

Abstract
The Drosophila embryo is an attractive model system for investigating the cellular and molecular basis of neuronal development. Here we describe the procedure for the visualization of Drosophila embryonic nervous system using antibodies to neuronal proteins. Since the entire embryonic peripheral nervous and central nervous systems are well characterized at the level of individual cells (Dambly-Chaudière et al., 1986; Bodmer et al., 1987; Bodmer et al., 1989), any aberrations to these systems can be easily identified using antibodies to different neuronal proteins. The developing embryos are collected at certain times to ensure that the embryos are in the proper developmental stages for visualization. After collection, the outer layers of the embryo, the chorion membrane and the vitelline envelope that surrounds the embryo, are removed before fixation. Embryos are then incubated with neuronal antibodies and visualized using fluorescently labeled secondary antibodies. Embryos at stages 12-17 are visualized to access the embryonic nervous system. At stage 12 the CNS germ band starts shortening and by stage 15 the definitive pattern of the commissure has been achieved. By stage 17 the CNS contracts and the PNS is fully developed (Campos-Ortega et al. 1985). Thus changes in the pattern of the PNS and CNS can be easily observed during these developmental stages.
Protocol
1. Preparation of Reagents
Prepare the PEMFA Buffer using 100mM Pipes, 2mM EGTA, and 1 mM MgSO4. Adjust the pH of the buffer to 7.0. The PEMFA buffer is filter sterilized and stored at 4°C.
Prepare PBT by adding 1ml of Triton X-100 to 100ml 1x PBS (Phosphate Buffered Saline) (pH 7.0). Store at 4°C.
Prepare 5% BSA (bovine serum albumin) with 0.01% NaAcide in 1x PBS (pH 7.0).
Prepare the saline solution using 0.7% NaCl and 0.03% Triton X-100 in deionized water.
Prepare the fixative by combining 4 parts PEMFA buffer with 1 part 37% Formaldehyde.
2. Collection of Embryos
Prepare a cage for the Drosophila strain of interest by placing >100 flies in a plastic bottle containing an adaptor with agar feeding plate.
Leave the cage of flies in the dark at room temperature for 72 hours, changing plates every 8-12 hours, so that the flies get acclimated to the cage and do not hold their eggs.
For the experiment collect embryos for 12 hours. Remove the Petri dish from the container and replace it. Take the removed Petri dish and wash the embryos off the dish using the saline solution and a clean paint brush.
Collect the embryos in a fine sieve. Rinse with saline to remove any excess yeast.
Using a spatula gently pick up the embryos and place them in a small glass vial with a cap. Add about 1ml of saline solution.
3. Removal of the Chorion and Vitelline Membranes and Fixation
Following the collection of embryos, prepare the Heptane-Fix solution by combining 50% heptane and 50% fixative. The solution will be layered with heptane on top and the fixative on the bottom.
In the glass vial wash the embryos by incubating them in the saline solution for 5 minutes. Remove excess saline using a Pasteur pipette.
Rinse the embryos with deionized water.
Dechorionate the embryos by incubating them in 50% bleach for 3 minutes.
Rinse the embryos thoroughly with deionized water.
Fix the embryos by incubating them for 30 minutes in the Heptane-Fix solution with gentle shaking.
Using a Pasteur pipette replace the fixative (bottom layer) with methanol leaving the heptane in the tube. Shake vigorously to remove the vitelline membrane. The devitellinized embryos will sink to the bottom of the tube.
Collect the embryos in a new glass vial using a Pasteur pipette. Rinse the embryos five times with methanol, each time incubate the embryos for 5 minutes.
4. Antibody Staining, Mounting, and Visualization
Rehydrate the embryos in a mixture of 50% PBT solution and 50% methanol for 5 minutes. Then rehydrate in 100% PBT solution for 5 minutes.
Block the embryos by incubating them in 5% BSA with 0.01% NaAcide solution for 1 hour at 4°C.
Embryos are now ready for antibody staining. Prepare the primary antibody by diluting it to the appropriate concentration using 5%BSA with 0.01% NaAcide solution. We use an antibody against the cysteine string protein (CSP), a vesicular synaptic protein and an anti-HRP or horse radish peroxidase antibody, which recognizes a neuronal specific-carbohydrate moiety present in the surface glycoproteins and labels all neurons.
Incubate the embryos overnight at 4°C in the primary antibody solution.
The following day, wash the embryos 10 times with PBT over the course of an hour.
Prepare the secondary antibody solution by diluting the appropriate concentration. We use Alexa secondary antibodies at a concentration of 1:200.
Incubate the embryos for two hours in 4°C in the secondary antibody solution.
Wash the embryos 10 times with PBT over the course of an hour.
Incubate the embryos overnight in Vectashield Mounting Medium.
Mount the embryos on a glass slide and seal the cover slip with nail polish. Visualize embryos using a fluorescence microscope.
5. Representative Results
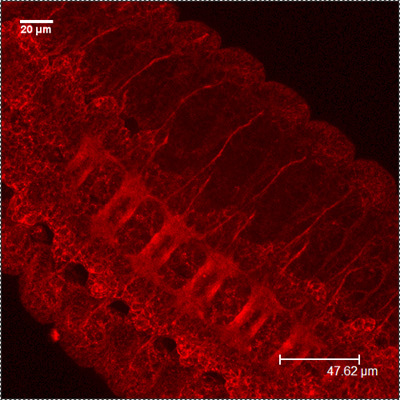 CSP
CSP HRP-FITC
HRP-FITC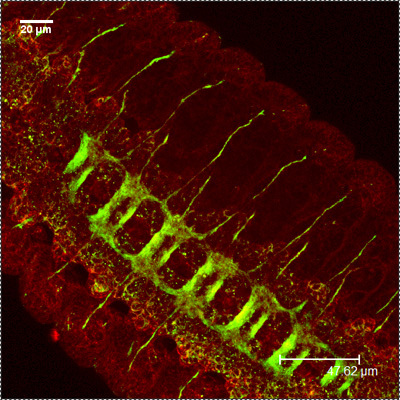 MergedFigure 1: A Representative images showing the embryonic CNS at stage 16-17, using antibodies against CSP (red) and HRP (green). Note the distinct staining of the commissure.
MergedFigure 1: A Representative images showing the embryonic CNS at stage 16-17, using antibodies against CSP (red) and HRP (green). Note the distinct staining of the commissure.
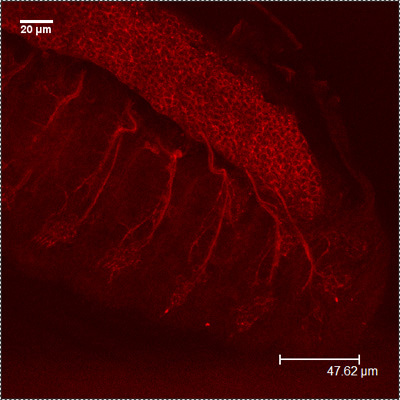 CSP
CSP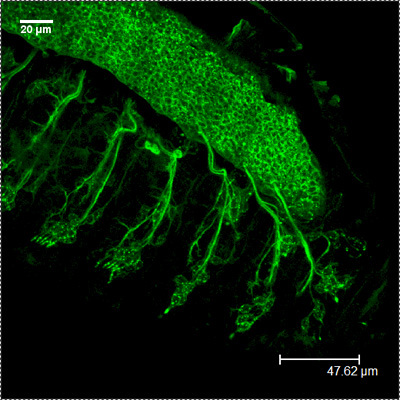 HRP-FITC
HRP-FITC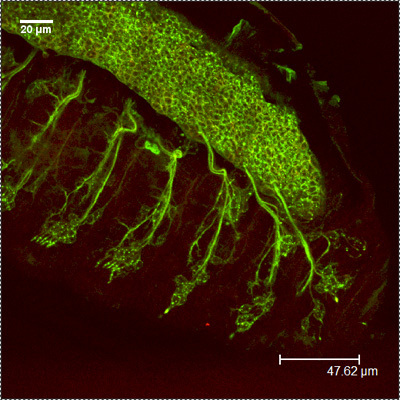 MergedFigure 2: A representative images showing the embryonic PNS at stage 16-17 using antibodies against CSP (red) and HRP (green). Note the repeated patterns of peripheral neurons.
MergedFigure 2: A representative images showing the embryonic PNS at stage 16-17 using antibodies against CSP (red) and HRP (green). Note the repeated patterns of peripheral neurons.
Discussion
The Drosophila embryo is a powerful system for investigating the cellular and molecular changes during neuronal development. In this protocol we have demonstrated how to prepare whole mount embryos for immunohistochemistry. We have adapted this protocol from the protocol described in Carroll and Scott, 1985. This protocol can also be adapted to test other neuronal antibodies, but optimization of antibodies should be done. Further, developmental defects of the embryonic PNS and CNS can be easily visualized by comparing embryos from different mutant lines. We have successfully used this protocol to visualize the PNS and CNS in embryos carrying motor protein mutants. Under high magnification we have also evaluated the axonal tracks within the embryonic PNS and CNS.
Disclosures
No conflicts of interest declared.
Acknowledgments
SG is supported by funds from the State University of New York at Buffalo and from John R. Oishei Foundation.
References
- Bodmer R, Barbel S, Sheperd S, Jack JW, Jan LY, Jan YN. Transformation of sensory organs by mutations of the cut locus of D. melanogaster. Cell. 1987;51:293–307. doi: 10.1016/0092-8674(87)90156-5. [DOI] [PubMed] [Google Scholar]
- Bodmer R, Carretto R, Jan YN. Neurogenesis of the peripheral nervous system in Drosophila embryos: DNA replication patterns and cell lineages. Neuron. 1989;3:21–32. doi: 10.1016/0896-6273(89)90112-8. [DOI] [PubMed] [Google Scholar]
- Campos-Ortega JA, Hartenstein V. The Embryonic Development of Drosophila melanogaster. New York: Springer-Verlag; 1985. [Google Scholar]
- Carroll SB, Scott MP. Localization of the fushi tarazu protein during Drosophila embryogenesis. Cell. 1985;43:47–57. doi: 10.1016/0092-8674(85)90011-x. [DOI] [PubMed] [Google Scholar]
- Dambly-Chaudière C, Ghysen A, Jan YN, Jan LY. Muscle connections between imaginal discs in Drosophila. Developmental Biology. 1986;113:288–294. doi: 10.1016/0012-1606(86)90164-8. [DOI] [PubMed] [Google Scholar]