

Abstract

Proteomic profiling of membrane proteins is of vital importance in the search for disease biomarkers and drug development. However, the slow pace in this field has resulted mainly from the difficulty to analyze membrane proteins by mass spectrometry (MS). The objective of this investigation was to explore and optimize solubilization of membrane proteins for shotgun membrane proteomics of the CD14 human monocytes by examining different systems that rely on: i) an organic solvent (methanol) ii) an acid-labile detergent 3-[3-(1,1-bisalkyloxyethyl)pyridin-1-yl]propane-1-sulfonate) (PPS), iii) a combination of both agents (methanol + PPS). Solubilization efficiency of different buffers was first compared using bacteriorhodopsin as a model membrane protein. Selected approaches were then applied on a membrane subproteome isolated from a highly enriched human monocyte population that was ~98% positive for CD14 expression by FACS analysis. A methanol-based buffer yielded 194 proteins of which 93 (48%) were mapped as integral membrane proteins. The combination of methanol and acid-cleavable detergent gave similar results; 203 identified proteins of which 93 (46 %) were mapped integral membrane proteins. However, employing PPS a total of 216 proteins of which 75 (35 %) were mapped integral membrane proteins. These results indicate that methanol unaided or in combination with PPS yielded significantly higher membrane protein identification/enrichment than the PPS alone.
Keywords: CD14 monocyte, Membrane proteins, Solubilization, Methanol, Detergents, LC-MS/MS
1. Introduction
Membrane proteins play an important role in the structural and functional organization of the cell and cellular organelles. They are important bio-effectors in many of physiological and pathological processes regulating dynamic pathways of eukaryotic mammalian cells [1]. Physiological processes involving signal transduction and their subsequent integration into distinct cellular pathways result in specific cellular responses (i.e. assembly of mitochondrial cytochrome c-oxidase [2]. Pathological processes are exemplified in dysregulation of programmed cell death through extrinsic apoptotic pathway (i.e. cancer) [3]. It has been estimated that 65% of all contemporary pharmaceuticals target membrane proteins while ~ 21 % of all open reading frames from sequenced human genome encode alpha-helical integral membrane proteins [4, 5]. Certain membrane proteins have important clinical roles. The status of several membrane proteins or their corresponding genes is key determinants in molecular oncology. They can be used as prognostic (an estimate of likely disease outcome or aggressiveness) or predictive (aid in treatment assignment) [6]. Relevant examples include: ErBb2/HER2 in breast cancer and EGFR in lung and colon cancer [6]. The critical role of membrane proteins in cellular biology and pathology along with their role in therapeutic modulation underscores the importance of a mass spectrometry (MS)-based proteomics in large-scale profiling of human membrane proteins [1].
Monocytes along with lymphocytes represent a category of leukocytes called agranulocytes, and are characterized by the absence of granules in their cytoplasm [7]. Human monocytes develop from myelo-monocytic stem cells in the bone marrow [8]. When released into the blood stream, they differentiate into two major subpopulations: the classical CD14 monocytes and the pro-inflammatory CD14/CD16 monocytes[9]. Monocytes that migrate further into tissues mature into macrophages [7]. Human circulating monocytes constitute ~ 5 % of the leukocytes present in peripheral blood. They play significant role in several highly regulated biological processes including: host defense, inflammation, and tumor surveillance [10, 11]. The regulation of these processes is facilitated by distinct cellular mechanisms including: phagocytosis, pinocytosis, chemotaxis and the release of cytokines[10]. At the molecular level these processes are not fully understood. Two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) analyses of the human monocyte proteome have been conducted, resulting in a detailed characterization of the monocyte cytosolic proteome [12, 13]. While analyses targeting lipid rafts of monocytic cell lines have been previously reported [14, 15], targeted shotgun analysis of the human circulating CD14 monocytes and corresponding membrane proteins should enable better understanding of their biological function, since cell line conditions do not always resemble those in vivo [16].
The ability to characterize membrane proteins using MS-based proteomics has lagged behind that of soluble proteins, mainly due to insolubility of integral membrane proteins in natural aqueous buffers [1, 17]. Based on their widespread use in membrane biochemistry, gel-based approaches utilizing chaotropes and detergents for membrane protein solubilization prior to 1D or 2D-PAGE have been coupled with MS and employed in membrane proteomics [18]. Low resolution of 1D-PAGE and typically poor yield of membrane proteins by 2D-PAGE , resulted in development of gel-free shotgun methods employing chaotropes (urea, guanidine) or classic detergents (i.e. SDS, CHAPS, Triton X-100) to solubilize membrane proteins [1]. However, chaotropes impede proteolysis while detergents interfere with separation and MS ionization efficiency of peptides and proteins [19, 20]. Although significant advances have been achieved in removal of detergents and chaotropes [21, 22], these steps require extensive manipulations and result in sample losses, which are particularly critical when starting with limited amounts of membrane proteins. To avoid negative effects caused by chaotropes and classical detergents, alternative chaotrope/ detergent-free shotgun approaches have been developed and employed for the MS analysis of membrane proteins [23-27]. Recently developed mass spectrometry-compatible surfactants have been increasingly used [28-30] in MS-based proteomics and proposed for shot-gun proteomics of membrane proteins [31-35]. Improved MS compatibility of these surfactants is based on their hydrolysis at low pH after digestion that results in minimal interference with downstream reversed-phase peptide separations and MS analysis[29].
Previously, we demonstrated the capability of 60% (v/v) buffered methanol (CH3OH) vs. non-solubilized membrane preparation to enrich integral membrane proteins for shot-gun proteomics by improving solubilization and digestion of complex membrane protein mixtures isolated from prokaryotic cells [36]. Subsequently we extended this approach, to eukaryotic mammalian systems [37]. The advantage of methanol-based vs. gel-based approach has been also demonstrated [38] and compared to other commonly used approaches[39-42]
The objective of this investigation was to optimize membrane protein solubilization and proteolysis prior to shotgun LC/MS analysis of the human monocyte membrane proteome. To accomplish this goal we compared different approaches that rely on i) 60% methanol in 50 mM ammonium bicarbonate (v/v), ii) 0.1% {3-[3-(1,1-bisalkyloxyethyl)pyridin-1-yl]propane-1-sulfonate (PPS) in 50 mM ammonium bicarbonate (v/v), iii) a combination of both, 60% methanol and 0.1 % PPS in 50 mM ammonium bicarbonate (v/v) and, iv) 8 M urea . These approaches were first tested on a single integral membrane protein to asses the solubilizing efficiency and completeness of tryptic digestion and then used for the solubilization and digestion of crude membrane preparation from a highly enriched human monocyte population. Our results demonstrate that methanol alone, or in combination with PPS, is more effective than PPS for the shotgun analysis of integral membrane proteins.
2. Materials and methods
2.1 Reagents
Bacteriorhodopsin from Halobacterium halobium (lyophilized purple membrane preparation) was obtained from Sigma (St. Louis, MO, USA). HPLC-grade methanol (CH3OH) was from EM Science (Darmstadt, Germany). Acid cleavable detergent: 3-[3-(1,1-bisalkyloxyethyl)pyridin-1-yl]propane-1-sulfonate (PPS) was purchased from Protein Discovery Inc. (Knoxville, TN). Ammonium bicarbonate (NH4HCO3), phenylmethylsulfonyl fluoride (PMSF), urea and formic acid (HCOOH), and 2-iodoacetamide (IAA) were obtained from Sigma (St. Louis, MO) Tris[2-carboxyethyl] phosphine (TCEP) Bond-Breaker ™ was from Pierce (Rockford, IL Fused-silica capillaries were acquired from Polymicro Technologies (Phoenix, AZ). All chemicals used were A.C.S. grade or higher, and all solvents used were HPLC grade or higher. Sequencing grade trypsin was obtained from Promega (Madison, WI). All solutions were prepared using water purified by a Nanopure II system (Dubuque, IA).
2.2 Bacteriorhodopsin solubilization and tryptic digestion
Equal aliquots of lyophilized bacteriorhodopsin (100 μg each) were solubilized at 1 μg/μL in five different buffer systems: (a) 50 mM NH4HCO3 buffer, (b) 50 mM NH4HCO3 buffer containing 60% (v/v) CH3OH, (c) 50 mM NH4HCO3 buffer containing 0.1% PPS, and (d) 50 mM NH4HCO3 buffer containing 60% (v/v) CH3OH and PPS to a final concentration of 0.1% (e) 50 mM NH4HCO3 buffer containing 8 M urea. Tryptic digestion was performed at an enzyme-to-protein ratio of 1:20. Solubilization of bacteriorhodopsin using 50 mM NH4HCO3 buffer was carried out using sonication in a water bath for 15 min followed by tryptic digestion for 20 h at 37 °C. Solubilization of bacteriorhodopsin using 60% (v/v) CH3OH was carried as previously described {Blonder, 2004 #66}. Briefly, bacteriorhodopsin was dissolved in 50 mM NH4HCO3 buffer containing 60% (v/v) CH3OH. After sonication in water bath for 15 min solubilized proteins were digested in the same buffer with trypsin at 37 °C for 20 hrs. Solubilization of bacteriorhodopsin using PPS was carried out using manufacturer’s recommended procedure. Briefly, lyophilized protein was vortexed in 50 mM NH4HCO3 buffer containing 0.1 % PPS followed by sonication in a water bath for 15 min. After incubation at 50 °C the sample was cooled at room temperature and digested with trypsin at 37 °C for 20 hrs. Solubilization of bacteriorhodopsin using 8 M urea was carried out using sonication in water bath for 15 min. Prior to proteolysis the buffer was diluted to 2M urea (final concentration) and digested with trypsin at 37 °C for 20 hrs. Aliquots of 20 μg of bacteriorhodopsin from each of the five digestates were removed at intervals of 0, 0.5, 4, 20 hrs and lyophilized. Solubilization and digestion efficiency of each buffering system was evaluated by SDS-PAGE analysis and visualized using Coomassie blue staining.
2.3 Isolation and Purification of Primary CD14 Human Monocytes
Primary human monocytes were isolated from peripheral blood of healthy donors in accordance with guidelines of the institutional board-approved research protocol. CD14+ monocytes were immuno-affinity purified to ~98% homogeneity as described elsewhere [43]. Briefly, mononuclear cells were separated from blood using standard gradient centrifugation with Ficoll-Hypaque (Pharmacia) followed by CD14+ monocyte purification using immuno-magnetic beads coated with anti CD14 MoAb (Miltenyi Biotech Inc., Auburn, CA) as previously described by Saikh et al [43]. Fluorescent antibody cell sorting (FACS) analysis of the purified population demonstrated that ~98% were positive for CD14 expression.
2.4 Monocytes membrane protein isolation, solubilization and proteolysis
Monocytes were lyzed using a combination of hypotonic lysis and sonication while crude membrane fraction was isolated by ultracentrifugation. Briefly, the cell pellet was resuspended in 50 mM NH4HCO3 containing 1 mM TCEP and 1 mM PMSF. Cells were homogenized in a Potter-Elvehjem homogenizer followed by ten cycles of 10 second sonication (20 % intensity) using Bronson microprobe sonicator. The homogenate was centrifuged at 5000 × g for 5 min to remove unbroken cells and cellular debris. The supernatant was alkylated using 5 mM IAA (final concentration) followed by ultracentrifugation at 100,000 × g for 1.5 hrs using a Beckman 50 Ti rotor. Supernatant was discarded and crude membrane fraction subjected to modified carbonate stripping treatment [44]. Briefly, membrane pellet was resuspended in 1 mL of 100 mM Na2CO3 (pH 11) and rotated for 2 hrs at 4 °C to eliminate peripheral membrane proteins. After ultracentrifugation at 100,000 × g for 1 hr the pellet was washed in d.d.H2O twice and lyophilized. Three equal aliquots of lyophilized membrane enriched monocyte fraction were resuspended at 1 mg/mL using the three buffer systems: (a) 50 mM NH4HCO3 buffer containing 60% (v/v) CH3OH, (b) 50 mM NH4HCO3 buffer containing PPS (c) 50 mM NH4HCO3 buffer containing 60% (v/v) CH3OH and 0.1% PPS. Samples were further solubilized and digested as described above (section 2.2). After digestion, samples containing PPS were acidified with HCl and incubated at 37 °C for additional one hour to hydrolyze PPS. All samples were lyophilized and dissolved in 0.1% TFA before LC-MS/MS analysis.
2.5 Nano-flow liquid chromatography-tandem mass spectrometry
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was performed using an Agilent 1100 nanoflow LC system coupled on-line with hybrid linear ion trap-FT-ICR instrument (LTQ-FT, Thermo Electron, San Jose, CA). Reversed-phase columns (75 μm i.d. × 10 cm fused silica capillary with a flame pulled tip) were slurry-packed in-house with 5 μm, 300 Å pore size C-18 stationary phase (Phenomenex, Torrence, CA). After sample injection (2 μg of peptides), the column was washed for 20 min with 98% mobile phase A (0.1% formic acid in water) at a flow rate of 0.5 μL/min. Peptides were eluted from the column using a linear gradient of 2% mobile phase B (0.1% formic acid in ACN) to 60% solvent B in 100 minutes at a flow rate of 0.25 μL/min, then to 98% B for an additional 10 min. The mass spectrometer was operated in a data-dependent mode to automatically switch between MS and MS/MS. The FT-ICR-MS survey scan (m/z ranges: 350-1800; 350-750, 740-1000 and 950-1800) was followed by seven MS/MS scans in which the most abundant peptide precursor ions detected in the preceding FT-ICR-MS survey scan were dynamically selected for collision induced dissociation (CID). The threshold of 200 ion counts was used for triggering an MS/MS scan. The normalized CID energy was 35 %; the electrospray voltage was set at 1.6 kV, and the voltage and temperature for the ion source capillary were set at 45 V and 160 °C, respectively.
2.6 Data analysis
All acquired raw data were searched independently against the human protein database (UniProt Human, release 09/2007), using SEQUEST (Thermo, San Jose, CA). The searches were carried out on a Beowulf 18-node parallel virtual machine cluster-computer. Dynamic modifications were added for the detection of the following: carboxyamidomethylated cysteine (+57 Da), and oxidized methionine (+16 Da). For the MS1 spectra acquired by FTICR-MS the monoisotopic precursor ion mass tolerance was set at 10 ppm Da while for the data dependent MS2 spectra, acquired by LIT—MS, the fragment ion tolerance was set at 0.5 Da. Only fully tryptic peptides with up to two miscleavages possessing delta correlation ΔCn ≥ 0.1 and charge state dependent cross correlation Xcorr of ≥ 2.0 for [M+H]1+, ≥ 2.3 for [M+2H]2+ and ≥ 3.75 for [M+3H]3+ were considered legitimately identified.
2.7 Transmembrane domain prediction and hydropathicity calculation
Alpha-helical transmembrane domains (TMD) were mapped using TMHMM [45] available at http://www.cbs.dtu.dk/services/TMHMM while protein grand average of hydropathicity (GRAVY) scores[46] were calculated using ProtParam tool available at the ExPASy Proteomics Server (http://www.expasy.org/tools/protparam.html).
3. Results and discussion
Along with recent advances in MS instrumentation it became evident that existing methods for the isolation, solubilization and proteolysis of membrane proteins are in need of further improvement and optimization [47]. While the mass spectrometer represents a critical component of any proteomic investigation, efficient upstream sample preparation is equally important for successful shotgun analysis of membrane proteins [17]. In addition to efficient isolation of membrane organelles/proteins the solubilization and digestion steps are of critical importance. It is vital to keep extracted membrane proteins solubilized and denatured throughout the proteolysis process [48].
Integral membrane proteins, tied to the membrane bilayer by alpha-helices or attached to it by fatty-acid modifications, require solubilizing conditions different from that of the water-soluble proteins. These characteristics make solubilization, denaturation, and proteolysis an intricate task. The optimization of these conditions for complex membrane protein mixtures is even more challenging as membrane proteins span a wide-range of hydrophobicities and numbers of transmembrane domains (TMDs). Typically, effective solubilization requires the use of solubilizing reagents including detergents, chaotropes or organic solvents to solubilize as large a percentage of the membrane proteome as possible. As an alternative to classic detergents (i.e. SDS, CHAPS), acid-cleavable detergents have recently been proposed for use in membrane proteomics primarily because of their minimal interference with LC separations and/or MS analysis [31, 49].
The objective of this study was to find a favorable solubilization and digestion approach for shotgun membrane proteomics of the CD14 human monocyte membrane proteome. We initially compared the efficiency of an i) acid-cleavable detergent (PPS), ii) an organic solvent system, iii) their mixture, and iv) a chaotrope (urea) to solubilize and digest a single integral membrane protein (bacteriorhodopsin). Bacteriorhodopsin, a prototypical water insoluble integral membrane protein purified from Halobacterium halobium was selected to test the effectiveness of the selected buffer systems. This hydrophobic (GRAVY = 0.723), 262 amino acid protein has seven TMDs and a distinct purple color [50, 51]. The 3D structure of bacteriorhodopsin has been resolved at a resolution of 1.55 Å, showing the bulk of protein embedded in the membrane bilayer with short interhelical loops, and short extramembrane N- and C-termini [52]. The purple color shows only when the membrane portion of the protein is intact, (i.e. naturally folded and embedded within the membrane bilayer).
To compare effectiveness, the following buffer systems were employed to solubilize and tryptically digest equal amounts of bacteriorhodopsin: (a) aqueous buffer (50 mM NH4HCO3) as a control, (b) organic-aqueous buffer (MeOH in 50 mM NH4HCO3), (c) detergent-based buffer (PPS in 50 mM NH4HCO3), (d) a combination of both, organic solvent + detergent (MeOH and PPS in 50 mM NH4HCO3) and (e) 8 M urea. After solubilization and tryptic digestion, aliquots of 20 μg of digestate were removed from each buffer at 0, 0.5, 4, 20 hrs intervals, analyzed by SDS-PAGE and visualized by Coomassie staining (Figure 1a-e).
Figure 1.
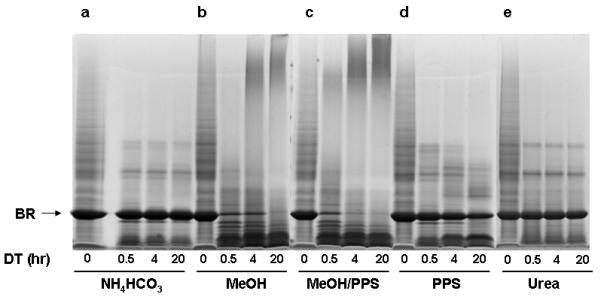
SDS-PAGE analysis/comparison of solubilization/digestion efficiencies of different approaches, applied on single hydrophobic multipass integral membrane protein bacteriorhodopsin (br). Aliquots of 20 μg were removed from each buffer at 0, 0.5, 4, 20 hour intervals during digestion (dt) and analyzed on a 4-12% SDS-PAGE followed by Coomassie staining: (a) 50 mM NH4HCO3 buffer, (b) 60% MeOH in 50 mM NH4HCO3 (v/v) buffer, (c) 0.1 % PPS in 50 mM NH4HCO3, (d) 60% MeOH/0.1% PPS in 50 mM NH4HCO3, (e) 8 M urea in 50 mM NH4HCO3.
On the basis of the staining pattern shown in Figure 1a, most of the membrane protein remained intact when digested in aqueous NH4HCO3 buffer. Importantly, we observed that the presence of the purple color of the bacteriorhodopsin solution persisted throughout the 20 h digestion period. This observation suggested that the intramembranous portion of bacteriorhodopsin remained intact since the membrane bilayer was not dissolved, rendering the intramembranous portion of the protein inaccessible to trypsin. In contrast, solubilization and digestion in a mixed organic aqueous buffer (Figure 1b) indicated complete bacteriorhodopsin digestion after 20 h, based on visual analysis. It suggested that dissolution of the membrane bilayer has been successfully achieved along with effective solubilization and denaturation of bacteriorhodopsin, allowing effective tryptic digestion [37]. It is important to note that immediately after CH3OH (60% v/v) was added to the aqueous bacteriorhodopsin suspension, the purple color faded suggesting the protein had been effectively denatured. The staining pattern depicting solubilization efficiency of the combination of CH3OH-based buffer and PPS (Figure 1c) indicates complete solubilization and digestion of bacteriorhodopsin after 20 h. The comparison of the density of bacteriorhodopsin bands in figures 1b and 1d suggests a synergistic solubilization/denaturation effect of organic solvent and PPS, allowing almost complete digestion after 4 hours. This finding is in agreement with the observation reported by Chen et al [35] and results reported by Bromberg and Klibanov[53]. The staining pattern depicted in Figure 1d shows that significant amount of intact protein remained within the PPS-based buffer even after 20 h of digestion at 37 °C. This suggests that the concentration of PPS used in this investigation is incapable of completely solubilizing or denaturing bacteriorhodopsin. The staining pattern of bacteriorhodopsin solubilized in urea is depicted in Figure 1e. It shows limited bacteriorhodopsin digestion, indicating insufficient solubilization of hydrophobic membrane proteins by urea in this experimental setting [17, 18].
In our second set of experiments we applied i) PPS, ii) methanol and iii) their combination on a membrane proteome isolated from human monocytes to assess the performance of these systems on a complex membrane protein mixture. Since the solubilization and the extent of bacteriorhodopsin proteolysis were poor in urea-based buffer (Figure 1e), this buffer system was not investigated further. Equal aliquots of crude membrane fraction from affinity-purified CD14 human monocytes were solubilized in each selected buffer system. After proteolysis, equal amounts of each digestate (2 μg) were analyzed in triplicate using high-resolution and high-precision LC-MS/MS. It should be pointed out that human monocytes show heterogeneity of protein expression between individuals and between different subpopulations within an individual[54, 55]. However, given the aim of our study, the important issue was to obtain a population of cells that are very highly enriched for what is typically classified as monocytes based on CD14 expression, and we used state-of-the-art technology to obtain an almost completely homogeneous population of monocytes as defined by that criterion. For a study aiming at defining different subpopulations of monocytes based on their membrane protein profiles, additional markers can be used to separate the subpopulations for analysis using our solubilization techniques. In this regard, it is also important to note that our use of primary cells allows us to investigate physiologically relevant conditions. In many cases, the latter cannot be achieved by use of cell lines given the transformations that they have undergone to achieve immortalization. Also, immortalized cell lines typically present a heterogeneous population due to the genomic instability that is common in a number of studied cases.
The number of peptides and proteins identified in the MS analysis using the three compared approaches are shown in Table 1. A total of 1355 peptides identified using the methanol-based approach (Supplementary table 1A) yielded identification of 194 proteins by ≥2 unique peptides (Supplementary Table 1B). A total of 1403 peptides identified using methanol/PPS combination (Supplementary Table 2A) allowed identification of 203 proteins by ≥2 unique peptides (Supplementary Table 2B). PPS alone permitted identification of 1370 tryptic peptides (Supplementary Table 3A) resulting in a total of 216 proteins identified by ≥2 unique tryptic peptides (Supplementary Table 3B). Based on the number of total peptide identifications it is evident that the extent of tryptic digestion is similar across all three approaches. This result is even more evident when the number of unique peptides and proteins identified by at least two peptides is compared (Table 1). Overall, the combination of methanol and PPS yielded the highest peptide/protein ratio of 4.48. The ratio for methanol alone and PPS alone was 3.98 and 3.59, respectively (Table 1). The enrichment of integral membrane proteins was assessed by mapping alpha-helical TMDs for each identified protein within the three datasets using TMHMM software [45].
Table 1.
Comprehensive results obtained using three distinctive solubilization approaches: methanol, methanol/PPS combination, and PPS alone
| MeOH | MeOH/PPS | PPS | ||
|---|---|---|---|---|
| Peptides: total IDs | a | 1355 | 1403 | 1370 |
| Peptides: unique IDs | b | 680 | 640 | 679 |
| Proteins: total IDs | c | 340 | 313 | 381 |
| Proteins: identified by ≥2 peptides | d | 194 | 203 | 216 |
| Peptide/protein ratio | a/c | 3.98 | 4.48 | 3.59 |
| IM* proteins: total IDs | e | 141 | 130 | 116 |
| Average GRAVYa | e | -0.096 | -0.102 | -0.007 |
| IM proteins ≥2 peptides | f | 93 | 93 | 75 |
| IM protein enrichment | f/d | 47.93% | 45.81% | 34.72% |
| Average TMD/protein | f | 2.7 | 2.8 | 2.3 |
| Hydrophobic IM proteins ≥2 pep | g | 39 | 43 | 30 |
| Hydrophobic IM protein (%) | g/d | 20.10% | 21.18% | 13.88% |
| Average GRAVYa | g | 0.26 | 0.32 | 0.24 |
All identified proteins and the entire human proteome database were analyzed using the ProtParam program (available at http://www.expasy.ch/sprot/sprot-top.html) to calculate the grand average of hydropathicity (GRAVY) for each protein.
We first preformed the analysis of the entire human proteome database (UniProt Human, release 09/2007) of 37714 entries which revealed that a total of 8056 (21.36 %) proteins contained at least one or more mapped TMD. The THMM analysis showed that methanol-based approach yielded a total of 141 integral membrane proteins possessing at least one mapped TMD, while the combination of methanol plus PPS and PPS alone yielded 130 and 116 integral membrane proteins, respectively (Table 1). For integral membrane proteins identified by at least two peptides the total number of identified integral membrane proteins was identical for methanol alone and the methanol/PPS combination (93 proteins each). PPS alone yielded 75 integral membrane protein identifications.
The enrichment of integral membrane proteins estimated by TMHMM software, from protein pools identified by at least two peptides, for methanol alone, methanol/PPS combination, and PPS alone was 47.93 %, 45.81 % and 34.72%, respectively (Table 1). The average number of TMDs per identified integral membrane protein was slightly higher for the methanol/PPS combination (2.8) than for methanol alone (2.7). PPS alone allowed only 2.3 TMDs to be mapped per identified membrane protein suggesting that PPS alone permitted lower protein sequence coverage than the methanol-based approaches. The overlap among the proteins identified using each buffer system is shown in Figure 2.
Figure 2.
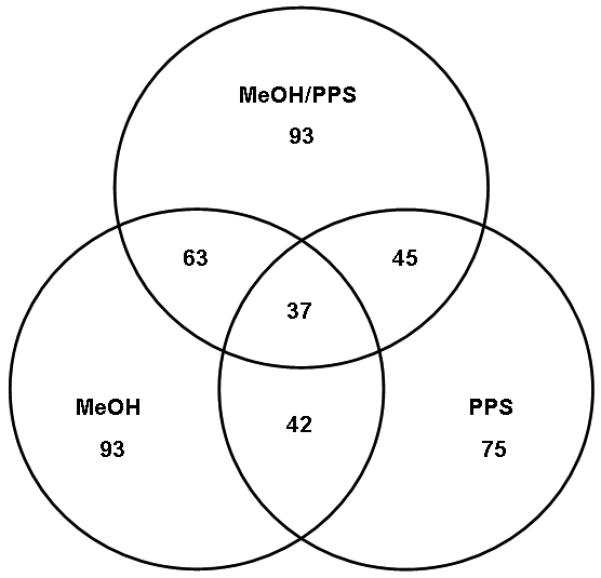
Triple Venn diagrams illustrating the relationship between the total numbers of integral membrane proteins identified by at least 2 peptides using methanol (MeOH), methanol/PPS combination (MeOH/PPS) and PPS alone.
These results indicate that each approach allowed the identification of different subsets of proteins due to different solubilizing conditions, showing respectable overlap of 67.74 % (63 proteins) between the proteins identified using the two methanol-based approaches. The overlap between the proteins identified using the two PPS-based approaches was lower (i.e. 60%). The enrichment of hydrophobic proteins was assessed by computing the grand average of hydropathicity (GRAVY) index, calculated for each identified protein within the three distinct datasets [46]. The GRAVY index is a global descriptor of protein solubility, and corresponds to the sum of hydrophobicity values for each of the amino-acids in the protein, normalized according to protein length (Note: proteins exhibiting positive GRAVY values were recognized as hydrophobic while proteins exhibiting negative GRAVY values were recognized as hydrophilic)[46]. The number of hydrophobic proteins identified in this investigation by at least two peptides is listed in Table 1. For the methanol-based approach the protein value was 39 (20.1 %), the methanol/PPS combination yielded 43 (21.18%), and 30 (13.88 %) for PPS. Figure 3 shows the GRAVY distributions for all proteins identified using the three compared approaches in relation to overall GRAVY distribution of the human genome. It is evident that methanol alone and methanol/PPS combination showed the ability to enrich for a greater proportion of hydrophobic proteins than PPS alone, which is in agreement with the results shown in Table 1. The correlation of hydrophilic and hydrophobic profiles of the identified proteins to the number of mapped TMDs is shown in Figure 4. Dot diagram analysis showed that methanol alone and the methanol/PPS combination allowed a greater number of integral membrane proteins with higher number of multiple TMDs to be identified than when PPS was used by itself. Overall, the results illustrate significantly higher enrichment of integral membrane proteins when 60% (v/v) methanol-based approaches were employed indicating that this solvent by itself or in combination with PPS represent a potent tool for MS-based membrane proteomics. The average TMD/protein ratio and average GRAVY for of hydrophobic integral membrane proteins suggest a synergistic effect of 60% (v/v) methanol/ 0.1 % PPS combination, which may increase the enrichment of hydrophobic integral membrane proteins. This is in agreement with previous observations [35, 49].
Figure 3.
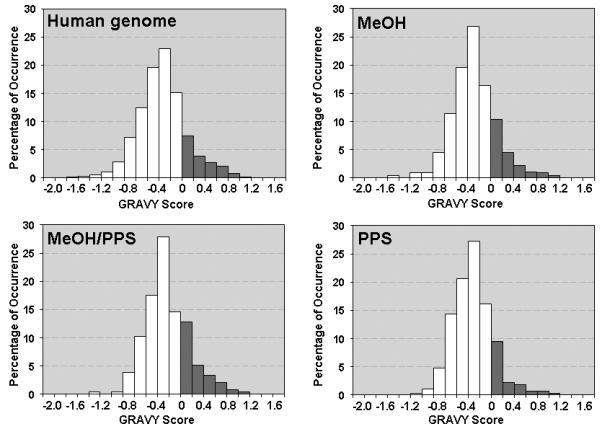
The relation of hydropathy profiles of the identified proteins using each compared methods and whole human proteome depicted by the GRAVY index analysis. The hydropathy plots for the human proteome, proteins identified using 60 % methanol based method, proteins identified using methanol/PPS combination and proteins identified by PPS only. Each histogram was generated by plotting the number of proteins per 0.2 GRAVY value increment.
Figure 4.
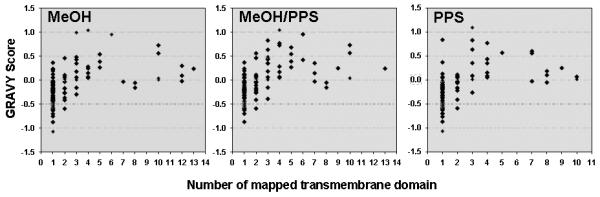
The relation of hydropathy profiles (GRAVY) and number of predicted TMDs (TMHMM) depicted by dot diagram for identified integral membrane proteins using methanol, methanol/PPS combination and PPS alone.
To increase the global understanding of membrane function within any cell type, increasing the coverage of membrane proteins identified is an absolute necessity. Blood monocytes play an important role in immuno-regulation and tumor surveillance. However, the molecular mechanisms underlying these biological processes are not well understood [13, 56]. A proteomic approach providing significant enrichment of integral membrane proteins should facilitate a better understanding of human monocyte functions in biological systems.
Recently, a global proteomic study of monocytes identified 164 proteins by coupling 2D gel electrophoresis and MS analysis [12]. However, only 12 membrane proteins were identified. By collating data obtained using compared approaches, we have been able to unambiguously identify a total of 165 unique integral membrane proteins, employing high precision and high resolution MS. Supplementary Table 4 contains broader characterization of these proteins including their function and involvement in human diseases.
Since we were investigating the CD14 human monocyte crude membrane fraction we first sought to determine if the CD14 (monocyte differentiation antigen) was positively identified. CD14 is a hydrophobic (GRAVY = 0.083) surface membrane protein attached to the plasma membrane via a phospholipid anchor [57]. Indeed, CD14 was identified by all of three approaches as shown in Table 2. The two methanol-based approaches allowed unambiguous identification of this protein through multiple peptides, while using PPS alone allowed only a single CD14 peptide to be identified. CD14 is a myelomonocytic differentiation antigen whose gene is located in the “critical” region of chromosome 5. This region is frequently deleted in certain myeloid leukemias [58]. Monocytes are active mediators of inflammation and infection processes [59] and have specificity for lipopolysaccharide (LPS) and other bacteria-wall-derived components. LPS signaling triggers a cascade that leads to cytokine production and shedding of the extracellular domain of CD14.
Table 2.
A list of peptides identifying monocyte differentiation antigen CD14, detected by LC-MS/MS using three distinctive approaches methanol, methanol/PPS combination and PPS alone
| MeOH | ||||||
| Protein name | MH+ a | Peptide | z b | Max XC c | Max ΔCN d | SC e |
| CD14 precursor | 1972.997 | R.AFPALTSLDLSDNPGLGER.G | 2 | 2.5298 | 0.51 | 1 |
| CD14 precursor | 1777.069 | R.LTVGAAQVPAQLLVGALR.V | 2 | 3.1222 | 0.4924 | 3 |
| CD14 precursor | 1933.171 | R.RLTVGAAQVPAQLLVGALR.V | 2 | 5.3189 | 0.7375 | 2 |
| CD14 precursor | 1797.006 | R.SWLAELQQWLKPGLK.V | 2 | 3.9901 | 0.5849 | 3 |
| MeOH/PPS | ||||||
| Protein name | MH+ | Peptide | z | Max XC | Max ΔCN | |
| CD14 precursor | 1142.668 | K.FPAIQNLALR.N | 2 | 2.7192 | 0.2607 | 2 |
| CD14 precursor | 1972.997 | R.AFPALTSLDLSDNPGLGER.G | 2 | 5.5742 | 0.5339 | 3 |
| CD14 precursor | 1777.069 | R.LTVGAAQVPAQLLVGALR.V | 2 | 3.3594 | 0.5768 | 3 |
| CD14 precursor | 1797.006 | R.SWLAELQQWLKPGLK.V | 2 | 2.8424 | 0.6037 | 1 |
| PPS | ||||||
| Protein name | MH+ | Peptide | z | Max XC | Max ΔCN | |
| CD14 precursor | 2138.189 | K.ITGTMPPLPLEATGLALSSLR.L | 2 | 2.5064 | 0.5238 | 4 |
Calculated mass for a given singly-charged peptide.
A charge state for a given peptide.
Maximal Xcorr observed for a given peptide. The cross-correlation score (Xcorr) of the peptide is based on the “fit” of the MS/MS data to the theoretical distribution of ions produced for the peptide.
Maximal ΔCN score observed for a given peptide. The ΔCN score represents a calculated “difference” between the top two Xcorr values for the given peptide.
Total spectral count for a given peptide.
The buffering of LPS is crucial during acute inflammatory and infectious processes[59]. The number of circulating CD14+ monocytes, and the expression of the CD14 marker by monocytes were found to be significantly lower in patients with septic shock and abnormal hepatocellular function [59, 60]. CD14 monocytes in patients with septic shock also exhibited a profound deficiency of TNF-α production, which is considered as hallmark of septic shock[59]. The molecular mechanism of this down-regulation is poorly understood. Thus, further study of CD14 holds a promise for developing a MS-based quantitative assay that might be used to measure CD14 concentration in responses to controlled experimentally evoked stimuli.
The ability to identify peptides residing within the TMD of membrane proteins is an indicator of the extent of a method’s solubilization capability. Microsomal glutathione S-transferase 3 (microsomal GST-3) is 17 kDa, multi-pass hydrophobic (GRAVY = 0.282) integral membrane protein, typically found embedded within endoplasmatic reticulum vesicles. In this study, microsomal GST-3 was identified using both methanol-based approaches but not when PPS was used alone (Table 3). Importantly, two identified peptides (R.IASGLGLAWIVGR.V and R.VLYAYGYYTGEPSKR.S) completely span the 3rd TMD as mapped by TMHMM, of which, R.VLYAYGYYTGEPSKR.S is a highly hydrophobic peptide (GRAVY = 1.308) and resides directly within the TMD. It is suggested that microsomal GST-3 plays significant role in cellular protection against oxidative stress and elimination of xenobiotics [61]. Recently it has been determined by subtractive hybridization screen and validated by northern analysis that after experimentally induced glucose deprivation in human neuroblastoma cells, microsomal GST-3 showed up-regulation by 4 fold [62]. This finding suggests an important role of microsomal GST-3 in the glucopenic response. Evidently, confident identification and significant enrichment of hydrophobic integral membrane proteins using 60% methanol alone or in combination with PPS as shown in Table 4, should allow more detailed characterization of the CD14 monocyte microsomal fraction, if used in the context of multidimensional shotgun analysis [48].
Table 3.
Peptides identifying Microsomal GST-3, identified by LC-MS/MS using methanol-based approaches
| MeOH | ||||||
| Gene name | MH+ a | Peptide | z b | Max XC c | Max ΔCN d | SC e |
| Microsomal GST-3 | 2974.386 | K.YKVEYPIMYSTDPENGHIFNC#IQR.A | 3 | 4.6173 | 0.6372 | 1 |
| Microsomal GST-3 | 1312.774 | R.IASGLGLAWIVGR.V | 2 | 3.4382 | 0.3644 | 5 |
| Microsomal GST-3 | 1766.875 | R.VLYAYGYYTGEPSKR.S | 2 | 3.3803 | 0.4787 | 3 |
| MeOH/PPS | ||||||
| Gene name | MH+ | Peptide | z | Max XC | Max ΔCN | SC |
| Microsomal GST-3 | 2974.386 | K.YKVEYPIMYSTDPENGHIFNC#IQR.A | 3 | 4.2044 | 0.6659 | 1 |
| Microsomal GST-3 | 1312.774 | R.IASGLGLAWIVGR.V | 2 | 3.4553 | 0.4516 | 5 |
| Microsomal GST-3 | 1766.875 | R.VLYAYGYYTGEPSKR.S | 2 | 3.4692 | 0.5654 | 4 |
Calculated mass for a given singly-charged peptide.
A charge state for a given peptide.
Maximal Xcorr observed for a given peptide. The cross-correlation score (Xcorr) of the peptide is based on the “fit” of the MS/MS data to the theoretical distribution of ions produced for the peptide.
Maximal ΔCN score observed for a given peptide. The ΔCN score represents a calculated “difference” between the top two Xcorr values for the given peptide.
Total spectral count for a given peptide.
Table 4.
A subset of multi-pass hydrophobic integral membrane proteins identified using methanol-based buffers
| Acc No | Protein Name | Helicesa | GRAVYb | Cellular location |
|---|---|---|---|---|
| P12235 | ADP/ATP translocase 1 | 3 | 0.059 | Mitochondrion inner membrane |
| P00846 | ATP synthase a chain | 6 | 0.952 | Mitochondrion inner membrane |
| Q6UW11 | ATWD578 | 3 | 0.181 | Integral to membrane |
| Q9NX76 | CKLF-like MARVEL transmembrane protein | 3 | 0.458 | Membrane |
| Q9BUN8 | Derlin-1 | 5 | 0.386 | Endoplasmic reticulum membrane |
| Q8TCJ2 | Dolichyl-diphosphooligosaccharide--protein | 10 | 0.038 | Endoplasmic reticulum membrane |
| P46977 | Dolichyl-diphosphooligosaccharide--protein | 13 | 0.238 | Endoplasmic reticulum membrane |
| Q9BW60 | Elongation of very long chain fatty acids protein 1 | 7 | 0.352 | Endoplasmic reticulum membrane |
| P33947 | ER lumen protein retaining receptor 2 | 4 | 0.722 | Endoplasmic reticulum membrane |
| Q9H3K2 | Growth hormone-inducible transmembrane protein | 6 | 0.418 | Membrane |
| O75352 | Mannose-P-dolichol utilization defect 1 protein | 5 | 0.679 | Membrane |
| O14880 | Microsomal glutathione S-transferase 3 | 4 | 0.282 | Microsome membrane |
| Q13423 | NAD(P) transhydrogenase, mitochondrial precursor | 12 | 0.299 | Mitochondrion inner membrane |
| P61619 | Protein transport protein Sec61 subunit alpha isoform 1 | 10 | 0.563 | Endoplasmic reticulum membrane |
| O43760 | Synaptogyrin-2 | 4 | 0.168 | Membrane |
| P30536 | Translocator protein | 5 | 0.266 | Mitochondrion membrane |
| Q9UNL2 | Translocon-associated protein subunit gamma | 4 | 0.066 | Endoplasmic reticulum membrane |
| Q9BVC6 | Transmembrane protein 109 precursor | 5 | 0.541 | Nucleus outer membrane |
| P57088 | Transmembrane protein 33 | 3 | 0.429 | Membrane |
| Q96CP4 | Transporter 1, ATP-binding cassette, sub-family B | 7 | 0.142 | Integral to membrane |
| Q6RW13 | Type-1 angiotensin II receptor-associated protein | 3 | 0.482 | Endoplasmic reticulum membrane |
| Q9H1C4 | UNC93 homolog B1 | 12 | 0.095 | Membrane |
| Q8N357 | Uncharacterized protein C2orf18 precursor | 10 | 0.561 | Membrane |
| P27449 | Vacuolar ATP synthase 16 kDa proteolipid subunit | 4 | 1.041 | Vacuole membrane |
A number of transmembrane domains mapped by TMHMM algorithm.
A grand average of hydropathicity (GRAVY) index calculated for a given protein.
In conclusion, the objective of this investigation was to optimize solubilizing conditions for solution-based shotgun membrane proteomics. We first compared different methods relying on: i) 60% buffered methanol ii) 0.1% cleavable detergent (PPS), iii) 60% buffered methanol/0.1% PPS combination and, iv) 8 M urea to solubilize bacteriorhodopsin. Because of inferior performance of urea-based buffer in present experimental setting further experiments were not performed. Thus, the remaining approaches were then used to solubilize and digest the CD14 human monocyte microsomal fraction. Using high precision MS coupled by nanoflow LC we determined that 60 % buffered methanol alone or in combination with acid-cleavable detergent (PPS) permitted better solubilization/digestion when compared to PPS alone. This was exemplified in significantly higher membrane protein identification/enrichment using both methanol-based approaches. Our results also suggest that the methanol/PPS combination may result in slightly higher enrichment of hydrophobic membrane proteins and might be beneficial for analysis of amount-limited membrane preparations.
Supplementary Material
ACKNOWLEDGEMENTS
This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract NO1-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the United States Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no competing financial interests.
REFERENCES
- [1].Wu CC, Yates JR. The application of mass spectrometry to membrane proteomics. Nat Biotechnol. 2003;21:262–7. doi: 10.1038/nbt0303-262. [DOI] [PubMed] [Google Scholar]
- [2].Fontanesi F, Soto IC, Horn D, Barrientos A. Assembly of mitochondrial cytochrome c-oxidase, a complicated and highly regulated cellular process. Am J Physiol Cell Physiol. 2006;291:C1129–47. doi: 10.1152/ajpcell.00233.2006. [DOI] [PubMed] [Google Scholar]
- [3].Ashkenazi A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat Rev Cancer. 2002;2:420–30. doi: 10.1038/nrc821. [DOI] [PubMed] [Google Scholar]
- [4].Hopkins AL, Groom CR. Opinion: The druggable genome. Nat Rev Drug Disc. 2004:1. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- [5].Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–38. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Milanezi F, Carvalho S, Schmitt FC. EGFR/HER2 in breast cancer: a biological approach for molecular diagnosis and therapy. Expert Rev Mol Diagn. 2008;8:417–34. doi: 10.1586/14737159.8.4.417. [DOI] [PubMed] [Google Scholar]
- [7].Nichols BA, Bainton DF, Farquhar MG. Differentiation of monocytes. Origin, nature, and fate of their azurophil granules. J Cell Biol. 1971;50:498–515. doi: 10.1083/jcb.50.2.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Louache F, Debili N, Marandin A, Coulombel L, Vainchenker W. Expression of CD4 by human hematopoietic progenitors. Blood. 1994;84:3344–55. [PubMed] [Google Scholar]
- [9].Ziegler-Heitbrock HW, Strobel M, Fingerle G, Schlunck T, et al. Small (CD14+/CD16+) monocytes and regular monocytes in human blood. Pathobiology. 1991;59:127–30. doi: 10.1159/000163629. [DOI] [PubMed] [Google Scholar]
- [10].Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–52. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ziegler-Heitbrock L. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J Leukoc Biol. 2007;81:584–92. doi: 10.1189/jlb.0806510. [DOI] [PubMed] [Google Scholar]
- [12].Jin M, Diaz PT, Bourgeois T, Eng C, et al. Two-dimensional gel proteome reference map of blood monocytes. Proteome Sci. 2006;4:16. doi: 10.1186/1477-5956-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bok E, Plazuk E, Hryniewicz-Jankowska A, Chorzalska A, et al. Lipid-binding role of betaII-spectrin ankyrin-binding domain. Cell Biol Int. 2007;31:1482–94. doi: 10.1016/j.cellbi.2007.06.014. [DOI] [PubMed] [Google Scholar]
- [14].Li N, Mak A, Richards DP, Naber C, et al. Monocyte lipid rafts contain proteins implicated in vesicular trafficking and phagosome formation. Proteomics. 2003;3:536–48. doi: 10.1002/pmic.200390067. [DOI] [PubMed] [Google Scholar]
- [15].Zhang N, Shaw AR, Li N, Chen R, et al. Liquid chromatography electrospray ionization and matrix-assisted laser desorption ionization tandem mass spectrometry for the analysis of lipid raft proteome of monocytes. Analytica Chimica Acta. 2008;627:82–90. doi: 10.1016/j.aca.2008.05.058. [DOI] [PubMed] [Google Scholar]
- [16].Stein WD, Litman T, Fojo T, Bates SE. A Serial Analysis of Gene Expression (SAGE) database analysis of chemosensitivity: comparing solid tumors with cell lines and comparing solid tumors from different tissue origins. Cancer Res. 2004;64:2805–16. doi: 10.1158/0008-5472.can-03-3383. [DOI] [PubMed] [Google Scholar]
- [17].Blonder J, Conrads TP, Veenstra TD. Characterization and quantitation of membrane proteomes using multidimensional MS-based proteomic technologies. Expert Rev Proteomics. 2004;1:153–63. doi: 10.1586/14789450.1.2.153. [DOI] [PubMed] [Google Scholar]
- [18].Santoni V, Molloy M, Rabilloud T. Membrane proteins and proteomics: Un amour impossible? Electrophoresis. 2000;21:1054–70. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1054::AID-ELPS1054>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- [19].Loo RR, Dales N, Andrews PC. The effect of detergents on proteins analyzed by electrospray ionization. Methods Mol Biol. 1996;61:141–60. doi: 10.1385/0-89603-345-7:141. [DOI] [PubMed] [Google Scholar]
- [20].Funk J, Li X, Franz T. Threshold values for detergents in protein and peptide samples for mass spectrometry. Rapid Commun Mass Spectrom. 2005;19:2986–8. doi: 10.1002/rcm.2142. [DOI] [PubMed] [Google Scholar]
- [21].Hixson KK, Rodriguez N, Camp DG, 2nd, Strittmatter EF, et al. Evaluation of enzymatic digestion and liquid chromatography-mass spectrometry peptide mapping of the integral membrane protein bacteriorhodopsin. Electrophoresis. 2002;23:3224–32. doi: 10.1002/1522-2683(200209)23:18<3224::AID-ELPS3224>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- [22].Nagaraj N, Lu A, Mann M, Wisniewski JR. Detergent-Based but Gel-Free Method Allows Identification of Several Hundred Membrane Proteins in Single LC-MS Runs. J Proteome Res. 2008;7:5028–32. doi: 10.1021/pr800412j. [DOI] [PubMed] [Google Scholar]
- [23].Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–7. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- [24].Wu CC, MacCoss MJ, Howell KE, Yates JR., 3rd A method for the comprehensive proteomic analysis of membrane proteins. Nat Biotechnol. 2003;21:532–8. doi: 10.1038/nbt819. [DOI] [PubMed] [Google Scholar]
- [25].Blackler AR, Speers AE, Ladinsky MS, Wu CC. A shotgun proteomic method for the identification of membrane-embedded proteins and peptides. J Proteome Res. 2008;7:3028–34. doi: 10.1021/pr700795f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chick JM, Haynes PA, Molloy MP, Bjellqvist B, et al. Characterization of the rat liver membrane proteome using peptide immobilized pH gradient isoelectric focusing. J Proteome Res. 2008;7:1036–45. doi: 10.1021/pr700611w. [DOI] [PubMed] [Google Scholar]
- [27].Dormeyer W, van Hoof D, Mummery CL, Krijgsveld J, Heck AJ. A practical guide for the identification of membrane and plasma membrane proteins in human embryonic stem cells and human embryonal carcinoma cells. Proteomics. 2008;8:4036–53. doi: 10.1002/pmic.200800143. [DOI] [PubMed] [Google Scholar]
- [28].Yu YQ, Gilar M, Lee PJ, Bouvier ES, Gebler JC. Enzyme-friendly, mass spectrometry-compatible surfactant for in-solution enzymatic digestion of proteins. Anal Chem. 2003;75:6023–8. doi: 10.1021/ac0346196. [DOI] [PubMed] [Google Scholar]
- [29].Norris JL, Porter NA, Caprioli RM. Combination detergent/MALDI matrix: functional cleavable detergents for mass spectrometry. Anal Chem. 2005;77:5036–40. doi: 10.1021/ac050460g. [DOI] [PubMed] [Google Scholar]
- [30].Umar A, Dalebout JC, Timmermans AM, Foekens JA, Luider TM. Method optimisation for peptide profiling of microdissected breast carcinoma tissue by matrix-assisted laser desorption/ionisation-time of flight and matrix-assisted laser desorption/ionisation-time of flight/time of flight-mass spectrometry. Proteomics. 2005;5:2680–8. doi: 10.1002/pmic.200400128. [DOI] [PubMed] [Google Scholar]
- [31].Norris JL, Porter NA, Caprioli RM. Mass spectrometry of intracellular and membrane proteins using cleavable detergents. Anal Chem. 2003;75:6642–7. doi: 10.1021/ac034802z. [DOI] [PubMed] [Google Scholar]
- [32].Arnold RJ, Hrncirova P, Annaiah K, Novotny MV. Fast proteolytic digestion coupled with organelle enrichment for proteomic analysis of rat liver. J Proteome Res. 2004;3:653–7. doi: 10.1021/pr034110r. [DOI] [PubMed] [Google Scholar]
- [33].Yu YQ, Gilar M, Gebler JC. A complete peptide mapping of membrane proteins: a novel surfactant aiding the enzymatic digestion of bacteriorhodopsin. Rapid Commun Mass Spectrom. 2004;18:711–5. doi: 10.1002/rcm.1374. [DOI] [PubMed] [Google Scholar]
- [34].Ruth MC, Old WM, Emrick MA, Meyer-Arendt K, et al. Analysis of membrane proteins from human chronic myelogenous leukemia cells: comparison of extraction methods for multidimensional LC-MS/MS. J Proteome Res. 2006;5:709–19. doi: 10.1021/pr050313z. [DOI] [PubMed] [Google Scholar]
- [35].Chen EI, Cociorva D, Norris JL, Yates JR. Optimization of mass spectrometry-compatible surfactants for shotgun proteomics. J Proteome Res. 2007;6:2529–38. doi: 10.1021/pr060682a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Blonder J, Goshe MB, Moore RJ, Pasa-Tolic L, et al. Enrichment of integral membrane proteins for proteomic analysis using liquid chromatography-tandem mass spectrometry. J Proteome Res. 2002;1:351–60. doi: 10.1021/pr0255248. [DOI] [PubMed] [Google Scholar]
- [37].Blonder J, Conrads TP, Yu LR, Terumuma A, et al. A detergent- and cyanogen bromide-free method for integral membrane proteomics: Application to Halobacterium purple membranes and the human epidermal membrane proteome. Proteomics. 2004;4:31–45. doi: 10.1002/pmic.200300543. [DOI] [PubMed] [Google Scholar]
- [38].Fischer F, Wolters D, Rogner M, Poetsch A. Toward the complete membrane proteome: high coverage of integral membrane proteins through transmembrane peptide detection. Mol Cell Proteomics. 2006;5:444–53. doi: 10.1074/mcp.M500234-MCP200. [DOI] [PubMed] [Google Scholar]
- [39].Zhang N, Chen R, Young N, Wishart D, et al. Comparison of SDS- and methanol-assisted protein solubilization and digestion methods for Escherichia coli membrane proteome analysis by 2-D LC-MS/MS. Proteomics. 2007;7:484–93. doi: 10.1002/pmic.200600518. [DOI] [PubMed] [Google Scholar]
- [40].Mitra SK, Gantt JA, Ruby JF, Clouse SD, Goshe MB. Membrane proteomic analysis of Arabidopsis thaliana using alternative solubilization techniques. J Proteome Res. 2007;6:1933–50. doi: 10.1021/pr060525b. [DOI] [PubMed] [Google Scholar]
- [41].Zhang H, Lin Q, Ponnusamy S, Kothandaraman N, et al. Differential recovery of membrane proteins after extraction by aqueous methanol and trifluoroethanol. Proteomics. 2007;7:1654–63. doi: 10.1002/pmic.200600579. [DOI] [PubMed] [Google Scholar]
- [42].Franzel B, Fischer F, Trotschel C, Poetsch A, Wolters D. The two-phase partitioning system--a powerful technique to purify integral membrane proteins of Corynebacterium glutamicum for quantitative shotgun analysis. Proteomics. 2009;9:2263–72. doi: 10.1002/pmic.200800766. [DOI] [PubMed] [Google Scholar]
- [43].Saikh KU, Khan AS, Kissner T, Ulrich RG. IL-15-induced conversion of monocytes to mature dendritic cells. Clin Exp Immunol. 2001;126:447–55. doi: 10.1046/j.1365-2249.2001.01672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fujiki Y, Hubbard AL, Fowler S, Lazarow PB. Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J Cell Biol. 1982;93:97–102. doi: 10.1083/jcb.93.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–80. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- [46].Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–32. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- [47].Mann M, Kelleher NL. Special Feature: Precision proteomics: The case for high resolution and high mass accuracy. Proc Natl Acad Sci U S A. 2008;105:18132–8. doi: 10.1073/pnas.0800788105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Blonder J, Chan KC, Issaq HJ, Veenstra TD. Identification of membrane proteins from mammalian cell/tissue using methanol-facilitated solubilization and tryptic digestion coupled with 2D-LC-MS/MS. Nat Protoc. 2006;1:2784–90. doi: 10.1038/nprot.2006.359. [DOI] [PubMed] [Google Scholar]
- [49].Chen EI, McClatchy D, Park SK, Yates JR., 3rd Comparisons of mass spectrometry compatible surfactants for global analysis of the mammalian brain proteome. Anal Chem. 2008;80:8694–701. doi: 10.1021/ac800606w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mollaaghababa R, Steinhoff HJ, Hubbell WL, Khorana HG. Time-resolved site-directed spin-labeling studies of bacteriorhodopsin: loop-specific conformational changes. M Biochemistry. 2000;39:1120–7. doi: 10.1021/bi991963h. [DOI] [PubMed] [Google Scholar]
- [51].Steinhoff HJ, Mollaaghababa R, Altenbach C, Hideg K, et al. Time-resolved detection of structural changes during the photocycle of spin-labeled bacteriorhodopsin. Science. 1994;266:105–7. doi: 10.1126/science.7939627. [DOI] [PubMed] [Google Scholar]
- [52].Luecke H, Schobert B, Richter HT, Cartailler JP, Lanyi JK. Structure of bacteriorhodopsin at 1.55 A resolution. J Mol Biol. 1999;291:899–911. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- [53].Bromberg LE, Klibanov AM. Detergent-enabled transport of proteins and nucleic acids through hydrophobic solvents. Proc Natl Acad Sci U S A. 1994;91:143–7. doi: 10.1073/pnas.91.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- [55].Hryniewicz-Jankowska A, Choudhary PK, Goodman SR. Variation in the monocyte proteome. Exp Biol Med. 2007;232:967–76. [PubMed] [Google Scholar]
- [56].Taylor PR, Gordon S. Monocyte heterogeneity and innate immunity. Immunity. 2003;19:2–4. doi: 10.1016/s1074-7613(03)00178-x. [DOI] [PubMed] [Google Scholar]
- [57].Simmons DL, Tan S, Tenen DG, Nicholson-Weller A, Seed B. Monocyte antigen CD14 is a phospholipid anchored membrane protein. Blood. 1989;73:284–9. [PubMed] [Google Scholar]
- [58].Goyert SM, Ferrero E, Rettig WJ, Yenamandra AK, et al. The CD14 monocyte differentiation antigen maps to a region encoding growth factors and receptors. Science. 1988;239:497–500. doi: 10.1126/science.2448876. [DOI] [PubMed] [Google Scholar]
- [59].de Werra I, Zanetti G, Jaccard C, Chiolero R, et al. CD14 expression on monocytes and TNF alpha production in patients with septic shock, cardiogenic shock or bacterial pneumonia. Swiss Med Wkly. 2001;131:35–40. doi: 10.4414/smw.2001.05883. [DOI] [PubMed] [Google Scholar]
- [60].Fernandez-Real JM, Lopez-Bermejo A, Broch M, Vendrell J, et al. Circulating soluble CD14 monocyte receptor is associated with increased alanine aminotransferase. Clinical Chem. 2004;50:1456–8. doi: 10.1373/clinchem.2003.030015. [DOI] [PubMed] [Google Scholar]
- [61].Jakobsson PJ, Morgenstern R, Mancini J, Ford-Hutchinson A, Persson B. Membrane-associated proteins in eicosanoid and glutathione metabolism (MAPEG). A widespread protein superfamily. Am J Resp Crit Care. 2000;161:S20–4. doi: 10.1164/ajrccm.161.supplement_1.ltta-5. [DOI] [PubMed] [Google Scholar]
- [62].Kobayashi K, Xin Y, Ymer SI, Werther GA, Russo VC. Subtractive hybridisation screen identifies genes regulated by glucose deprivation in human neuroblastoma cells. Brain Res. 2007;1170:129–39. doi: 10.1016/j.brainres.2007.07.042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.