

Abstract
One of the most important goals in neuroscience is to understand the molecular cues that instruct early stages of synapse formation. As such it has become imperative to develop objective approaches to quantify changes in synaptic connectivity. Starting from sample fixation, this protocol details how to quantify synapse number both in dissociated neuronal culture and in brain sections using immunocytochemistry. Using compartment-specific antibodies, we label presynaptic terminals as well as sites of postsynaptic specialization. We define synapses as points of colocalization between the signals generated by these markers. The number of these colocalizations is quantified using a plug in Puncta Analyzer (written by Bary Wark, available upon request, c.eroglu@cellbio.duke.edu) under the ImageJ analysis software platform. The synapse assay described in this protocol can be applied to any neural tissue or culture preparation for which you have selective pre- and postsynaptic markers. This synapse assay is a valuable tool that can be widely utilized in the study of synaptic development.
Protocol
Solutions to Prepare:
- Antibody Buffer:
- 150 mM NaCl
- 50 mM Tris-Base (Fisher, Cat. No: BP152-5, 50 mM) - 1.21 g
- 1% BSA (Sigma, Cat. No: A2153, 1%) - 2.0 g
- 100 mM L-lysine (Sigma, Cat. No: L-1137, 100 mM) - 3.65 g
- Adjust pH to 7.4
- 0.04% Azide
- Adjust volume to 200 ml with distilled H2O.
- Filter through 0.22μm filter (Millipore, Cat. No: SCGPU02RE).
- PFA Diluant:
- 168 ml 0.5 M Na2HPO4 (dibasic)
- 72 ml 0.5M Na2HPO4 (monobasic)
- 660 ml distilled H2O
- 4% PFA fixative for cultured neurons (solution #2):
- 10 ml 16% PFA solution (Electron Microscopy Sciences, Cat. No: 15711)
- 30 ml 4% PFA diluant (solution #2)
- 4% PFA in PBS:
- 4 g PFA (Electron Microscropy Sciences, Cat. No: 19210)
- 100 ml PBS (Invitrogen, Cat. No: 20012-027)
- Heat to 40°C, stir overnight.
- Filter through a 0.22μm filter (Millipore, Cat. No: SCGPU02RE)
- Blocking buffer (Total volume (for 24-well plate) = (#coverslips+1) x 200μl)
- 50% Antibody buffer (solution #1)
- 50% Normal goat serum (Gibco, Cat. No: 16210)
- 0.2% Triton X-100 (Roche Diagnostics Gmbh, Cat. No: 9002-93-1)
- 30% Sucrose in PBS
- 30 g sucrose (MP Biomedicals, Inc., Cat. No: 821713)
- 70 ml PBS (Invitrogen, Cat. No: 20012-027)
- Mix with a stirbar until sucrose is in solution.
- Bring the volume up to 100ml with PBS
- Filter through a 0.22 μm filter (Millipore, Cat. No: SCGPU02RE)
Preparation of Neuronal Cultures:
The protocol described here is applicable to any primary neuronal cultures grown on 12 mm glass coverslips (Karl Hecht, No. O, Cat. No: 99010) in 24-well plates (Falcon, 35-3047). For example, in our laboratory we culture rat retinal ganglion cells (RGCs) purified from rat retina harvested from P5-7 animals1,2. Cells are grown on glass coverslips coated with poly-d-lysine (Sigma, Cat. No: P6407) and mouse laminin (Cultrex, Cat. No: 3400-010-01). We utilize this culture preparation in a couple of different ways for our synapse assay. One manipulation that we perform involves culturing RGCs either in the presence or absence of astrocyte-secreted synaptogenic factors. Alternatively, we also employ these different treatment conditions in experiments where RGCs have been transfected to overexpress a protein of interest. In the latter case, we co-transfect the cells with a cell label (e.g. GFP or tdTomato). These different experimental approaches affect how one performs certain steps of a synapse assay, which we clarify below.
1. Fixing Dissociated Purified RGCs
Remove culture media from the RGC-containing wells and add 500 μl (for a 24 well plate) 4% paraformaldehyde (PFA) prewarmed to 37°C to each well. Allow cells to fix for 7 minutes at room temperature.
Following fixation rinse cells 3 times with phosphate-buffered saline (PBS) (Invitrogen, Cat. No: 20012-027). IMPORTANT: Cells should never be left without liquid in the wells, once a buffer is removed from a well it should be promptly replaced by the next buffer. At this point, cells are ready for immunostaining.
2. Blocking Unspecific Binding Sites on the RGCs
Prepare blocking buffer containing 50% normal goat serum (NGS, Gibco, Cat. No: 16210) and 0.2% Triton X-100 (Roche Diagnostics Gmbh, Cat. No: 9002-93-1). After removing PBS from each well, add 200 μl of the blocking buffer to each well and block for 30 minutes at room temperature.
Remove blocking buffer and rinse 3 times with PBS.
3. Applying Primary Antibody Solution
IMPORTANT: Be sure to choose primary antibodies for your pre and postsynaptic markers that are obtained from different species.
Prepare a primary antibody dilution in 90% antibody buffer, 10% NGS solution containing the pre and postsynaptic antibody pair of your choice. For example rabbit anti-synapsin (presynaptic marker) (1:750, cytosolic domain, Synaptic Systems) and mouse anti-Homer (postsynaptic marker) (1:500, mouse, Synaptic Systems). IMPORTANT: Centrifuge primary antibody dilution for 5 minutes at maximum speed in a bench top centrifuge to remove any precipitated antibody.
Add 200 μl primary antibody solution to each well.
Incubate overnight at 4°C. The plate should be placed in a bigger container which is humidified to prevent drying of the primary solution. BREAK POINT: Cells can stay in primary antibody mixture for up to 3 days prior to continuing with the protocol.
The following day, remove primary antibody from each well and rinse wells 3 times with PBS.
4. Applying Secondary Antibody Solution
Prepare a secondary antibody solution containing your secondary antibodies diluted 1:1000 in antibody buffer containing 10% NGS. IMPORTANT: Centrifuge secondary antibody dilution for 5 minutes at maximum speed in a bench top centrifuge to remove precipitated antibody. Skipping this step results in high secondary antibody background. Untransfected cells: In the absence of a cell label, use Alexa-594 and Alexa-488 conjugated secondaries for labeling pre and postsynaptic markers respectively. For example we use goat anti-rabbit Alexa594 to label the anti-synapsin antibody and goat anti mouse Alexa-488 conjugated secondary to label the anti-homer antibody. IMPORTANT: Use Alexa-488 conjugated secondaries for primary antibodies with weaker signal. Transfected cells: Choose secondary antibodies that accommodate the excitation-emission spectrum of your fluorescent cell label. For example, when we label transfected cells with tdTomato we use Alexa-647 conjugated goat anti-rabbit to recognize the anti-synapsin antibody and Alexa-488 conjugated goat anti-mouse to recognize the anti-homer antibody. Also, the use of NGS in this protocol is a result of our choice of secondary antibodies produced in goat.
Add 200 μl secondary antibody solution to each well.
After incubating for two hours at room temperature in a dark location, rinse 3 to 4 times with PBS.
5. Mounting Coverslips
Mount coverslips into Vectashield mounting medium with DAPI (Vector Laboratories Inc., Cat. No: H-1200) onto glass slides (VWR scientific, Cat. No: 48311-703).
Gently apply clear nail polish around the edges of the coverslip and allow to dry for at least 30 minutes in a dry, dark place. IMPORTANT: Avoid nudging or moving the coverslips during application of the nail polish since this would result in sheering of the cells.
6. Imaging
For imaging, a fluorescence microscope equipped with a camera capable of taking pictures at 4 different channels is necessary to be able to image both synaptic markers, your cell fill and nuclei (DAPI/optional). Cells should be imaged using an oil immersion 63x objective. We image using the Zeiss AxioImager fluorescence microscope with the Zeiss Plan-APOCHROMAT 63x/1.4 Oil DIC ∞/0.17 objective.
Select cells that are at least two cell diameters away from their nearest neighbors. To avoid bias and ensure randomness of cell selection if visualizing untransfected cells, select cells in the DAPI channel, then take images in all channels. If visualizing transfected cells select cells in the channel corresponding to the fluorescent label to identify transfected cells (e.g. tdTomato).
Acquire your images: Untransfected cells: For each selected cell, obtain 8 bit images in the GFP and Texas Red channels. The superimposed pseudocolored image should have your pre- and postsynaptic markers in red and green, respectively. Transfected cells: For each selected cell obtain 8 bit images in the GFP, Texas Red and Cy5 (or Cy5.5) channels. The superimposed pseudocolored image should have your pre- and postsynaptic markers in red and green, respectively. Pseudo color your cell fill in blue.
7. Image Analysis and Co-localized Puncta Quantification
We use Puncta Analyzer program for quantification co-localized synaptic puncta. Puncta Analyzer plug in is written by Bary Wark, and is available upon request (c.eroglu@cellbio.duke.edu). Puncta Analyzer runs in ImageJ 1.26 (http://rsbweb.nih.gov/ij/, newer versions of ImageJ cannot run the application). To install Puncta Analyzer simply place the downloaded application folder in the "Plugins" folder in the ImageJ 1.26 directory.
Open one of your images using ImageJ. Use one of the selection tools in the ImageJ menu to determine the region of interest (ROI). We regularly use the circular selection tool to select a region approximately one-cell diameter radially around the soma of interest.
With your region of interest (ROI) selected, go to the plugins menu and select "Puncta Analyzer".
In the "Analysis Options" window that appears, select "Red Channel", "Green Channel", the first "Subtract Background" and "Set results file...." Click "OK". You will be asked to define a location to save your results in. These results can be exported to Excel for further analysis.
In the window that appears next, make sure a rolling ball radius of 50 is selected and uncheck the "White Background" option (this modification is not required but is often preferred by users of the application for ease of visualization). Click "OK".
A new window will appear alongside a mask corresponding with your red channel image. Adjust the threshold until you feel that the red mask corresponds as well as possible to as many discrete individual puncta without introducing too much noise. This is one of the most subjective steps of this protocol, so take care to develop a consistent approach. Click "Done". Set the minimum puncta size to 4 pixels and modify nothing else. Click "OK".
Repeat the previous step, this time for the green channel.
Once you complete the previous step, the plugin will provide quantification corresponding to puncta in each channel separately and to colocalized puncta between the two channels.
BRAIN SECTIONS:
The synapse assay can be applied to cryosections from brain, and to any other nervous system tissue (such as spinal cord or retina) provided that there is a suitable pre- and postsynaptic marker pair (with antibodies that work well in sections) that can be utilized to identify the synapses you wish to quantify. The synapse assay can reveal the temporal regulation of synapse formation in a given brain region and can quantify effects on synaptic connectivity in transgenic animals or in a sample that has been manipulated in some other fashion.
1. Harvesting Brain Tissue from Mice
All animal procedures should be done in concordance with IACUC animal protocols.
Euthanize mice by exsanguination and perfusion with PBS. Perfusion with PBS is crucial for removal of blood and will reduce background signal for the staining procedures. IMPORTANT: Do not perfuse with fixatives such as 4% PFA. This will negatively affect staining results.
2. Fixation
Fix the whole brain in 4% PFA in PBS at 4°C overnight. The next day rinse the brains 3 times with PBS.
Cryoprotect the brains by placing them into 30% sucrose in PBS. The tissue will initially float. Keep at 4°C until the tissue sinks to the bottom. At that stage the cryoprotection is complete.
3. Embedding/Cryosectioning
Embed brains at desired orientation for sectioning (e.g. sagittal or coronal) in a 2:1 solution of 30% sucrose:OCT (Tissue-Tek, Cat. No: 4583) in PBS. Freeze embedded brains on a flat surface of dry ice. Frozen embedded brains can be placed in freezer bags and kept at -80 up to a year before sectioning.
Cryosection the tissue into 12-16μm sections and mount on glass slides (Sigma, Cat. No: S4651). Slides can be stored up to a week at -80°C prior to staining.
Slides to be stained should be dried at 37°C for 30 minutes and rinsed 1x with PBS to remove residual OCT.
4. Blocking Sections
Block sections in 20% normal goat serum (NGS) in PBS for one hour at room temperature. IMPORTANT: Do not include Triton X-100 at this stage. Prior to addition of the blocking solution, you may create a hydrophobic barrier around the sections on your slide using an Elite PAP Pen (DBS, Cat. No: K039)
5. Applying Primary Antibodies
Dilute your primary antibodies in PBS with 0.3% Triton and 10% NGS.
In brain sections we use PSD-95 (Zymed, Rabbit, 1:500) to label glutamatergic postsynaptic compartments and VGlut1 or VGlut2 (Chemicon, guinea pig, 1:2500) to label glutamatergic presynaptic terminals. Centrifuge primary antibody dilution for 5 minutes at maximum speed in a bench top centrifuge to remove precipitated antibody if present.
Incubate sections in primary antibody solution for 36-60 hours at 4°C.
6. Applying Secondary Antibodies
Wash the primary antibody off by immersing the slides 3x in PBS for 15 minutes each. After this step make sure to protect your slides from direct light.
Dilute secondary antibodies at a dilution of 1:200 in the same buffer as described for primary antibodies. For example for VGlut/PSD95 staining we use goat anti guinea-pig Alexa 488 (Vglut) and goat anti rabbit Alexa 594 (PSD-95) (Invitrogen).
Incubate sections in secondary antibody solution for 2 hours at room temperature in the dark.
Wash the unbound secondary antibodies off the slides by immersing the slides 4x in PBS for 15 minutes each.
7. Mounting
Add small drops of Vectashield mounting medium with DAPI (Vector Laboratories Inc.) onto glass slides (VWR scientific) and then cover the slides with coverslips (VWR Scientific, No. 1.5, 48393 241). Apply nail polish to inhibit movement of the coverslips.
8. Imaging
IMPORTANT: A confocal microscope with at least 3 channels is required for the imaging described here. We image on a Leica SP5 laser-scanning confocal microscope using a 63x oil immersion objective.
Synaptic brain regions of interest should be selected for scanning consistently between sections. For example, in our synapse assay of retinal ganglion cell synapses onto superior collicular neurons, we image the outer superior collicular region adjacent to the inferior colliculus which encompasses the synaptic layer that receives retinocollicular terminals7,8,9. We image and quantify the synapses in the upper 150 μm synaptic zone of the superior colliculus where RGCs are known to establish synaptic contacts9.
For each section, in both the 488 and 594 channels, we image serial optical sections at 0.33 μm intervals over a total depth of 5 μm for a total of 15 optical sections. At least 3 sections from each animal must be imaged and at least 3 animals must be included from a given experimental condition.
Maximum intensity projections (MIPs) are generated from groups of 3 consecutive sections yielding 5 MIPs representing 1μm of depth each. These MIPs are quantified.
9. Image Quantification
May be performed exactly as described for RGCs, however the shape and size of the ROI will change as a function of which region of the images you wish to quantify.
Keys to Success:
Purified RGCs:
Before fixing purified RGCs (or other dissociated culture) make sure to pre-warm 4% PFA in a water bath set to 37°C for ~20-25 minutes (store at 4°C at all other times, do not use PFA dilutions older than 7 days).
Particularly for cultured neurons, it is advisable to not use a vacuum aspirator during washes for all rinse steps. Gentler methods such as using a Pasteur pipette and a suction bulb noticeably improve the quality of your staining.
Take care not to let your cells dry out during your rinse steps.
Avoid nudging or moving your coverslips during application of nail polish as doing so can result in shearing of the cells on your coverslip.
Spin down all primary and secondary antibodies to eliminate any precipitated antibodies that will negatively impact staining.
Be sure to select primary antibodies for your pre- and postsynaptic markers raised in different species. Failure to do so will eliminate your ability to discriminate between the two signals after application of secondary antibodies.
Brain Sections:
Do not perfuse your mice with fixatives such as PFA.
Do not include Triton-X 100 in your blocking solution for brain sections. Doing so damages the quality of staining for presynaptic markers, particularly presynaptic markers that are associated with synaptic vesicles.
Use of a confocal microscope for imaging is required for image acquisition that is of good enough quality to perform a synapse analysis.
Representative Results:
The synapse assay described above is designed to capture changes in synaptic connectivity in vitro and in vivo. In our lab, we utilize the synapse assay to determine the effect of either individual or multiple astrocyte-secreted molecules on synapse formation. We commonly perform this synapse assay on purified RGCs that we culture in vitro.
A well described effect of chronic application of astrocyte-conditioned media (ACM) in purified RGC cultures is a multiple-fold increase in the number of synapses that are formed between RGCs3,4,5,6. Figure 1A and 1B show representative images of untransfected purified RGCs treated with either basal growth media or with ACM. After staining for excitatory pre- and postsynaptic markers, the ACM-induced increase in synapse formation is qualitatively evident (Figure 1A, 1B). Indeed, this finding has been corroborated by several studies that have used electrophysiology to show that ACM-induced synapses are functional and electron microscopy to show that the synapses are ultrastructurally normal3,4,6. Other work in our laboratory has identified the calcium channel subunit α2δ-1 as the neuronal receptor for a strongly synaptogenic astrocyte-secreted molecule, thrombospondin3. Here we include the results of an experiment in purified RGCs that was performed to determine whether increased levels of α2δ-1 further enhances ACM-induced synapse formation (Figure 3).
RGCs were cotransfected with either an empty vector or with a construct encoding α2δ-1 with a separate construct encoding tdTomato after 5 days in vitro (DIV5). Following chronic treatment with either ACM or GM, RGCs were fixed and stained on DIV 11. Following immunolabeling of pre- and postsynaptic markers of excitatory synapses we see a qualitatively obvious increase in synapse number in purified RGCs expressing either an empty vector (pcDNA3.1) or α2δ-1 (Fig 1C, 1D). We quantify the synaptogenic effect of ACM using Puncta analyzer to count synapse number (Fig 2) for at least 15 neurons from each condition. A sample of this size allows us to calculate the average number of synapses per neuron and find a statistically significant, ~10-fold increase in synapses formed by ACM-treated neurons that are expressing an empty vector (Fig 3, gray bars). Furthermore, we show that overexpression of α2δ-1 leads to a significant potentiation of ACM-induced synapse formation (~20-fold. Fig 3, black bars).
In addition to cultured neurons, we can quantify synaptic density in different brain regions using this technique. The superior colliculus is a brain structure that receives retinocollicular projections, originating from RGCs in the retina7,8,9. Over postnatal development, the number of excitatory synapses formed by RGCs onto their targets in the superior colliculus dramatically increases from P7 to P217,8,9.
Using our quantification technique, we show that the number of retinocollicular synapses formed in the superior colliculus increases from P7 to P14. To do this, we quantified synaptic density at P7 and again at P14. We stain the superior colliculus for PSD-95 (postsynaptic) and for VGlut2 (presynaptic marker specific to RGC synapses in the superior colliculus). The outer synaptic region of the superior colliculus was imaged (Fig 4A) and co-localized VGlut2/PSD-95 synapses were quantified (Fig 4B, 4C) from at least 3 sections per animal and from at least 3 animals per time point. Quantification of the resulting data clearly demonstrates an over three-fold significant increase in the number of synapses between P7 and P14. These results are in agreement with previously published findings utilizing electron microscopy (Fig 4D)9.
In conclusion, the method of synapse number quantification we describe here is a useful tool to determine synapse number and density in culture and in neural tissues, enabling us to study the effects of manipulating synapse formation in vitro or in vivo.
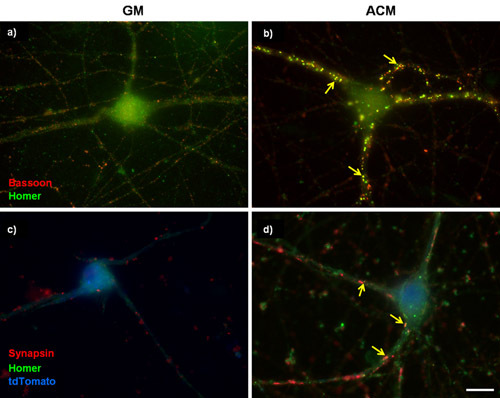 Figure 1: Representative synaptic staining in purified RGCs. (A and B) 3 days in vitro (DIV) RGCs were cultured either in (A) basal growth media (GM) or in (B) pro-synaptogenic mouse astrocyte conditioned media (ACM) for an additional 6 days. The cells were then labeled for bassoon (presynaptic, red) and homer (postsynaptic, green). Mouse ACM strongly stimulates synapse formation between RGCs as determined by the increase in the number of co-localized bassoon and homer puncta. (C and D) 5DIV RGCs were transfected with a plasmid to overexpress the calcium channel subunit α2δ-1. Transfected cells were identified with a tdTomato cell fill (blue) and have been stained for presynaptic marker synapsin and postsynaptic marker homer. Arrows indicate synapses. Scale bar represents 20 μm.
Figure 1: Representative synaptic staining in purified RGCs. (A and B) 3 days in vitro (DIV) RGCs were cultured either in (A) basal growth media (GM) or in (B) pro-synaptogenic mouse astrocyte conditioned media (ACM) for an additional 6 days. The cells were then labeled for bassoon (presynaptic, red) and homer (postsynaptic, green). Mouse ACM strongly stimulates synapse formation between RGCs as determined by the increase in the number of co-localized bassoon and homer puncta. (C and D) 5DIV RGCs were transfected with a plasmid to overexpress the calcium channel subunit α2δ-1. Transfected cells were identified with a tdTomato cell fill (blue) and have been stained for presynaptic marker synapsin and postsynaptic marker homer. Arrows indicate synapses. Scale bar represents 20 μm.
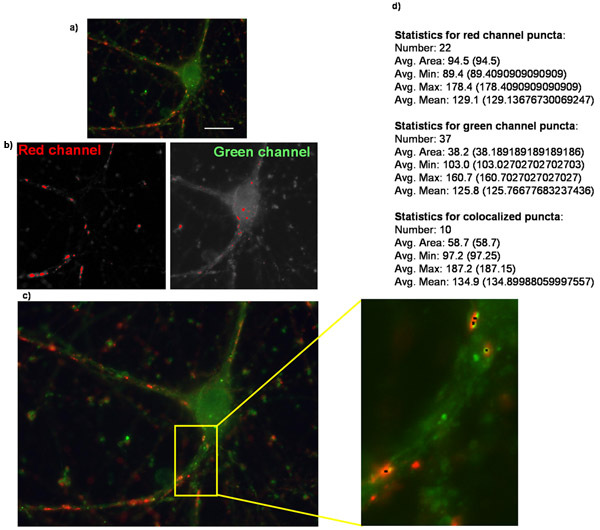 Figure 2: Quantification of synapse number using Puncta Analyzer. Shown here is an example of (a) an original image of a purified retinal ganglion cell overexpressing the thrombospondin receptor α2δ-1 and is stained for presynaptic marker synapsin and postsynaptic marker homer. (b) Images corresponding to the masks created in each channel when analyzing puncta in Puncta Analyzer. (c) Puncta Analzyer will create an image such as the one shown here in which puncta are indicated by small black dots (inset). Also shown is (d) the numerical output of the application where puncta number along with several other parameters are provided in textual/numerical form (bold text added for emphasis).
Figure 2: Quantification of synapse number using Puncta Analyzer. Shown here is an example of (a) an original image of a purified retinal ganglion cell overexpressing the thrombospondin receptor α2δ-1 and is stained for presynaptic marker synapsin and postsynaptic marker homer. (b) Images corresponding to the masks created in each channel when analyzing puncta in Puncta Analyzer. (c) Puncta Analzyer will create an image such as the one shown here in which puncta are indicated by small black dots (inset). Also shown is (d) the numerical output of the application where puncta number along with several other parameters are provided in textual/numerical form (bold text added for emphasis).
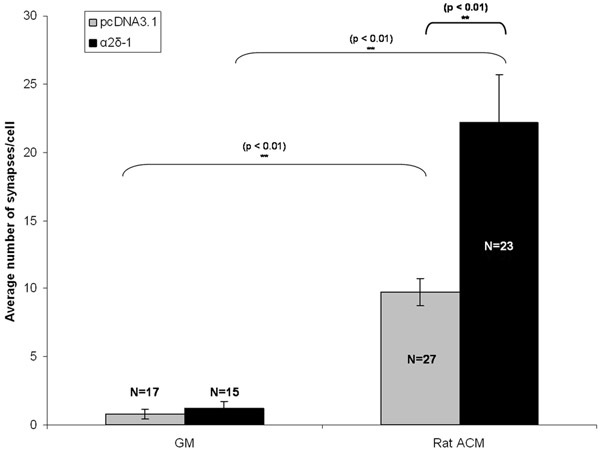 Figure 3: Representative result for synapse assay. Shown here is the synaptogenic effect of chronic treatment of RGCs with astrocyte-conditioned media (ACM) in transfected RGCs expressing either empty vector (pcDNA3.1, gray bars) or the thrombospondin receptor α2δ-1. Error bars represent the SEM. GM = Growth Media. ACM = (Rat) Astrocyte Contitioned Media.
Figure 3: Representative result for synapse assay. Shown here is the synaptogenic effect of chronic treatment of RGCs with astrocyte-conditioned media (ACM) in transfected RGCs expressing either empty vector (pcDNA3.1, gray bars) or the thrombospondin receptor α2δ-1. Error bars represent the SEM. GM = Growth Media. ACM = (Rat) Astrocyte Contitioned Media.
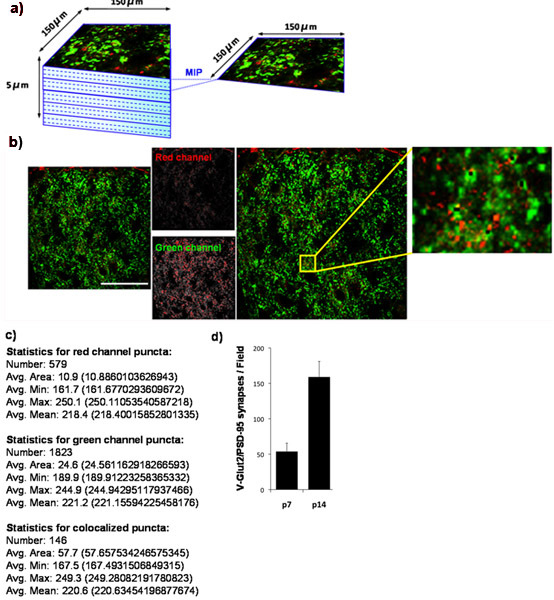 Figure 4: Quantification of synaptic density in mouse superior colliculus. (a) To determine the developmental changes in the number of glutamatergic synapses established by RGCs onto its superior collicular targets in the rodent brain, we stained cryosections from mouse brain with antibodies against the presynaptic marker VGlut2 (green) and the postsynaptic marker, PSD-95, (red). We imaged the outer 150 x 150 μm region of the mouse superior colliculus (SC) corresponding to the synaptic target region for RGCs by using a laser scanning confocal microscope. A z-stack for each SC section was collected for a total depth of 5 μm (15 x 0.33 μm optical sections). Maximum image projections (MIPs) were generated for groups of 3 consecutive optical sections yielding 5 MIPs/section each representing 1 μm of depth. Shown below is a representative MIP taken from superior colliculus of a P14 WT mouse. The presynaptic marker, VGlut2, is shown in green and the postsynaptic marker, PSD-95, is shown in red. (c) Numerical output produced by Puncta Analyzer. (d) Quantification of the analysis of synapse number for at least 3 sections per animal with 3 animals per time point (error bars represent SEM). Scale bar represents 50 μm.
Figure 4: Quantification of synaptic density in mouse superior colliculus. (a) To determine the developmental changes in the number of glutamatergic synapses established by RGCs onto its superior collicular targets in the rodent brain, we stained cryosections from mouse brain with antibodies against the presynaptic marker VGlut2 (green) and the postsynaptic marker, PSD-95, (red). We imaged the outer 150 x 150 μm region of the mouse superior colliculus (SC) corresponding to the synaptic target region for RGCs by using a laser scanning confocal microscope. A z-stack for each SC section was collected for a total depth of 5 μm (15 x 0.33 μm optical sections). Maximum image projections (MIPs) were generated for groups of 3 consecutive optical sections yielding 5 MIPs/section each representing 1 μm of depth. Shown below is a representative MIP taken from superior colliculus of a P14 WT mouse. The presynaptic marker, VGlut2, is shown in green and the postsynaptic marker, PSD-95, is shown in red. (c) Numerical output produced by Puncta Analyzer. (d) Quantification of the analysis of synapse number for at least 3 sections per animal with 3 animals per time point (error bars represent SEM). Scale bar represents 50 μm.
Discussion
The synapse assay described above is based in the context of our experimental goals, wherein we focus largely on excitatory projections of RGCs, either in purified culture or in brain section. We have provided a reference table listing antibodies that work well for labeling excitatory synapses (Table 1).
This synapse assay can be adapted to quantify synapse number of any neuronal population or any other synaptic subtype for which there is a selective pre- and postsynaptic marker. For example, the synapse assay described here can be applied to studying GABA-ergic (as opposed to glutamatergic) synapses by using appropriate pre- and postsynaptic markers10,11.
This synapse assay is an important tool for labs investigating changes in synaptic connectivity between neurons. While it is necessary to corroborate findings of this assay with electron microscopy and electrophysiology, it nevertheless offers a relatively easy way to answer important questions about the molecular cues that modulate a neurons' ability to form synapses.
Disclosures
No conflicts of interest declared.
Acknowledgments
Puncta Analyzer Plug-in for Image J was written by Barry Wark (current address: Physion Consulting) in the lab of Ben A. Barres (Stanford University).
Funding;
Alfred P. Sloan Foundation
The Esther A. and Joseph Klingenstein Fund, Inc.
Broad Biomedical Research Foundation
Cure for Huntington s Disease Initiative
References
- Barres BA, Silverstein BE, Corey DP, Chun LLY. Immunological, morphological, and electrophysiological variation among retinal ganglion cells purified by panning. Neuron. 1988;1:791–803. doi: 10.1016/0896-6273(88)90127-4. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- Eroglu C. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science. 277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. 2001;291:657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- Christopherson KS. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Godement P, Salaun J, Imbert M. Prenatal and postnatal development of retinocollicular projections in the mouse. J. Comp. Neurol. 1985;230:552–575. doi: 10.1002/cne.902300406. [DOI] [PubMed] [Google Scholar]
- Schmidt JT. Formation of retinotopic connections: selective stabilization by an activity-dependent mechanism. Cell. Mol. Neurobiol. 1985;5:65–84. doi: 10.1007/BF00711086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs GM, Jacobson M, Caviness VS. Postnatal changes in arborization patterns of murine retinocollicular axons. J. Comp. Neurol. 1986;246:395–408. doi: 10.1002/cne.902460308. [DOI] [PubMed] [Google Scholar]
- Elmariah SB, Oh EJ, Hughes EG, Balice-Gordon RJ. Astrocytes regulate inhibitory synapse formation via Trk-mediated modulation of postsynaptic GABAA receptors. J. Neurosci. 2005;25:3638–3650. doi: 10.1523/JNEUROSCI.3980-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes EG, Elmariah SB, Balice-Gordon RJ. Astrocyte secreted proteins selectively increase hippocampal GABAergic length, branching and synaptogenesis. Moll. Cell. Neurosci. 2010;43:136–145. doi: 10.1016/j.mcn.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]