

Abstract
Spinal muscular atrophy (SMA) is caused by loss of the survival motor neuron 1 gene (SMN1) and retention of the SMN2 gene, resulting in reduced SMN. SMA mice can be rescued with high expression of SMN in neurons, but when is this high expression required? We have developed a SMA mouse with inducible expression of SMN to address the temporal requirement for high SMN expression. Both embryonic and early postnatal induction of SMN resulted in a dramatic increase in survival with some mice living greater than 200 days. The mice had no marked motor deficits and neuromuscular junction (NMJ) function was near normal thus it appears that induction of SMN in postnatal SMA mice rescues motor function. Early postnatal SMN induction, followed by a 1-month removal of induction at 28 days of age, resulted in no morphological or electrophysiological abnormalities at the NMJ and no overt motor phenotype. Upon removal of SMN induction, five mice survived for just over 1 month and two female mice have survived past 8 months of age. We suggest that there is a postnatal period of time when high SMN levels are required. Furthermore, two copies of SMN2 provide the minimal amount of SMN necessary to maintain survival during adulthood. Finally, in the course of SMA, early induction of SMN is most efficacious.
INTRODUCTION
Proximal spinal muscular atrophy (SMA) is a leading genetic cause of infant and early childhood mortality (1). SMA is a recessive disorder that results in degeneration of motor neurons and atrophy of muscle (2). The survival motor neuron protein is encoded by two genes, SMN1 and SMN2, which lie adjacent to each other (3–5). SMA is caused by loss or mutation of the SMN1 gene and retention of the SMN2 gene (3,6). The SMN1 and SMN2 genes differ by one key nucleotide, a C-to-T transition in exon 7, which alters an exon splice modulator (7–10). This single nucleotide causes the majority of the transcript from the SMN2 gene to lack exon 7 (SMN▵7), whereas SMN1 produces full-length SMN transcripts (11,12). The SMN▵7 protein does not oligomerize efficiently and is rapidly degraded, thus causing a reduction in SMN levels, which are insufficient for the correct function and ultimately survival of motor neurons (13–15). The phenotypic severity of SMA is modulated by the SMN2 copy number and thus amount of SMN produced by the SMN2 gene (4,5,16–18). Missense mutations in SMN occur in both mild and severe SMA patients. Missense mutations in severe SMA patients often disrupt SMN's ability to efficiently oligomerize and thus the protein is rapidly degraded (13–15,19). In some missense mutations found in mild SMA patients, the SMN protein is still able to interact with, or complement, the SMN produced by SMN2 and thus produces more functional SMN complexes (20).
SMN is a 38 kDa ubiquitously expressed protein present in the cytoplasm and nucleus, where it often accumulates in structures called gems (4,21). SMN forms a complex with gemins 2–8 and unrip which enables the assembly of Sm proteins onto snRNAs (6,22–24). SnRNAs have a critical function in the splicing of genes and, as might be expected, complete loss of Smn is embryonic lethal (25). SMA mouse models have been produced by expressing SMN2 in mice that lack functional mouse Smn (26,27). As in humans, a high copy number of SMN2 results in complete rescue in mice, whereas two copies of SMN2 results in SMA mice that die at PND05 (26). Furthermore, expression of human SMN lacking exon 7 in SMA mice, or the use of a deletion allele that removes mouse Smn exon 7, results in slightly milder mice that live for 10–14 days (27,28).
The snRNP assembly function is disrupted in SMA. The degree of assembly activity inversely correlates with severity of SMA mice, as well as the ability of specific transgenic lines to correct SMA (29,30). The decrease in assembly results in decreased levels of certain UsnRNA, in particular the U11 and U12, which are important in splicing minor introns found in a subset of genes (29,30). Although splicing changes have been reported in various tissues from SMA mice, it is not clear whether these alterations have a direct role in causing SMA, or are a consequence of the advanced stage of disease in the mouse (30). The reported splice alterations could be secondary changes due to the moribund state of the mouse, spurious changes due to the number of tests performed or indirect changes that are not in the critical SMA pathway (6). Indeed further studies on younger animals have not revealed splicing dysregulation in the spinal cord of SMA animals (31) and certainly not all tissues require high SMN levels (32). Thus, it is unclear how splicing changes in, for instance, the kidney relate to SMA. Furthermore, the critical splicing events that occur within the motor neuron, and the consequence of these changes, remain to be determined in SMA. SMN has also been found in axons and may be critical in the transport of mRNA, such as β-actin, to the growth cones of axons (33). Thus, axon transport has been suggested to be the critical pathway disrupted in SMA (33). In motor neuron cultures from severe SMA mice, as well as knockdown of SMN in zebrafish axons, defects such as shorter axons are observed (33–35). However in SMA mice, motor axons show no growth defect (36) although there are alterations at the neuromuscular junction (NMJ) (28,36–39). It is currently unclear what the biochemical activity of SMN in axons is and as such it has not been assayed for alteration in SMA (6). Thus the exact function(s) of SMN that is/are disrupted to cause SMA remains unresolved (6).
Regardless of the exact mechanism of SMN's function, an attractive strategy for development of therapeutics in SMA is to increase SMN levels. A series of compounds have been developed that increase the amount of SMN produced by SMN2 by various mechanisms (40–47). Antisense oligonucleotides directed against specific elements in the SMN2 gene can significantly increase the amount of SMN in the required tissues (48–51). Gene therapy with AAV vectors that encode SMN or express a construct that alters the splicing of SMN2 can significantly increase SMN levels and rescue SMA mice (52–54). Indeed, self-complementary AAV9-SMN (scAAV9-SMN) has a remarkable impact on the SMA phenotype with survival extended beyond 300 days, as compared with 14 days for untreated mice, clearly demonstrating the importance of restoring SMN levels in SMA (52,55,56). Two elements that are critical to understanding the specific function of SMN in SMA as well as developing effective therapeutic strategies are the temporal and spatial requirements for high SMN levels. We have previously addressed the spatial requirement demonstrating that first, high levels of SMN are required in neurons to obtain correction of SMA. Second, not all tissues require high SMN levels and third, two copies of SMN2 do produce sufficient SMN for most tissues (32). In the current paper, we address the temporal requirement of high SMN levels in SMA mice using an inducible transgenic approach. We developed SMA mice containing a construct that expresses SMN only upon administration of doxycycline. We demonstrate that embryonic or early postnatal induction of SMN results in substantial rescue of SMA mice. However, later postnatal induction resulted in reduced rescue, indicating that for severe SMA there is a critical window of SMN induction that is most effective for rescue. The mice with restored SMN develop normal NMJs. In SMA mice that had been rescued by postnatal induction of SMN, we halted SMN induction to determine the long-term requirement for high SMN expression. In most cases, the mice survived for 1 month and then showed a precipitous decline, but no substantial physiological abnormality of the NMJ or clear evidence of a neuromuscular phenotype was observed. This could indicate that the level of SMN in the autonomic nervous system is critical at this latter time point. In two female SMA animals that were larger at birth, PND01 SMN induction followed by removal of SMN induction at PND28 resulted in survival of fertile breeding animals without any further induction of SMN. We suggest that in SMA mice, two copies of SMN2 is the minimum SMN level required for survival, and in these mice, a slightly higher SMN level at birth results in sufficient SMN for adult neurons. This could indicate that type II and type III SMA patients might have sufficient SMN for survival but not enough SMN during a specific developmental window thus resulting in SMA.
RESULTS
Generation of SMN inducible lines
To determine when high levels of SMN are required to rescue SMA mice, we expressed SMN under the tetracycline response element (TRE) and a minimal cytomegalovirus (CMV) promoter. The SMN construct is diagramed in Figure 1 and consists of two minimal CMV promoters and the TRE element. Thus upon activation, one CMV promoter drives SMN and the other drives luciferase expression. The bidirectional constructs were microinjected into FVB/N oocytes. A total of 20 founders were obtained using two constructs. Two founders that expressed SMN were used for further experiments. The lines were tested for SMN expression using the Camk2a-tTA (57), which expresses the tetracycline transactivator (tTA) under the forebrain Calcium Calmodulin-dependent kinase II (Camk2a) promoter (Camk2a-tTA, Jackson stock no. 003010) (57). This results in expression of high levels of SMN in the brain. The two transgenic lines used expressed SMN and were responsive to doxycycline when administered to the animals. The Camk2a-tTA line, or other available tTA lines including SMN promoter-tTA lines we made, did not result in rescue of SMA mice. We thus investigated the ROSA26rtTA (ROSA26rtTA, EGFP) (58) line (Jackson stock no. 005572), which has been used previously to drive expression in neural tissue (58). This line expresses the reverse tetracycline transactivator (rtTA) under the ROSA26 locus upon removal of a floxed cassette containing translation stop sites flanked by Cre. The ROSA26 locus is ubiquitously expressed (58). The line was first bred with a Sox2-Cre line to remove a stop cassette between the ROSA26 promoter and rtTA. (59). The resulting line was then crossed with SMN2+/+; SMN▵7+/+; Smn+/− mice to obtain offspring that were SMN2+/−; ROSA26rtTA; +/− SMN▵7+/−. The ROSA26 locus and SMN2 transgene insertion lie relatively close to each other so we performed crosses to obtain a recombinant chromosome containing both SMN2 and ROSA26rtTA on the same chromosome. Briefly, we crossed ROSA26rtTA/SMN2 mice against wild-type mice and scored for the presence of both the SMN2 and ROSA26rtTA transgene in the progeny. One of 176 offspring contained the desired recombinant chromosome. This mouse was then bred to obtain mice homozygous for SMN2; ROSA26rtTA; SMN▵7, heterozygous for Smn and contained Luci-TRE-SMN (which was also bred to homozygosity in some mice). These mice were used to generate SMA mice that contained the inducible transgene.
Figure 1.

Diagram of inducible Luciferase (Luci) Tetracycline response element (TRE) Survival motor neuron (SMN) transgenic construct and reverse tetracycline trans activator (rtTA) inserted into the ROSA26 locus with the floxed stop cassette removed (58). IRES, internal ribosome entry sequence; EGFP, enhanced green fluorescent protein; PminCMV, minimal cytomegalovirus promoter; Luc, Luciferase; SA, splice acceptor site; rtTA, reverse tetracycline trans activator; Dox, doxycycline.
Expression of SMN from the inducible transgene
To determine the expression of SMN from the inducible transgene SMN2+/+; ROSA26rtTA+/+; SMN▵7+/+; Luci-TRE-SMN; Smn+/−, mice were characterized by quantitative western blot analysis and immunohistochemistry using a human specific SMN antibody (48). We also performed RT–PCR and real time RT–PCR to determine the relative amount of SMN mRNA produced upon induction. Figure 2 shows the luciferase activity in neonatal brain (Fig. 2A) and spinal cord (Fig. 2B) after doxycycline food is introduced to the mother. Induction occurs at high levels after 48 h in these neonates. We then performed western blots using a human specific SMN antibody and a luciferase antibody in mice at 3, 5 and 10 days after induction. Figure 2C and D show the SMN expression in brain and spinal cord of induced mice over this range. Induction is clearly detectable at postnatal day 3 (PND03) and SMN levels and luciferase continue to increase at PND10. The quantification of the western blots is shown in Figure 2E and F. The results of real time RT–PCR for amount of full-length SMN containing exon 7 in neonatal brain and spinal cord at 0, 3, 5 and 10 days postinduction is shown in Figure 2G and H. Full-length SMN is induced at PND03 and continues to increase at PND10 in a similar manner to SMN protein. We next examined induction of SMN in adult carrier animals (Smn+/−) that were fed doxycycline directly in their food. Figure 3A and B show the results of SMN induction beginning at PND28 and at 2, 3, 5 and 10 days after induction in brain and spinal cord tissue in western blots using the human specific SMN antibody (48). Thus, the expression pattern is similar in adult and neonatal animals with clear induction of SMN at PND03 that steadily increases to maximum induction 10 days after doxycycline dosing. Quantative analysis of SMN expression relative to actin and luciferase expression relative to actin was also performed at each time point in brain (Fig. 3C) and spinal cord (Fig. 3D) tissue. We also examined induction of SMN in tissues outside the nervous system. All tissues examined showed SMN induction as would be expected from the ROSA26rtTA locus (Fig. 3E). Last, we examined expression of SMN in motor neurons using immunohistochemistry. As shown in Figure 3G, induction of SMN by doxycycline results in a clear increase in SMN expression in motor neurons as well as gems within those motor neurons. SMN expression in high copy SMN2 spinal cord (Fig. 3F) and non-induced spinal cord (Fig. 3H) samples are shown for comparison. Thus SMN is induced in the locations required for correction of SMA.
Figure 2.

Analysis of SMN induction in neonatal mice. (A and B) Doxycycline was administered to the mother at PND0 (birth of the pups) and luciferase activity measured in the (A) brain and (B) spinal cord of the neonates at the indicated time after the start of induction. D indicates the number of days after the start of induction. (C and D) Western blots of (C) brain and (D) spinal cord tissue of the neonates for quantification of luciferase (Luci), SMN and actin. Non-I indicates mice of the same genotype as induced mice but without induction, ▵7 indicates ▵7 carrier mice (SMN2+/+;Smn+/−; SMN▵7+/+) without the inducible transgene. All induced mice are heterozygous for mouse Smn (Smn+/−) and SMN is detected with a human specific antibody. (E and F) Quantification of luciferase to actin ratio (grey bars) and SMN to actin ratio (white bars) from western blots of neonatal mice post-induction shown in (C) and (D). (n= 24 mice at day 3, n= 17 mice at day 17, n= 19 mice at day 10). (G and H) Real-time RT–PCR of full-length human SMN mRNA relative to cyclophilin mRNA after induction of SMN in (G) brain and (H) spinal cord tissue. (n= 4 mice for Non-I, n= 4 mice at day 3, n= 5 mice at day 5, n= 7 mice at day 10).
Figure 3.

Analysis of SMN induction in adult mice. (A and B) Doxycycline was administered to the mice at 28 days of age and then western blots of (A) brain and (B) spinal cord tissue of adult mice for quantification of luciferase (luci), SMN and actin performed. Non-I indicates mice of the same genotype as induced mice but without induction, ▵7 indicates ▵7 carrier mice (SMN2+/+; Smn+/−;SMN▵7+/+) without the inducible transgene. All induced mice are heterozygous for mouse Smn (Smn+/−) and SMN is detected with a human specific antibody. (C and D) Quantification of SMN to actin ratio (white bars), and luciferase to actin ratio (gray bars), from western blots of adult mice post-induction shown in (A) and (B). (n= 3 mice for brain and n= 3 mice for spinal cord). (E) Western blot of a series of tissues harvested 10 days after SMN induction. Induction of SMN and luciferase is observed in all tissues tested. (F–H) Immunostaining of human SMN in motor neurons of spinal cord sections from (F) high-copy SMN2 mice, (G) mice induced to express SMN for 28 days and (H) non-induced mice. Immunostaining is performed with a human specific SMN antibody and animals were sacrificed at 28 days of age. Arrows indicate gems, scale bar is 50 µm.
Induction of SMN expression rescues SMA mice
We next tested the ability of the inducible system to rescue SMA mice by first inducing SMN in the embryonic stage. Mothers were fed doxycycline when embryos were at 13 days of gestation (E13) to induce SMN in embryonic mice. The survival curves for SMA induced and non-induced mice of the same genotype are shown in Figure 4A. The non-induced SMA mice (SMN2+/+; SMN▵7+/+; Smn−/−; ROSA26tTA+/+; Luci-TRE-SMN +) died on average at PND14 and showed no extension of lifespan compared with mice lacking the inducible transgenes. In contrast, induction of SMN in SMA mice from E13 with doxycycline resulted in substantial rescue of SMA with some mice living over 200 days.
Figure 4.

Survival analysis of induced mice. (A) Survival curve of SMA mice induced for SMN at embryonic day 13 compared with non-induced mice of the same genotype. Non-induced SMA mice lived an average of 13 days and none lived past 18 days (n= 29 mice). Induced mice lived on average for 132 ± 32 days and some lived passed 200 days (n= 14 mice), Log Rank P ≤ 0.001 compared with non-induced mice. (B) Survival curve of SMA mice induced for SMN at PND0/PND01 compared with non-induced mice of the same genotype. The average survival of mice with PND0/PND01 induction was 86 days with a number of mice living past 200 days (n= 38 mice), Log Rank P ≤ 0.001 compared with non-induced mice. The non-induced SMA mice lived an average of 13 days and none lived past 18 days (n= 29). When induction was started at PND02 some mice did show extended survival (mean 25 ± 9, max 151 days) but a lower and less pronounced response was observed (n= 15 mice). (C and D) Western blot analysis showing SMA mice with SMN and luciferase induction (+Dox) and SMA mice that had SMN induced for 28 days and then doxycycline was removed for 10 days (−Dox) in (C) brain and (D) spinal cord tissue. Human specific SMN antibody was used (48). Non-I indicates animals of the same genotype apart from the mouse Smn locus (Smn+/−) that were not fed doxycycline. ▵7 indicates ▵7 SMA carrier mice. WT indicates wild-type mice. (E) Weight curve of carrier Smn+/− mice (n= 20 mice) and rescued Smn−/− SMA mice (n= 10 mice) with SMN induction. Notice the reduced weight of rescued mice although they do steadily increase in weight.
We then examined induction of SMN in the postnatal period by starting induction of SMN at birth (PND0/PND01) and at PND02. The induction, as indicated in the previous section, takes 3 days to result in significant levels of SMN. Thus SMN expression is detectable at PND03 when started at birth (PND0/PND01), and PND05 when started at PND02. As shown in Figure 4B, induction at birth (PND03 SMN expression) had a substantial impact on survival with the average life span being 86 days and some mice living over 200 days. The variability in survival may be a reflection of variable SMN induction. This survival was not statistically different from that resulting from embryonic induction. Thus postnatal induction of SMN has a major impact on survival of SMA mice. A later induction at PND02 (PND05 SMN expression) resulted in an effect on survival with one mouse living for 151 days. However, the effect was less dramatic than that observed with earlier induction, indicating that early postnatal induction of SMN is more effective in rescuing survival. Figure 4 shows SMN levels in the brain (Fig. 4C) and spinal cord (Fig. 4D) of rescued SMA mice on western blots indicating that there was substantial induction of luciferase and SMN expression upon administration of doxycycline. Removal of doxycycline for 10 days after 28 days of administration revealed levels of SMN and luciferase expression similar to non-induced, ▵7 and wild-type mice (Fig. 4C and D). Figure 4E shows the weight increase of SMA mice. In general, SMA rescued mice were smaller than their littermates but showed normal activity until close to death when they showed reduced movement. In some cases, a small amount of ear necrosis occurred but no abnormality of tail or feet was observed. Upon death, some mice that survived for over 100 days were found to have abnormalities of the gut with an impacted bowel and pockets of fluid and gas (pneumoperitoneum). This was not observed in control sibs also on doxycycline food.
In SMA mice that develop with low levels of SMN, synaptic transmission at the NMJ is impaired (38,60,61). The deficits include reduction in endplate current (EPC) amplitude (a measure of strength of the synapse) that is primarily due to reduction in quantal content (the number of vesicles released). Repetitive stimulation reveals an increase in facilitation, which is consistent with reduced probability of vesicle release. We tested whether postnatal induction of SMN in SMA mice resulted in restoration of NMJ function. We recorded from the tibialis anterior (TA) of three SMA mice in which SMN had been induced by feeding the mice doxycycline. Each recording was compared with an age-matched mouse heterozygous for the mouse Smn locus (Smn+/−) and housed with the SMA mice and containing the transgenes. We found no statistically significant difference in EPC or miniature endplate current (MEPC) amplitude between control and SMN rescued endplates (Fig. 5A and Table 1). During development of the NMJ in SMN deficient mice, there is a slowed switch from expression of embryonic AChRs to adult AChRs. This manifests as a prolongation of the time constant of MEPC decay. We measured time constant of MEPC decay in the two groups and obtained nearly identical values. There was, however, a slight tendency towards reduced depression during repetitive stimulation in SMN rescued endplates (Fig. 5A, P < 0.05). This is consistent with a slight reduction in probability of vesicle release, and is similar to the finding during repetitive stimulation seen in P10 SMA mice of the SMN2+/+; ▵7SMN+/+; Smn−/− genotype. Overall, function of the NMJ appeared to be near normal, with only a modest reduction in the probability of vesicle release. In addition to electrophysiological changes at the NMJ, altered morphology of the NMJ have been reported in SMA mice with accumulation of neurofilament and lack of mature, fully developed NMJs (36–38,62). As shown in Figure 5B–G, NMJs in transverse abdominis muscle of SMA rescued animals show full maturity, proper innervation and do not show abnormal accumulation of neurofilament in the NMJ or the nerve. As the mice had no marked motor deficits and NMJ function was near normal it appears that induction of SMN in postnatal SMA mice rescues motor function.
Figure 5.

Correction of neuromuscular phenotype in SMA mice with SMN induction. (A) Shown are the endplate current (EPC) evoked by nerve stimulation, the miniature endplate current (MEPC) thought to represent the post-synaptic response to acetylcholine in one synaptic vesicle and the response to repetitive stimulation of the nerve at 50 Hz for both a control endplate and an endplate from a mouse where SMN expression was rescued by the doxycycline-driven construct. There is no significant difference between the control and the SMN rescued NMJ in either EPC or MEPC amplitude. There is slightly more depression during a 50 Hz train of stimuli for the control endplate. (B–G) There is no significant difference between the control (B–D) and the SMN rescued (E–G) NMJ morphology, size or innervation pattern. (B and E) Anti-neurofilament 160, (C and F) alpha-bungarotoxin, (D and G) merged image. Scale bar is 20 µm.
Table 1.
Electrophysiological analysis of rescued SMA mice.
| Control | SMN rescue on doxycycline | Control | 1 month after doxycycline withdrawal | |
|---|---|---|---|---|
| EPC amplitude (nA) | 72.0 ± 4.6 | 60.0 ± 6.3 | 93.7 ± 5.9 | 84.7 ± 1.5 |
| MEPC amplitude (nA) | 1.11 ± 0.03 | 1.12 ± 0.02 | 1.28 ± 0.09 | 1.37 ± 0.05 |
| MEPC time constant of decay (ms) | 1.21 ± 0.08 | 1.22 ± 0.07 | 1.23 ± 0.03 | 1.21 ± 0.05 |
| Quantal content | 66.2 ± 6.4 | 55.5 ± 5.2 | 76.1 ± 9.1 | 62.8 ± 2.4 |
| P10/P1 during a 50 Hz train | 0.63 ± 0.06* | 0.83 ± 0.02* | .67 ± 0.03 | .70 ± 0.08 |
| MEPC frequency (Hz) | 1.5 ± 0.02 | 1.3 ± 0.3 | 1.9 ± 0.1 | 3.7 ± 1.3 |
P10/P1 is a measure of depression during repetitive stimulation and is calculated by dividing the amplitude of the 10th endplate current in a train of stimuli by the amplitude of the first endplate current. For groups, n= 3, all values are shown ±SEM.
EPC, endplate current; MEPC, miniature endplate current.
*P < 0.05 for control versus SMN rescue.
Decay of SMN induction upon doxycycline removal
We next investigated the decay of SMN expression conferred by withholding doxycycline in induced mice. The decay of luciferase expression after removal of doxycycline was first examined in brain and spinal cord tissue. Figure 6A and B shows the decay of luciferase in mice that had doxycycline food removed and thus induction of expression of the transgene terminated. The decay of luciferase expression in brain (Fig. 6A) and spinal cord (Fig. 6B) occurs gradually with loss of luciferase expression observed 10 days after removal of the doxycycline food. We next followed SMN and luciferase decay of expression in brain (Fig. 6C) and spinal cord (Fig. 6D) tissue by western blot analysis. Induction with doxycycline food was performed starting at PND0 until PND28 and then doxycycline was removed. Tissue was collected 2, 3, 5 and 10 days after removal. SMN and luciferase expression returned to close to baseline levels 10 days after removal. Quantification of the luciferase to actin expression and SMN to actin expression from western blot analysis of the brain (Fig. 6E) and spinal cord (Fig. 6F) is shown. Thus 10 days after doxycycline removal, the SMN expression observed is only from the SMN2 transgene. The SMN mRNA levels were also assayed by real-time RT–PCR 15 days after removal of doxycycline and showed low SMN mRNA levels compared with the induced samples in both brain (Fig. 6G) and spinal cord (Fig. 6H).
Figure 6.

Decay of SMN induction upon doxycycline removal. (A and B) Doxycycline was removed and luciferase (Luci) activity measured at 2, 3, 5 and 10 days after removal of SMN induction in (A) brain (B) spinal cord. The mice where induced for 28 days starting at PND0. (n= 3 for brain, n= 3 spinal cord). (C and D) Western blot analysis of SMN, luciferase and actin protein after removal of SMN induction in (C) brain and (D) spinal cord using a human specific monoclonal antibody. –D: days after doxycycline removal. Non-I: non-induced mice and ▵7: ▵7 carrier mice that lack the inducible transgene. (E and F) Quantification of western blots showing the luciferase to actin ratio (gray bars) and the SMN to actin ratio (white bars) and at 2, 3, 5 and 10 days after removal of doxycycline in (E) brain (n= 3 mice) and (F) spinal cord (n= 3 mice). (G and H) Real-time RT–PCR of full-length human SMN mRNA relative to cyclophilin mRNA after removal of SMN induction for 15 days (–15D, n= 5 mice) compared with 28-day induction (I, n= 8 mice) and non-induced mice (Non-I, n= 6 mice) in (G) brain and (H) spinal cord tissue.
We next examined the effect of removal of SMN induction on SMA mice. SMA mice containing the inducible transgene (SMN2+/+; SMN▵7+/+; Smn−/−; Luci-TRE-SMN) where induced at birth (thus induction of SMN at PND03) and kept on doxycycline food until PND28. At PND28, the doxycycline induction was removed and the mice observed. When SMN induction was removed from SMA mice they behaved normally and there was no overt phenotype. In most cases (n= 5), the SMA mice without induction lived for just over a month and then showed rapid decline and death. In the second group of mice (n= 2), where the animals were in general larger at birth, removal of SMN induction had no effect. The mice were phenotypically normal and have survived beyond 8 months of age. The mice were still alive at the time of the preparation of this manuscript. These long-surviving mice were female and gave birth to litters that contained the expected number of SMA animals. Thus, two copies of SMN2 likely express SMN just in the range required in these mice in adult life. Furthermore, two copies of SMN2 can produce sufficient SMN for all cells later in life but not during a critical post-natal period.
To determine the effect of reduction in levels of SMN after development of the NMJ is complete, we performed electrophysiology 1 month following withdrawal of doxycycline induction of SMN in SMA mice. Each recording was compared with an age-matched mouse heterozygous for mouse Smn+/− but containing the same transgenes and housed under the same conditions. One of the three mice recorded had become ill and was no longer walking at the time of euthanasia. Despite this mouse appearing very weak, there were no differences in neuromuscular transmission between Smn−/− adult mice where induction of SMN had been withdrawn and heterozygous controls 1 month after removal of doxycycline (Fig. 7A, Table 1). The amplitude of the endplate current was similar in the two groups as was the number of vesicles released and the response to repetitive stimulation. It thus appears that function of the NMJ is not dependent in adult mice on the continued presence of SMN for periods of up to 1 month. Furthermore, immunostaining of NMJs in the transverse abdominis revealed normal fully developed NMJs with innervation and no neurofilament accumulation again revealing normal NMJs even after SMN induction removal for 1 month in SMA animals (Fig. 7B–G). Thus high SMN levels are required between birth and postnatal day 28 for motor neurons and not in adult stages were SMN2 produces sufficient SMN for this system.
Figure 7.
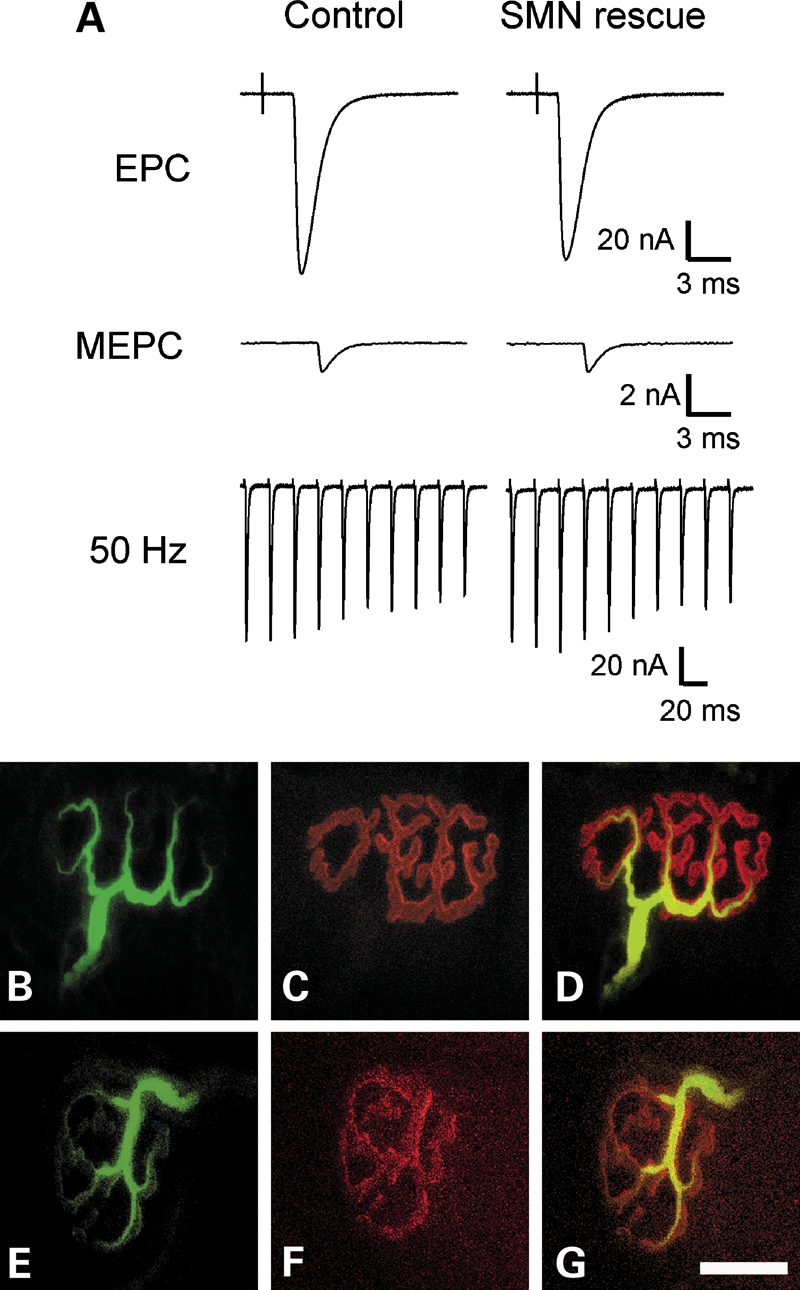
No neuromuscular phenotype in SMA mice with removal of SMN induction. (A) Shown are the EPC, MEPC and the response to 50 Hz stimulation for both a control endplate and an endplate from a mouse where SMN expression was rescued by the doxycycline-driven construct, but doxycycline was removed 1 month earlier. (B–G) There is no significant difference between the control (B–D) and the SMN rescued (E–G) NMJ morphology, size or innervation pattern. (B and E) Anti-neurofilament 160, (C and F) alpha-bungarotoxin, (D and G) merged image. Scale bar is 20 µm.
DISCUSSION
The SMN protein complexes with a series of other proteins gemins 2–8 and unrip to form the SMA complex which functions in assembly of Sm proteins onto snRNAs (6,22–24). Complete loss of SMN either in an organism or a particular tissue is lethal as would be expected as assembled snRNPs are essential for correct splicing of genes required in all cells (6,25,27,63–65). SMA is caused by loss of SMN1 and retention of SMN2, which leads to reduced levels of both full-length SMN mRNA and SMN protein (4,5). The copy number of SMN2 and the ability of SMN2 to produce full-length mRNA inversely correlate with phenotypic severity (16–18). But when and where are levels of SMN above that produced by two copies of SMN2 needed to correct SMA? Indeed this becomes particularly important with the use of Cre drivers to either remove or activate SMN as the timing of Cre expression in the tissue of interest will be critical. For instance, the cholineacetyltransferase (ChAT) promoter does not reach full activity until postnatal day 7 in mice and thus may not be an optimal driver to test the importance of SMN in developing motor neurons (66,67). Indeed SMN driven by the ChAT promoter had no impact on severe SMA mice (SMN2+/+ Smn−/−) (unpublished data). We have previously shown that high SMN levels in the nervous system obtained with the Prion promoter did result in rescue of severe SMA mice, whereas high expression in skeletal muscle using the human skeletal actin promoter did not (32). Thus high levels of SMN above that produced by SMN2 is important in tissues where the Prion promoter is expressed which is the nervous system but does include tissue outside the nervous system such as the neuroendocrine glands and the combined expression of SMN in muscle and nerve as being important cannot be excluded.
Another important question with respect to SMN pathophysiology is which cells in the nervous system are most critical? Is high SMN expression in motor neurons sufficient or do other neural cell types play a role? In this regard, removal of Smn exon 7 using the Oligo-Cre driver to give a non-functional Smn specifically in motor neurons in the presence of SMN2 results in a SMA phenotype indicating the importance of high SMN in motor neurons (68). However, the phenotype of these mice is not as severe as would be expected. This could be due to the requirement of high levels of SMN in neurons that Oligo-Cre is not expressed in, such as interneurons, or in cells outside of the nervous system that contribute to the SMA phenotype (68). Alternatively, the Cre driver may leave a sufficient number of motor neurons untouched that can then innervate muscle sufficiently. Interestingly, recently interneurons have been extensively examined in SMA mice (69). Studies show that the output of the motor neurons upon interneuron stimulation is substantially reduced suggesting that the motor circuit is disrupted in SMA mice (69). Furthermore, reduced input to the motor neuron was demonstrated as less vesicular glutamate transporter 1 puncta were present on motor neurons (60,69). However, it appears that this phenotype is retained in the Oligo-Cre exon 7 deleted mice implying that it is related to reduced SMN levels in the motor neuron (68). Further studies are needed to define exactly where high SMN levels, above that produced by two copies of SMN2, are required in SMA mice. In these studies, careful consideration of when and where the Cre drivers express will be critical to interpreting the results.
SMN functions in assembly of snRNPs, and this function is disrupted in SMA animals. There is a consistant correlation of snRNP activity with severity of the mice. Mild mice (SMNA2G; SMN2+/+; Smn−/−) show considerably greater activity than severe (SMN2+/+; Smn−/−) or ▵7 (SMN2+/+; SMN▵7+/+; Smn−/−) mice (29). Furthermore, SMN containing the A111G missense mutation corrects one-copy SMN mice (SMN2+/−; Smn−/−) as well as snRNP assembly activity (20,30). Reduced SMN levels in SMA mice leads to alteration in the snRNP profile and the amount of particular snRNPs (20,29,30). Given the alteration of snRNP levels in SMA mice, one would predict changes in splicing of certain sensitive genes.
Exon array studies have revealed multiple changes in spinal cord tissue of SMA mice as well as multiple other tissues such as the kidney (30). However, these studies were performed in mice relatively close to death and it is hard to determine which changes are a consequence of the state of the mouse as opposed to altered snRNP levels. As indicated earlier, transgenic studies show that high levels of SMN are not required for renal and hepatic maintenance and normal function. The Prion-SMN transgene does not increase SMN levels in these tissues but does correct the SMA phenotype (32). Indeed, it is most likely that the changes in these tissues have little consequence in SMA disease.
SMN has also been detected in axons and is believed to be important in the transport of mRNAs to the axon tip (70). However, it is not clear what exactly SMN does in this process making it difficult to assay for defects in SMA mice. Finally, it is not clear why motor neuron axons would be particularly affected by alterations in mRNA transport. Here we show that the critical window of high SMN expression is a postnatal period which is not when β-actin would be most important for growth of axons. We have not detected abnormal growth of motor axons in SMA mice during embryogenesis (36). Recently removal of β-actin from motor neurons was shown to have no major impact thus making it unlikely that a disruption of β-actin is a critical event, at least in motor neurons, in SMA (71).
To define specific SMN targets, one must consider which changes are critical before the end state of the disease. Furthermore, no studies to date have studied changes in motor neurons as opposed to whole spinal cord (31). Thus the direct targets of SMN deficiency and the mechanism of how reduced SMN causes SMA have yet to be defined. The studies in the current paper would indicate that the critical time period for any putative causal splicing change in SMA would be between birth and postnatal day 28. Interestingly, this period is when many changes occur in the splicing pattern of genes (72).
We have investigated the temporal requirement for high levels of SMN, above the level produced by two copies of SMN2, in SMA mice. Remarkable rescue of SMA ▵7 mice has occurred with early introduction of scAAV9-SMN at P0 or P1 (52) via the facial vein. When high-titer virus is used, scAAV9 crosses the blood–brain barrier resulting in considerable transduction of motor neurons and rescue of SMN ▵7 SMA mice beyond a year of age (52,55,56,73,74). Furthermore, normal function of the NMJ is found in these rescued mice (52) similar to that observed with rescued mice in the current study. Delayed introduction (PND05) of scAAV9 into the circulation results in some rescue of SMA, but survival is greatly diminished at later time points thereafter (PND10) (52). One possible reason for this is the reduced transduction of motor neurons in older mice (52,73). Alternatively, SMN may have to be introduced early in the disease to have an effect on survival.
In the current paper, we have used a doxycycline inducible transgene to determine when high levels of SMN are required. In agreement with studies using scAAV9-SMN, we find early induction of SMN at PND03-PND04 gives maximum effect on survival. Later induction (PND06) results in some rescue but there is a significantly reduced response. Thus, this data predict any SMN-inducing strategy for treatment of SMA is best introduced early in the disease course, especially in severe SMA mice. The question remains open as to whether SMN-inducing therapies will be effective during the plateau phase of SMA type II and III (75–77). However, it has been reported from motor neuron estimation studies that a functional motor neuron drop occurs early followed by more gradual motor neuron loss, possibly due to ageing (75–77). Is there a critical period in all SMAs when SMN-inducing therapies need to be introduced to have a clinical effect? Certainly it will be important to consider this when moving therapeutics tested in mice into human clinical trials.
We have shown that, after induction of SMN in the postnatal period, motor neuron electrophysiology is nearly normal and NMJs are fully developed. Indeed, even after removal of doxycycline for a month, there is still no marked abnormality of the NMJ. Thus high levels of SMN over the early postnatal period completely correct the motor neuron. This indicates that high SMN levels are only required over a limited postnatal time frame for correct function of NMJs. Furthermore, in two female SMA mice, which were larger at birth, the removal of SMN induction at PND28 resulted in no detectable defects. These females have given birth to multiple litters and have lived out beyond 8 months of age. This indicates that two copies of SMN2 can in some cases produce sufficient SMN for all tissues in adult life, but this level of SMN is close to the limit of what is required, thus resulting in two groups of animals. This may indicate that after early correction of SMN levels in mild type II and III SMA patients there will not be a requirement for continual high levels of SMN expression. Heart defects have recently been reported in SMA mice most likely arising due to low SMN in the autonomic nervous system (78–80). It is not known if autonomic nervous system defects occur in SMA in man. However, it should be noted that the mouse has a high heart rate and thus the defects seen in mouse may be reflected in different autonomic systems in man.
In conclusion, postnatal induction of SMN postnatally rescues SMA mice and an ∼4-week postnatal period of high SMA is sufficient for long-term rescue of motor defects. Thus, a short-term induction of SMN at the correct time and in the correct cell type can have a major impact on survival in murine SMA.
MATERIALS AND METHODS
Generation of transgenic mice
Luci-TRE-SMN transgene
The SMN cDNA containing exons 1–8 was excised from pcDNA3 (81) with NheI and XhoI and subcloned into pBI-L between the NheI and SalI sites (Clontech). The pBI-L -SMN vector has luciferase driven by a minimal CMV promoter in one direction, and SMN driven by a minimal CMV promoter in the other direction. The TRE element lies between the two promoters and thus activation with rtTA drives expression of both luciferase and SMN simultaneously. The second construct was engineered to remove 31 bp from the 5′ end of vector and contain more of the SMN exon 8 untranslated region. The previously described HSA-SMN construct was cut with BglII and XhoI and the pcDNA3-SMN plasmid was cut with BglII and XhoI. The HSA-SMN BglII/XhoI fragment was ligated to the cut pcDNA3-SMN plasmid to create pcDNA3-SMN (B). Thirty-one base pairs were removed from the 5′ end by first cutting pcDNA3-SMN (B) with HindIII and BamHI. The following oligonucleotides were annealed (AGCTTGCGGCCCGCTAGCAG and ACGCCGGCGATCGTCCTAG) and ligated to the cut pcDNA3-SMN (B) plasmid. The resulting construct was cut with NheI and XhoI and subcloned into pBI-L as described earlier.
The constructs were linearized with AseI and the 6.3 kb construct was injected into the male pronucleus of FVB/N mice. Twenty founders were obtained and analyzed for the expected transmission of the transgene and a single insertion by Southern blotting. Those animals were crossed to mice that expressed CamK2ka-tTA (57) (Jackson Laboratories stock no. 003010) to check for mice that had high levels of doxycycline responsive SMN expression in the brain. The mice that showed positive expression where then crossed to SMA mice (SMN2+/+; Smn+/−; SMN▵7+/+) to obtain SMN2+/+; Smn+/−; SMN▵7+/+; Luci-TRE-SMN+/+ mice. These mice were crossed with SMN2+/+;Smn+/−;SMN▵7; ROSA26rTA+/+ generated in the next section. The final genotype of the mice was SMN2+/+;Smn+/−;SMN▵7+/+; ROSA26rTA+/+;Luci-TRE-SMN either +/+ or +/−. Two lines, in particular, were followed as they showed the capability of rescuing the SMA phenotype (see in what follows). All breeding and subsequent use of animals in this study were approved by the IACUC of The Ohio State University, Columbus, OH.
ROSA26rtTA
The ROSA26rtTA (Jackson Laboratories stock no. 005572) (58) was obtained and crossed with Sox2-Cre mice to remove the stop cassette between the ROSA26 promoter and rtTA. ROSA26rtTA mice lacking the stop cassette were then crossed with mice containing SMN2 (26) to obtain mice that had SMN2 and ROSA26rtTA. The SMN2 insertion lies relatively close to the ROSA26 locus on the same chromosome (82); therefore, we crossed the mice to wild-type FVB/N mice and identified a recombination event that placed SMN2 and ROSA26rtTA on the same chromosome. One of 178 offspring was positive for both transgenes. This mouse was then bred to obtain the SMN2+/+;SMN▵7+/−; Smn+/−; ROSA26rtTA+/+ mice.
Genotyping of transgenes
The SMN2, Smn knockout allele and SMN▵7 alleles were genotyped as previously described (28). The ROSA26rtTA was amplified with ROSA26F aagtcgctctgagttgttatcag and rtTA-R cgggttgttaaaccttcgattccg which is positive if the rtTA is present. To determine homozygozity of the ROSA26rtTA transgene, the same ROSA26F primer was used with ROSA26R ggagcgggagaaatggatatga. This amplifies when an intact ROSA26 is present but not when ROSA26rtTA is homozygous. The Luci-TRE-SMN transgene was detected with Luci-TRE-Vec-F gatcctctagtcagctgacgcgt and SMN exon3R cagtgtaaaccacaacacagg, SMN exon6F tcccatatgtccagattctcttg and SMN exon8R tcaactgcctcaccaccgtgc as well as luciferaseF acttgactggcgacgtaatccacg and luciferaseR cgcttccatcttccagggatacga. The SMN exon6F and SMN exon8F primers also detect the SMN▵7 transgene but the two transgenes can be distinguished by size (presence or absence of SMN exon 7). The following PCR conditions were used for amplification 94°C for 3 min, then 30 cycles of 95°C for 30 s, 60°C for 30 s, 72°C for 1 min, final extension 72°C for 3 min.
Doxycycline administration for induction of SMN
For embryonic induction, timed pregnancies were set up as described previously (36). At E13, a water bottle containing 500 µg/ml of doxycycline (Sigma) was introduced. Detailed protocols can be found at http://openwetware.org/wiki/Conklin. The administration was continued with weekly water bottle changes until the birth of the pups. Then doxycycline food (200 mg/kg) (Bioserv) was introduced to the cage and the doxycycline water removed. For postnatal induction, high-concentration doxycycline food (6 g/kg) (Bioserv) was administered to the mother thus the pups received doxycycline in the mother's milk. High-concentration doxycycline food (6 g/kg) was found to severely affect birth rates if administered to pregnant mice, however, we did not detect any other adverse effects in SMA or control animals receiving high doxycycline food after birth.
Luciferase assay
Tissues were dissected from the mouse, homogenized in five volumes of 10 mm Tris, pH7.5 and then 80 µl of lysate mixed with 20 µl of buffer (Promega E397A). The sample was frozen and thawed twice, centrifuged and supernatant collected. Supernatant containing 50 µg of protein was mixed with 100 µl of luciferase reagent (Promega E1483) just prior to measurement in a luminometer (Lumat LB9507 Berthold).
Western blot
Western blot analysis of adult and neonatal tissue was performed as previously described (28,32). Briefly, five volumes of blending buffer was added to the tissue sample (5% SDS, 62.5 mm Tris pH 6.8, 5 mm EDTA) and the sample homogenized. The protein level in the sample was determined, an equal volume of loading buffer added (62.5 mm Tris pH 6.8, 20% glycerol, 200 mm DTT, 0.2% bromophenol blue) and 50 µg of protein per lane loaded onto a 12.5% SDS polyacrylamide gel. The separated proteins were transferred to a 0.2 µm pore size PVDF membrane (BR 128950261, Bio-Rad) using Genie Electrophoretic Blotter system (Invitrogen) as previously described (28,32). Blots were blocked for 1 h in 5% milk, PBS-Tween 0.05% and incubated with primary antibody [hSMN-KH (48) 1:20 for 4 h and/or overnight, mAb anti-luciferase antibody (L2164, Sigma) 1:1000 for 1 h, mAb mouse-anti β-actin (A5441, Sigma) 1:50 000 for 1 h]. Qualitative blots were incubated with anti-mouse HRP secondary antibody (Jackson ImmunoResearch) (1:10 000) and developed with the ECL system (GE biosciences) according to the manufacturer's instructions as described previously (28). For all quantitative western blots, the LI-COR Odyssey System (Biosciences) was used. Blots were incubated with goat anti-mouse IRDye 800CW antibody (926-32210, LI-COR), 1:7000 for 1 h. Detection was performed using the LI-COR Odyssey Imaging System (Biosciences) and quantification was determined using Odyssey Infrared Imaging System Application Software (Biosciences).
Immunohistochemistry
Spinal cord sections: PND28 mice on doxycycline were perfused with 4% paraformadhyde in PBS, the spinal cord was isolated and cryopreserved in 20% sucrose, PBS overnight. Tissue was frozen in OTC medium (TissueTek) in liquid nitrogen cooled isopentane and 14 µm spinal cord cryostat sections were obtained. Sections were blocked in 0.5% goat serum, 1% Tween-20 and incubated with hSMN-KH antibody ascites fluid (1:100) for 1 h, washed and incubated with goat anti-mouse Alexa488 (Invitrogen) 1:1000 for 30 min. Sections were mounted in Vectashield (Vector Labs). Whole mount TVA: Transverse abdominis muscles were isolated from animals sacrificed for electrophysiology and fixed in 4% paraformaldehyde in PBS for 24 h. TVA muscles were teased apart, blocked in 0.5% goat serum, 1% Tween-20 and incubated in neurofilament 160 (Millipore) 1:500 overnight, washed overnight and incubated with goat anti-mouse Alexa488 (Invitrogen) 1:1000 and alpha-bungarotoxin conjugated to Alexa594 (Invitrogen) 1:1000 for 2 h. Whole muscle fibers were mounted in Vectashield (Vector Labs). All images were captured with the Leica TCS_SL scanning confocal microscope system using an inverted Leica DMIRE2 microscope and PMT detectors. Images were captured at room temperature with the 40× HCX Plan Apo CS oil, NA = 1.25 objective. A Z-Galvo stage was used to obtain Z-series stacks of ∼30 images each. Image acquisition, overlays, scale bars and measurements were produced with the Leica Confocal Software v2.61 and subsequential image processing was performed with Adobe Photoshop CS2.
RT–PCR and real-time PCR analysis
RNA was isolated from tissues homogenized using Trizol (Invitrogen) and purified using the RNeasy kit (Qiagen) according to the manufacturer's instructions. RT–PCR was performed as previously described (32). Real-time PCR was performed as previously described using identical probes in a Taqman assay (83).
Electrophysiology
The recording chamber was continuously perfused with Ringer's solution containing the following (in mmol/l): 118 NaCl, 3.5 KCl, 2 CaCl2, 0.7 MgSO4, 26.2 NaHCO3, 1.7 NaH2PO4 and 5.5 glucose, pH 7.3–7.4 (20–22°C, equilibrated with 95% O2 and 5% CO2). Endplate recordings were performed as previously described (84–86). Briefly, after dissection, the TA muscle was partially bisected and folded apart to flatten the muscle. Muscle strips were stained with 10 µm 4-Di-2ASP [4-(4-diethylaminostyryl)-N-methylpyridinium iodide] (Molecular Probes) and imaged with an upright epifluorescence microscope. All of the endplates were imaged and impaled within 100 µm. We used two-electrode voltage clamp to measure EPC and MEPC amplitude. Muscle fibers were crushed away from the endplate band and voltage clamped to −45 mV to avoid movement after nerve stimulation. For all experiments, quantal content was calculated by dividing peak EPC current amplitude by peak MEPC current amplitude.
FUNDING
This work was supported by the National Institutes of Health, R01 HD060586 to A.H.M.B., RC2 NS069476 to A.H.M.B. and M.M.R., the Miracles for Madison fund and the Cade & Katelyn Cure SMA fund. T.T.L. was supported by funding from Muscular Dystrophy Association, 135614 to A.H.M.B. The initial transgenic construction was supported by the National Institutes of Health, R01 NS038650 to A.H.M.B. Additional support provided by National Institutes of Health, P30NS045758.
ACKNOWLEDGEMENTS
We thank Dr Yimin Hua and Dr Adrian Krainer for the generous gift of the human specific KH (SMN) antibody as well as many interesting discussions. We thank Dr Brian Kaspar and all members of his laboratory in particular Dr Kevin Foust for many interesting discussions.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Roberts D.F., Chavez J., Court S.D. The genetic component in child mortality. Arch. Dis. Child. 1970;45:33–38. doi: 10.1136/adc.45.239.33. doi:10.1136/adc.45.239.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crawford T.O., Pardo C.A. The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. doi:10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- 3.Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M., et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. doi:10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 4.Coovert D.D., Le T.T., McAndrew P.E., Strasswimmer J., Crawford T.O., Mendell J.R., Coulson S.E., Androphy E.J., Prior T.W., Burghes A.H. The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 1997;6:1205–1214. doi: 10.1093/hmg/6.8.1205. doi:10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 5.Lefebvre S., Burlet P., Liu Q., Bertrandy S., Clermont O., Munnich A., Dreyfuss G., Melki J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 1997;16:265–269. doi: 10.1038/ng0797-265. doi:10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 6.Burghes A.H., Beattie C.E. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009;10:597–609. doi: 10.1038/nrn2670. doi:10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lorson C.L., Hahnen E., Androphy E.J., Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl Acad. Sci. USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. doi:10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monani U.R., Lorson C.L., Parsons D.W., Prior T.W., Androphy E.J., Burghes A.H., McPherson J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. doi:10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 9.Cartegni L., Krainer A.R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 2002;30:377–384. doi: 10.1038/ng854. doi:10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 10.Kashima T., Manley J.L. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 2003;34:460–463. doi: 10.1038/ng1207. doi:10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 11.Gennarelli M., Lucarelli M., Capon F., Pizzuti A., Merlini L., Angelini C., Novelli G., Dallapiccola B. Survival motor neuron gene transcript analysis in muscles from spinal muscular atrophy patients. Biochem. Biophys. Res. Commun. 1995;213:342–348. doi: 10.1006/bbrc.1995.2135. doi:10.1006/bbrc.1995.2135. [DOI] [PubMed] [Google Scholar]
- 12.Parsons D.W., McAndrew P.E., Monani U.R., Mendell J.R., Burghes A.H., Prior T.W. An 11 base pair duplication in exon 6 of the SMN gene produces a type I spinal muscular atrophy (SMA) phenotype: further evidence for SMN as the primary SMA-determining gene. Hum. Mol. Genet. 1996;5:1727–1732. doi: 10.1093/hmg/5.11.1727. doi:10.1093/hmg/5.11.1727. [DOI] [PubMed] [Google Scholar]
- 13.Lorson C.L., Strasswimmer J., Yao J.M., Baleja J.D., Hahnen E., Wirth B., Le T., Burghes A.H., Androphy E.J. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat. Genet. 1998;19:63–66. doi: 10.1038/ng0598-63. doi:10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- 14.Lorson C.L., Androphy E.J. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum. Mol. Genet. 2000;9:259–265. doi: 10.1093/hmg/9.2.259. doi:10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- 15.Burnett B.G., Munoz E., Tandon A., Kwon D.Y., Sumner C.J., Fischbeck K.H. Regulation of SMN protein stability. Mol. Cell Biol. 2009;29:1107–1115. doi: 10.1128/MCB.01262-08. doi:10.1128/MCB.01262-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McAndrew P.E., Parsons D.W., Simard L.R., Rochette C., Ray P.N., Mendell J.R., Prior T.W., Burghes A.H. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 1997;60:1411–1422. doi: 10.1086/515465. doi:10.1086/515465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prior T.W., Krainer A.R., Hua Y., Swoboda K.J., Snyder P.C., Bridgeman S.J., Burghes A.H., Kissel J.T. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am. J. Hum. Genet. 2009;85:408–413. doi: 10.1016/j.ajhg.2009.08.002. doi:10.1016/j.ajhg.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vezain M., Saugier-Veber P., Goina E., Touraine R., Manel V., Toutain A., Fehrenbach S., Frebourg T., Pagani F., Tosi M., et al. A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum. Mutat. 2010;31:E1110–E1125. doi: 10.1002/humu.21173. doi:10.1002/humu.21173. [DOI] [PubMed] [Google Scholar]
- 19.Monani U.R., Pastore M.T., Gavrilina T.O., Jablonka S., Le T.T., Andreassi C., DiCocco J.M., Lorson C., Androphy E.J., Sendtner M., et al. A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J. Cell Biol. 2003;160:41–52. doi: 10.1083/jcb.200208079. doi:10.1083/jcb.200208079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Workman E., Saieva L., Carrel T.L., Crawford T.O., Liu D., Lutz C., Beattie C.E., Pellizzoni L., Burghes A.H. A SMN missense mutation complements SMN2 restoring snRNPs and rescuing SMA mice. Hum. Mol. Genet. 2009;18:2215–2229. doi: 10.1093/hmg/ddp157. doi:10.1093/hmg/ddp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Q., Dreyfuss G. A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 1996;15:3555–3565. [PMC free article] [PubMed] [Google Scholar]
- 22.Meister G., Buhler D., Pillai R., Lottspeich F., Fischer U. A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat. Cell. Biol. 2001;3:945–949. doi: 10.1038/ncb1101-945. doi:10.1038/ncb1101-945. [DOI] [PubMed] [Google Scholar]
- 23.Pellizzoni L., Yong J., Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002;298:1775–1779. doi: 10.1126/science.1074962. doi:10.1126/science.1074962. [DOI] [PubMed] [Google Scholar]
- 24.Pellizzoni L. Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep. 2007;8:340–345. doi: 10.1038/sj.embor.7400941. doi:10.1038/sj.embor.7400941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schrank B., Gotz R., Gunnersen J.M., Ure J.M., Toyka K.V., Smith A.G., Sendtner M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl Acad. Sci. USA. 1997;94:9920–9925. doi: 10.1073/pnas.94.18.9920. doi:10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monani U.R., Sendtner M., Coovert D.D., Parsons D.W., Andreassi C., Le T.T., Jablonka S., Schrank B., Rossol W., Prior T.W., et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. doi:10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 27.Hsieh-Li H.M., Chang J.G., Jong Y.J., Wu M.H., Wang N.M., Tsai C.H., Li H. A mouse model for spinal muscular atrophy. Nat. Genet. 2000;24:66–70. doi: 10.1038/71709. doi:10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 28.Le T.T., Pham L.T., Butchbach M.E., Zhang H.L., Monani U.R., Coovert D.D., Gavrilina T.O., Xing L., Bassell G.J., Burghes A.H. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. doi:10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 29.Gabanella F., Butchbach M.E., Saieva L., Carissimi C., Burghes A.H., Pellizzoni L. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE. 2007;2:e921. doi: 10.1371/journal.pone.0000921. doi:10.1371/journal.pone.0000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Z., Lotti F., Dittmar K., Younis I., Wan L., Kasim M., Dreyfuss G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133:585–600. doi: 10.1016/j.cell.2008.03.031. doi:10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baumer D., Lee S., Nicholson G., Davies J.L., Parkinson N.J., Murray L.M., Gillingwater T.H., Ansorge O., Davies K.E., Talbot K. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet. 2009;5:e1000773. doi: 10.1371/journal.pgen.1000773. doi:10.1371/journal.pgen.1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gavrilina T.O., McGovern V.L., Workman E., Crawford T.O., Gogliotti R.G., DiDonato C.J., Monani U.R., Morris G.E., Burghes A.H. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum. Mol. Genet. 2008;17:1063–1075. doi: 10.1093/hmg/ddm379. doi:10.1093/hmg/ddm379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rossoll W., Jablonka S., Andreassi C., Kroning A.K., Karle K., Monani U.R., Sendtner M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J. Cell Biol. 2003;163:801–812. doi: 10.1083/jcb.200304128. doi:10.1083/jcb.200304128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jablonka S., Beck M., Lechner B.D., Mayer C., Sendtner M. Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J. Cell Biol. 2007;179:139–149. doi: 10.1083/jcb.200703187. doi:10.1083/jcb.200703187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McWhorter M.L., Monani U.R., Burghes A.H., Beattie C.E. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J. Cell Biol. 2003;162:919–932. doi: 10.1083/jcb.200303168. doi:10.1083/jcb.200303168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGovern V.L., Gavrilina T.O., Beattie C.E., Burghes A.H. Embryonic motor axon development in the severe SMA mouse. Hum. Mol. Genet. 2008;17:2900–2909. doi: 10.1093/hmg/ddn189. doi:10.1093/hmg/ddn189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kariya S., Park G.H., Maeno-Hikichi Y., Leykekhman O., Lutz C., Arkovitz M.S., Landmesser L.T., Monani U.R. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008;17:2552–2569. doi: 10.1093/hmg/ddn156. doi:10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kong L., Wang X., Choe D.W., Polley M., Burnett B.G., Bosch-Marce M., Griffin J.W., Rich M.M., Sumner C.J. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J. Neurosci. 2009;29:842–851. doi: 10.1523/JNEUROSCI.4434-08.2009. doi:10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murray L.M., Comley L.H., Thomson D., Parkinson N., Talbot K., Gillingwater T.H. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008;17:949–962. doi: 10.1093/hmg/ddm367. doi:10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- 40.Butchbach M.E., Singh J., Thorsteinsdottir M., Saieva L., Slominski E., Thurmond J., Andresson T., Zhang J., Edwards J.D., Simard L.R., et al. Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum. Mol. Genet. 2010;19:454–467. doi: 10.1093/hmg/ddp510. doi:10.1093/hmg/ddp510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Avila A.M., Burnett B.G., Taye A.A., Gabanella F., Knight M.A., Hartenstein P., Cizman Z., Di Prospero N.A., Pellizzoni L., Fischbeck K.H., et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J. Clin. Invest. 2007;117:659–671. doi: 10.1172/JCI29562. doi:10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Narver H.L., Kong L., Burnett B.G., Choe D.W., Bosch-Marce M., Taye A.A., Eckhaus M.A., Sumner C.J. Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition. Ann. Neurol. 2008;64:465–470. doi: 10.1002/ana.21449. doi:10.1002/ana.21449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andreassi C., Jarecki J., Zhou J., Coovert D.D., Monani U.R., Chen X., Whitney M., Pollok B., Zhang M., Androphy E., et al. Aclarubicin treatment restores SMN levels to cells derived from type I spinal muscular atrophy patients. Hum. Mol. Genet. 2001;10:2841–2849. doi: 10.1093/hmg/10.24.2841. doi:10.1093/hmg/10.24.2841. [DOI] [PubMed] [Google Scholar]
- 44.Singh J., Salcius M., Liu S.W., Staker B.L., Mishra R., Thurmond J., Michaud G., Mattoon D.R., Printen J., Christensen J., et al. DcpS as a therapeutic target for spinal muscular atrophy. ACS Chem. Biol. 2008;3:711–722. doi: 10.1021/cb800120t. doi:10.1021/cb800120t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Riessland M., Ackermann B., Forster A., Jakubik M., Hauke J., Garbes L., Fritzsche I., Mende Y., Blumcke I., Hahnen E., et al. SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum. Mol. Genet. 2010;19:1492–1506. doi: 10.1093/hmg/ddq023. doi:10.1093/hmg/ddq023. [DOI] [PubMed] [Google Scholar]
- 46.Hastings M.L., Berniac J., Liu Y.H., Abato P., Jodelka F.M., Barthel L., Kumar S., Dudley C., Nelson M., Larson K., et al. Tetracyclines that promote SMN2 exon 7 splicing as therapeutics for spinal muscular atrophy. Sci. Transl. Med. 2009;1:5ra12. doi: 10.1126/scitranslmed.3000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mattis V.B., Ebert A.D., Fosso M.Y., Chang C.W., Lorson C.L. Delivery of a read-through inducing compound, TC007, lessens the severity of a spinal muscular atrophy animal model. Hum. Mol. Genet. 2009;18:3906–3913. doi: 10.1093/hmg/ddp333. doi:10.1093/hmg/ddp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hua Y., Sahashi K., Hung G., Rigo F., Passini M.A., Bennett C.F., Krainer A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24:1634–1644. doi: 10.1101/gad.1941310. doi:10.1101/gad.1941310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh N.N., Shishimorova M., Cao L.C., Gangwani L., Singh R.N. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 2009;6:341–350. doi: 10.4161/rna.6.3.8723. doi:10.4161/rna.6.3.8723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burghes A.H., McGovern V.L. Antisense oligonucleotides and spinal muscular atrophy: skipping along. Genes Dev. 2010;24:1574–1579. doi: 10.1101/gad.1961710. doi:10.1101/gad.1961710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Passini M.A., Bu J., Richards A.M., Kinnecom C., Sardi S.P., Stanek L.M., Hua Y., Rigo F., Matson J., Hung G., et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011;3:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foust K.D., Wang X., McGovern V.L., Braun L., Bevan A.K., Haidet A.M., Le T.T., Morales P.R., Rich M.M., Burghes A.H., et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010;28:271–274. doi: 10.1038/nbt.1610. doi:10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Passini M.A., Bu J., Roskelley E.M., Richards A.M., Sardi S.P., O'Riordan C.R., Klinger K.W., Shihabuddin L.S., Cheng S.H. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J. Clin. Invest. 2010;120:1253–1264. doi: 10.1172/JCI41615. doi:10.1172/JCI41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meyer K., Marquis J., Trub J., Nlend Nlend R., Verp S., Ruepp M.D., Imboden H., Barde I., Trono D., Schumperli D. Rescue of a severe mouse model for spinal muscular atrophy by U7 snRNA-mediated splicing modulation. Hum. Mol. Genet. 2009;18:546–555. doi: 10.1093/hmg/ddn382. doi:10.1093/hmg/ddn382. [DOI] [PubMed] [Google Scholar]
- 55.Valori C.F., Ning K., Wyles M., Mead R.J., Grierson A.J., Shaw P.J., Azzouz M. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med. 2010;2:35ra42. doi: 10.1126/scitranslmed.3000830. [DOI] [PubMed] [Google Scholar]
- 56.Dominguez E., Marais T., Chatauret N., Benkhelifa-Ziyyat S., Duque S., Ravassard P., Carcenac R., Astord S., de Moura A.P., Voit T., et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum. Mol. Genet. 2010;20:681–693. doi: 10.1093/hmg/ddq514. doi:10.1093/hmg/ddq514. [DOI] [PubMed] [Google Scholar]
- 57.Mayford M., Bach M.E., Huang Y.Y., Wang L., Hawkins R.D., Kandel E.R. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. doi:10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- 58.Belteki G., Haigh J., Kabacs N., Haigh K., Sison K., Costantini F., Whitsett J., Quaggin S.E., Nagy A. Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res. 2005;33:e51. doi: 10.1093/nar/gni051. doi:10.1093/nar/gni051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hayashi S., Lewis P., Pevny L., McMahon A.P. Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech. Dev. 2002;119(Suppl. 1):S97–S101. doi: 10.1016/s0925-4773(03)00099-6. doi:10.1016/S0925-4773(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 60.Ling K.K., Lin M.Y., Zingg B., Feng Z., Ko C.P. Synaptic defects in the spinal and neuromuscular circuitry in a mouse model of spinal muscular atrophy. PLoS ONE. 2010;5:e15457. doi: 10.1371/journal.pone.0015457. doi:10.1371/journal.pone.0015457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ruiz R., Casanas J.J., Torres-Benito L., Cano R., Tabares L. Altered intracellular Ca2+ homeostasis in nerve terminals of severe spinal muscular atrophy mice. J. Neurosci. 2010;30:849–857. doi: 10.1523/JNEUROSCI.4496-09.2010. doi:10.1523/JNEUROSCI.4496-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Murray L.M., Lee S., Baumer D., Parson S.H., Talbot K., Gillingwater T.H. Pre-symptomatic development of lower motor neuron connectivity in a mouse model of severe spinal muscular atrophy. Hum. Mol. Genet. 2010;19:420–433. doi: 10.1093/hmg/ddp506. doi:10.1093/hmg/ddp506. [DOI] [PubMed] [Google Scholar]
- 63.Frugier T., Tiziano F.D., Cifuentes-Diaz C., Miniou P., Roblot N., Dierich A., Le Meur M., Melki J. Nuclear targeting defect of SMN lacking the C-terminus in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2000;9:849–858. doi: 10.1093/hmg/9.5.849. doi:10.1093/hmg/9.5.849. [DOI] [PubMed] [Google Scholar]
- 64.Cifuentes-Diaz C., Frugier T., Tiziano F.D., Lacene E., Roblot N., Joshi V., Moreau M.H., Melki J. Deletion of murine SMN exon 7 directed to skeletal muscle leads to severe muscular dystrophy. J. Cell Biol. 2001;152:1107–1114. doi: 10.1083/jcb.152.5.1107. doi:10.1083/jcb.152.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vitte J.M., Davoult B., Roblot N., Mayer M., Joshi V., Courageot S., Tronche F., Vadrot J., Moreau M.H., Kemeny F., et al. Deletion of murine Smn exon 7 directed to liver leads to severe defect of liver development associated with iron overload. Am. J. Pathol. 2004;165:1731–1741. doi: 10.1016/S0002-9440(10)63428-1. doi:10.1016/S0002-9440(10)63428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Naciff J.M., Behbehani M.M., Misawa H., Dedman J.R. Identification and transgenic analysis of a murine promoter that targets cholinergic neuron expression. J. Neurochem. 1999;72:17–28. doi: 10.1046/j.1471-4159.1999.0720017.x. doi:10.1046/j.1471-4159.1999.0720017.x. [DOI] [PubMed] [Google Scholar]
- 67.Misawa H., Nakata K., Toda K., Matsuura J., Oda Y., Inoue H., Tateno M., Takahashi R. VAChT-Cre. Fast and VAChT-Cre.Slow: postnatal expression of Cre recombinase in somatomotor neurons with different onset. Genesis. 2003;37:44–50. doi: 10.1002/gene.10224. doi:10.1002/gene.10224. [DOI] [PubMed] [Google Scholar]
- 68.Park G.H., Maeno-Hikichi Y., Awano T., Landmesser L.T., Monani U.R. Reduced survival of motor neuron (SMN) protein in motor neuronal progenitors functions cell autonomously to cause spinal muscular atrophy in model mice expressing the human centromeric (SMN2) gene. J. Neurosci. 2010;30:12005–12019. doi: 10.1523/JNEUROSCI.2208-10.2010. doi:10.1523/JNEUROSCI.2208-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mentis G.Z., Blivis D., Liu W., Drobac E., Crowder M.E., Kong L., Alvarez F.J., Sumner C.J., O'Donovan M.J. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron. 2011;69:453–467. doi: 10.1016/j.neuron.2010.12.032. doi:10.1016/j.neuron.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rossoll W., Bassell G.J. Spinal muscular atrophy and a model for survival of motor neuron protein function in axonal ribonucleoprotein complexes. Results Probl. Cell. Differ. 2009;48:289–326. doi: 10.1007/400_2009_4. doi:10.1007/400_2009_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cheever T.R., Olson E.A., Ervasti J.M. Axonal regeneration and neuronal function are preserved in motor neurons lacking ss-actin in vivo. PLoS ONE. 2011;6:e17768. doi: 10.1371/journal.pone.0017768. doi:10.1371/journal.pone.0017768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li Q., Lee J.A., Black D.L. Neuronal regulation of alternative pre-mRNA splicing. Nat. Rev. Neurosci. 2007;8:819–831. doi: 10.1038/nrn2237. doi:10.1038/nrn2237. [DOI] [PubMed] [Google Scholar]
- 73.Foust K.D., Nurre E., Montgomery C.L., Hernandez A., Chan C.M., Kaspar B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. doi:10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Duque S., Joussemet B., Riviere C., Marais T., Dubreil L., Douar A.M., Fyfe J., Moullier P., Colle M.A., Barkats M. Intravenous Administration of Self-complementary AAV9 Enables Transgene Delivery to Adult Motor Neurons. Mol. Ther. 2009;17:1187–1196. doi: 10.1038/mt.2009.71. doi:10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Swoboda K.J. Seize the day: Newborn screening for SMA. Am. J. Med. Genet. A. 2010;152A:1605–1607. doi: 10.1002/ajmg.a.33519. doi:10.1002/ajmg.a.33519. [DOI] [PubMed] [Google Scholar]
- 76.Swoboda K.J., Prior T.W., Scott C.B., McNaught T.P., Wride M.C., Reyna S.P., Bromberg M.B. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann. Neurol. 2005;57:704–712. doi: 10.1002/ana.20473. doi:10.1002/ana.20473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lewelt A., Krosschell K.J., Scott C., Sakonju A., Kissel J.T., Crawford T.O., Acsadi G., D'Anjou G., Elsheikh B., Reyna S.P., et al. Compound muscle action potential and motor function in children with spinal muscular atrophy. Muscle Nerve. 2010;42:703–708. doi: 10.1002/mus.21838. doi:10.1002/mus.21838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bevan A.K., Hutchinson K.R., Foust K.D., Braun L., McGovern V.L., Schmelzer L., Ward J.G., Petruska J.C., Lucchesi P.A., Burghes A.H., et al. Early heart failure in the SMN{Delta}7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum. Mol. Genet. 2010;19:3895–3905. doi: 10.1093/hmg/ddq300. doi:10.1093/hmg/ddq300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heier C.R., Satta R., Lutz C., DiDonato C.J. Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum. Mol. Genet. 2010;19:3906–3918. doi: 10.1093/hmg/ddq330. doi:10.1093/hmg/ddq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shababi M., Habibi J., Yang H.T., Vale S.M., Sewell W.A., Lorson C.L. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum. Mol. Genet. 2010;19:4059–4071. doi: 10.1093/hmg/ddq329. doi:10.1093/hmg/ddq329. [DOI] [PubMed] [Google Scholar]
- 81.Le T.T., Coovert D.D., Monani U.R., Morris G.E., Burghes A.H. The survival motor neuron (SMN) protein: effect of exon loss and mutation on protein localization. Neurogenetics. 2000;3:7–16. doi: 10.1007/s100480000090. doi:10.1007/s100480000090. [DOI] [PubMed] [Google Scholar]
- 82.Gogliotti R.G., Lutz C., Jorgensen M., Huebsch K., Koh S., Didonato C.J. Characterization of a commonly used mouse model of SMA reveals increased seizure susceptibility and heightened fear response in FVB/N mice. Neurobiol. . Dis. 2011;43:142–151. doi: 10.1016/j.nbd.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Heier C.R., Gogliotti R.G., DiDonato C.J. SMN transcript stability: could modulation of messenger RNA degradation provide a novel therapy for spinal muscular atrophy? J. Child Neurol. 2007;22:1013–1018. doi: 10.1177/0883073807305669. doi:10.1177/0883073807305669. [DOI] [PubMed] [Google Scholar]
- 84.Wang X., Engisch K.L., Li Y., Pinter M.J., Cope T.C., Rich M.M. Decreased synaptic activity shifts the calcium dependence of release at the mammalian neuromuscular junction in vivo. J. Neurosci. 2004;24:10687–10692. doi: 10.1523/JNEUROSCI.2755-04.2004. doi:10.1523/JNEUROSCI.2755-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang X., Engisch K.L., Teichert R.W., Olivera B.M., Pinter M.J., Rich M.M. Prolongation of evoked and spontaneous synaptic currents at the neuromuscular junction after activity blockade is caused by the upregulation of fetal acetylcholine receptors. J. Neurosci. 2006;26:8983–8987. doi: 10.1523/JNEUROSCI.2493-06.2006. doi:10.1523/JNEUROSCI.2493-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang X., Pinter M.J., Rich M.M. Ca2+ dependence of the binomial parameters p and n at the mouse neuromuscular junction. J. Neurophysiol. 2010;103:659–666. doi: 10.1152/jn.00708.2009. doi:10.1152/jn.00708.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]