Chen GQ, Gouaux E. Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature. 1998;395:913–917. doi: 10.1038/27692. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Mayer M, Gouaux E. Tuning activation of the AMPA-sensitive GluR2 ion channel by genetic adjustment of agonist-induced conformational changes. Proc Natl Acad Sci U S A. 2003;100:5736–5741. doi: 10.1073/pnas.1037393100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balannik V, Menniti FS, Paternain AV, Lerma J, Stern-Bach Y. Molecular mechanism of AMPA receptor noncompetitive antagonism. Neuron. 2005;48:279–288. doi: 10.1016/j.neuron.2005.09.024. [DOI] [PubMed] [Google Scholar]
- Birdsey-Benson A, Gill A, Henderson LP, Madden DR. Enhanced efficacy without further cleft closure: re-evaluating twist as a source of agonist efficacy in AMPA receptors. J Neurosci. 2010;30:1463–1470. doi: 10.1523/JNEUROSCI.4558-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerrum EJ, Biggin PC. Rigid body essential x-ray crystallography: distinguishing the bend and twist of glutamate receptor ligand binding domains. Proteins. 2008;72:434–446. doi: 10.1002/prot.21941. [DOI] [PubMed] [Google Scholar]
- Boulter J, Hollmann M, O'Shea-Greenfield A, Hartley M, Deneris E, Maron C, Heinemann S. Molecular cloning and functional expression of glutamate receptor subunit genes. Science. 1990;249:1033–1037. doi: 10.1126/science.2168579. [DOI] [PubMed] [Google Scholar]
- Fenwick MK, Oswald RE. On the mechanisms of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor binding to glutamate and kainate. J Biol Chem. 2010;285:12334–12343. doi: 10.1074/jbc.M109.086371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez J, Du M, Parameshwaran K, Suppiramaniam V, Jayaraman V. Role of dimer interface in activation and desensitization in AMPA receptors. Proc Natl Acad Sci U S A. 2010;107:9891–9896. doi: 10.1073/pnas.0911854107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, O'Shea-Greenfield A, Rogers SW, Heinemann S. Cloning by functional expression of a member of the glutamate receptor family. Nature. 1989;342:643–648. doi: 10.1038/342643a0. [DOI] [PubMed] [Google Scholar]
- Holm MM, Naur P, Vestergaard B, Geballe MT, Gajhede M, Kastrup JS, Traynelis SF, Egebjerg J. A binding site tyrosine shapes desensitization kinetics and agonist potency at GluR2. A mutagenic, kinetic, and crystallographic study. J Biol Chem. 2005;280:35469–35476. doi: 10.1074/jbc.M507800200. [DOI] [PubMed] [Google Scholar]
- Horning MS, Mayer ML. Regulation of AMPA receptor gating by ligand binding core dimers. Neuron. 2004;41:379–388. doi: 10.1016/s0896-6273(04)00018-2. [DOI] [PubMed] [Google Scholar]
- Jin R, Banke TG, Mayer ML, Traynelis SF, Gouaux E. Structural basis for partial agonist action at ionotropic glutamate receptors. Nat Neurosci. 2003;6:803–810. doi: 10.1038/nn1091. [DOI] [PubMed] [Google Scholar]
- Keinänen K, Wisden W, Sommer B, Werner P, Herb A, Verdoorn TA, Sakmann B, Seeburg PH. A family of AMPA-selective glutamate receptors. Science. 1990;249:556–560. doi: 10.1126/science.2166337. [DOI] [PubMed] [Google Scholar]
- Madden DR, Cheng Q, Thiran S, Rajan S, Rigo F, Keinänen K, Reinelt S, Zimmermann H, Jayaraman V. Stereochemistry of glutamate receptor agonist efficacy: engineering a dual-specificity AMPA/kainate receptor. Biochemistry. 2004;43:15838–15844. doi: 10.1021/bi048447y. [DOI] [PubMed] [Google Scholar]
- Maltsev AS, Oswald RE. Hydrophobic side chain dynamics of a glutamate receptor ligand binding domain. J Biol Chem. 2010;285:10154–10162. doi: 10.1074/jbc.M109.088641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mano I, Lamed Y, Teichberg VI. A venus flytrap mechanism for activation and desensitization of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors. J Biol Chem. 1996;271:15299–15302. doi: 10.1074/jbc.271.26.15299. [DOI] [PubMed] [Google Scholar]
- Mosbacher J, Schoepfer R, Monyer H, Burnashev N, Seeburg PH, Ruppersberg JP. A molecular determinant for submillisecond desensitization in glutamate receptors. Science. 1994;266:1059–1062. doi: 10.1126/science.7973663. [DOI] [PubMed] [Google Scholar]
- Nayeem N, Mayans O, Green T. Conformational flexibility of the ligand-binding domain dimer in kainate receptor gating and desensitization. J Neurosci. 2011;31:2916–2924. doi: 10.1523/JNEUROSCI.4771-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partin KM, Patneau DK, Winters CA, Mayer ML, Buonanno A. Selective modulation of desensitization at AMPA versus kainate receptors by cyclothiazide and concanavalin A. Neuron. 1993;11:1069–1082. doi: 10.1016/0896-6273(93)90220-l. [DOI] [PubMed] [Google Scholar]
- Partin KM, Patneau DK, Mayer ML. Cyclothiazide differentially modulates desensitization of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor splice variants. Mol Pharmacol. 1994;46:129–138. [PubMed] [Google Scholar]
- Patneau DK, Vyklicky L, Jr, Mayer ML. Hippocampal neurons exhibit cyclothiazide-sensitive rapidly desensitizing responses to kainate. J Neurosci. 1993;13:3496–3509. doi: 10.1523/JNEUROSCI.13-08-03496.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plested AJ, Mayer ML. AMPA receptor ligand binding domain mobility revealed by functional cross linking. J Neurosci. 2009;29:11912–11923. doi: 10.1523/JNEUROSCI.2971-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priel A, Kolleker A, Ayalon G, Gillor M, Osten P, Stern-Bach Y. Stargazin reduces desensitization and slows deactivation of the AMPA-type glutamate receptors. J Neurosci. 2005;25:2682–2686. doi: 10.1523/JNEUROSCI.4834-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Trussell LO. The kinetics of the response to glutamate and kainate in neurons of the avian cochlear nucleus. Neuron. 1992;9:173–186. doi: 10.1016/0896-6273(92)90232-3. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stern-Bach Y, Stevens CF. The tetrameric structure of a glutamate receptor channel. Science. 1998;280:1596–1599. doi: 10.1126/science.280.5369.1596. [DOI] [PubMed] [Google Scholar]
- Sommer B, Köhler M, Sprengel R, Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67:11–19. doi: 10.1016/0092-8674(91)90568-j. [DOI] [PubMed] [Google Scholar]
- Soto D, Coombs ID, Kelly L, Farrant M, Cull-Candy SG. Stargazin attenuates intracellular polyamine block of calcium-permeable AMPA receptors. Nat Neurosci. 2007;10:1260–1267. doi: 10.1038/nn1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern-Bach Y, Russo S, Neuman M, Rosenmund C. A point mutation in the glutamate binding site blocks desensitization of AMPA receptors. Neuron. 1998;21:907–918. doi: 10.1016/s0896-6273(00)80605-4. [DOI] [PubMed] [Google Scholar]
- Sun Y, Olson R, Horning M, Armstrong N, Mayer M, Gouaux E. Mechanism of glutamate receptor desensitization. Nature. 2002;417:245–253. doi: 10.1038/417245a. [DOI] [PubMed] [Google Scholar]
- Tomita S, Adesnik H, Sekiguchi M, Zhang W, Wada K, Howe JR, Nicoll RA, Bredt DS. Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature. 2005;435:1052–1058. doi: 10.1038/nature03624. [DOI] [PubMed] [Google Scholar]
- Turetsky D, Garringer E, Patneau DK. Stargazin modulates native AMPA receptor functional properties by two distinct mechanisms. J Neurosci. 2005;25:7438–7448. doi: 10.1523/JNEUROSCI.1108-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]