

Abstract
The regulation of cellular adhesion to the extracellular matrix (ECM) is essential for cell migration and ECM remodeling. Focal adhesions are macromolecular assemblies that couple the contractile F-actin cytoskeleton to the ECM. This connection allows for the transmission of intracellular mechanical forces across the cell membrane to the underlying substrate. Recent work has shown the mechanical properties of the ECM regulate focal adhesion and F-actin morphology as well as numerous physiological processes, including cell differentiation, division, proliferation and migration. Thus, the use of cell culture substrates has become an increasingly prevalent method to precisely control and modulate ECM mechanical properties.
To quantify traction forces at focal adhesions in an adherent cell, compliant substrates are used in conjunction with high-resolution imaging and computational techniques in a method termed traction force microscopy (TFM). This technique relies on measurements of the local magnitude and direction of substrate deformations induced by cellular contraction. In combination with high-resolution fluorescence microscopy of fluorescently tagged proteins, it is possible to correlate cytoskeletal organization and remodeling with traction forces.
Here we present a detailed experimental protocol for the preparation of two-dimensional, compliant matrices for the purpose of creating a cell culture substrate with a well-characterized, tunable mechanical stiffness, which is suitable for measuring cellular contraction. These protocols include the fabrication of polyacrylamide hydrogels, coating of ECM proteins on such gels, plating cells on gels, and high-resolution confocal microscopy using a perfusion chamber. Additionally, we provide a representative sample of data demonstrating location and magnitude of cellular forces using cited TFM protocols.
Protocol
1. Activating the coverslip surface
Coverslips (#1.5, 22x40 mm) are cleaned using a series of soap and ethanol washes in a previously described protocol (Waterman-Storer, 1998) to clean and remove dust.
Place coverslips in a stainless steel holder rack, such that coverslips are spaced apart and not touching.
In chemical fume hood (nitrile gloves and goggles recommended), dilute full strength 3-aminopropyltrimethoxysilane in isopropanol for a final concentration of 2% (2ml silane /100 ml isopropanol) to fill a square glass dish (~350ml volume). Due to reactivity with plastic, use a glass Pasteur pipette to apply 3-aminopropyltrimethoxysilane to the isopropanol.
Fully immerse coverslips from 1.2 into this solution for 10 min while gently stirring on a stir plate in the fume hood.
Wash coverslips by immersing in ddH2O (4 exchanges of water). Allow 10 min soaking time for the final exchange, with stirring. Amino-silane containing solutions should be disposed as hazardous waste.
Dry coverslips in incubator at warm temperature (~37°C) for 10 minutes in a dust free environment.
Cool to room temperature.
In the fume hood, immerse coverslips in 1% glutaraldehyde solution in ddH2O in a glass square dish on stir plate for 30 minutes.
Wash by 3 exchanges of ddH2O for 10 minutes per exchange, with stirring. Dispose of glutaraldehyde as hazardous waste.
Dry at room temperature, covering with aluminum foil to avoid dust from sticking to the coverslips.
Store in a dry place, away from dust, for up to 2 months.
2. Preparation of polyacrylamide (PAA) gel
Prepare stock solutions of acrylamide/bis-acrylamide mix from 40% acrylamide and 2% bis-acrylamide, following Table 1. We maintain several stock solutions that are optimized for PAA gels of different stiffness; examples are listed in Table 1 and 2. Stock solutions can be kept for several years, as long as they are maintained in a darkened bottle at 4 C.
Working solutions containing the final desired concentrations of acrylamide/bis-acrylamide are obtained from stock solutions. For example, we prepare a working solution of 7.5% acrylamide/0.10% bis-acrylamide in ddH2O for making 2.8kPa PAA gels.
Degas acrylamide solution in a vacuum chamber for 20 min, to reduce oxygen within the solution which prevents PAA polymerization.
Prepare 10% ammonium persulfate (APS) solution (0.5g/5mL). Use fresh working stock within 3 days. Alternatively, stocks can be frozen to be used at later dates.
While acrylamide is degassing, wipe a 1x3" microscope glass slide with Rain-X wipes vigorously to make glass slide surface hydrophobic. To remove excess Rain-X, wipe glass slide with damp Kimwipe. Set glass slide aside, covered.
Remove acrylamide solution from vacuum chamber and add fluorescent beads (1% by volume, 5 μl for the working solution listed in Table 1). Add 0.75μl TEMED and 2.5 μl 10% APS, which will initiate gel polymerization. Mix well by pipetting for ~5 sec, to minimize the introduction of bubbles,
Apply 10-12 μl of the acrylamide solution to hydrophobic microscope slide (prepared in step 2.4) and place activated 22x40mm coverslip on top of the droplet. Gel solution should coat entire coverslip. Smooth out any bubbles that may appear within the solution. Allow the gel solution to polymerize at room temperature for ~10 min.
The completion of polymerization can be assessed by inverting remaining working solution in microcentrifuge tube. Also, the polymerized gel may pull away from coverslip edges. Immediately after macroscopic polymerization is observed, separate the coverslip from the glass slide. Using the fine tip of a pair of tweezers or a razor blade edge, carefully remove coverslip, with gel attached, from microscope slide surface and immerse gel in ddH2O, to maintain hydration.
3. Coupling extracellular matrix (ECM) proteins to the PAA gel
Three distinct methods can be used to attach ECM protein either to the top surface of the PAA gel (3.1 and 3.2) or incorporating ECM protein within the gel volume (3.3). Here, we discuss the coupling of fibronectin to PAA gels to result in a surface ligand density that is equivalent to the amount adsorbed on glass after incubation with 10 μg/mL fibronectin solution for 1 hour. Considerations for choosing a method are detailed in the discussion.
- Cross-linking ECM protein to PAA gel surface by Sulfo-SANPAH Reactive amines on proteins are covalently attached to the PAA gel surface by the heterobifunctional cross-linker Sulfo-SANPAH
- Prepare 40 μl working aliquots of Sulfo-SANPAH by dissolving Sulfo-SANPAH powder in anhydrous dimethyl sulfoxide (DMSO) (20 μl per mg of Sulfo-SANPAH). Flash freeze stocks in liquid nitrogen and store at -80°C for later use.
- Remove ddH2O from gel surface using a coverslip spinner (< 2sec). Avoid drying the gel.
- Dilute Sulfo-SANPAH-DMSO aliquots in ddH2O (2mg/ml, pH 7) immediately before use and coat gel surface (~200 μl). Note that the reactivity half-life of Sulfo-SANPAH is short (~5 min) at room temperature in water; therefore, these steps should be done at a rapid pace.
- Expose gel surface to UV light in a UV cross-linker oven (8W, 254 nm wavelength at a distance of 2-3 inches for 1.5 min). Sulfo-SANPAH will change in color from orange to brown.
- Dip UV-treated coverslips in a beaker with fresh ddH2O and remove excess water from gel surface using a coverslip spinner (<2sec).
- Pipette ~50 μL of cold 1mg/ml Fibronectin (FN) (in PBS, pH 7.4) on Parafilm in Petri dish container. Invert coverslip on top of FN drop, gel side exposed to FN.
- React at room temperature for 1-2 hours or at 4°C overnight.
- Place coverslips in 6 cm tissue culture dishes containing PBS (pH 7.4), enough to cover coverslip, under sterile conditions in tissue culture hood.
- Wash extensively with several washes (3-5) of PBS (pH 7.4), under sterile conditions.
- Sterilize the coverslips by use of germicidal lamp in tissue culture hood for 30 min.
- Incubate coverslips in cell media for 30-45 min prior to plating cells.
- Cross-linking ECM to the PAA gel surface by hydrazine hydrate Carbohydrate groups on proteins are oxidized and coupled to the gel using hydrazine hydrate.
- Prepare polyacrylamide gels as described in section 2.
- Place PAA gel coverslips in plastic Petri dish in a fume hood and using gloves pipette approximately 1 ml of undiluted hydrazine hydrate onto the surface of each PAA gel and incubate for at least two hours, but no longer than 24 hours
- Add ddH2O to the Petri dish; Remove hydrazine hydrate solution and dispose as hazardous waste.
- Add 5% acetic acid to the Petri dish to immerse coverslip. Cover and incubate for one hour.
- Remove the acetic acid and wash with ddH2O. Incubate in ddH2O for one hour. The coverslips are now activated and ready to cross-link oxidized Fibronectin (FN).
- Dilute 10 μl of 1 mg/ml FN solution in 940 μl of 50 mM sodium acetate buffer (pH 4.5) in a dark micro centrifuge tube, making a final concentration of 10 μg/ml.
- Make stock of 20X sodium meta-periodate by adding 80 mg of sodium meta-periodate to 1 ml of 50 mM sodium acetate buffer (pH 4.5).
- Add 50 μl of 20X sodium meta-periodate stock to the FN solution prepared in 3.2.6, such that final working concentration are 10 μg/ml FN and 4 μg/ml sodium meta-periodate. Incubate in the dark tube at room temperature for 30 minutes.
- Remove excess ddH2O from activated gel surface prepared in 3.2.5 using a coverslip spinner (< 2 sec). Avoid drying the gel.
- Pipet ~500 μl of FN solution onto activated gel surface and incubate for 1 hr at room temperature.
- Place coverslips in dishes containing PBS (pH 7.4), enough to cover coverslip.
- Wash extensively with several washes (3-5) of PBS (pH 7.4).
- Sterilize the coverslips by use of germicidal lamp in tissue culture hood for 30 min.
- Incubate coverslips in cell media for 30-45 min prior to plating cells.
- Bulk conjugation of ECM protein in PAA gel by Acryloyl-X, succinimidyl ester This protocol is done prior to 2.5. Reactive amines on proteins are coupled to an acrylamide monomer with NHS ester chemistry and then co-polymerized into the bulk of the PAA gel.
- Conjugate ECM protein of choice to Acryloyl-X per the manufacturer's instructions. Stock solutions of conjugated protein should be stored at 4°C.
- Calculate the volume of PAA working solution required for gel fabrication (e.g. 10 uL per coverslip).
- Subtract 50 μL (e.g. 10% volume) of water from amount listed in the working solution recipe in Table 1 and initiate polymerization.
- Remove volume calculated in 3.3.2 and add ECM/Acryloyl-X solution to 10% volume. (e.g. 1 μL of ECM/Acryloyl-X to 9 μL of PAA solution). This step should be performed rapidly, as gel is polymerizing.
- Complete steps 2.6 and 2.7 as described previously.
- Wash extensively with several washes (3-5) of PBS (pH 7.4), under sterile conditions.
- Sterilize the coverslips by use of germicidal lamp in tissue culture hood for 30 min.
- Incubate coverslips in cell media for 30-45 min prior to plating cells.
4. Loading coverslip into confocal imaging chamber
These steps are conducted after cells have been allowed to spread on ECM-coated gel substrate (~6-12 hrs). To assemble the confocal imaging chamber (RC-30WA), it is useful to consult the Warner Instruments website for guidance.
Warm cell media and 0.5% or 0.25% trypsin and load into a 5ml or 10ml syringe.
Load a 22x30mm coverslip onto the Top Coverslip holder of a Warner Instruments confocal imaging chamber (RC-30WA) using vacuum grease to keep the coverslip in place.
Place a chamber forming rubber gasket on top of the coverslip, allowing access to both inlet and outlet polyethylene tubing. This will allow a spacing of 150-1000 μm between the top coverslip and the gel-coated 22x40mm coverslip, depending on the size of gasket used.
Load syringes onto inlet tubing, via connector kit, and check that media flows through tubing and onto Top Coverslip and no bubbles are apparent in the flow lines.
Apply vacuum grease onto base of chamber and load gel-coated 22x40mm coverslip, cell-side up. Apply warm media to cells.
Place Top Coverslip holder onto chamber base, with Chamber Gasket separating it from the gel-coated coverslip. Make sure that the Locating Pins within the chamber base sit within the Locating Holes in the Top Coverslip.
Apply the Pressure Plate to the chamber base and use the Pressure Plate Wrench to screw in and secure the Pressure Plate.
Check the flow of media through the tubing and chamber to monitor any potential leaks within the chamber and to eliminate any media-free zones on the cell surface. Note: using a small gauge needle to draw a slight vacuum while infusing with media can help eliminate media-free zones.
Apply the confocal imaging chamber to the Stage Adapter situated within a microscope holder for imaging.
Image fluorescently-labeled protein and fluorescent beads embedded within the gel substrate on a confocal fluorescence microscope.
To obtain an image of unstrained bead positions within the gel, perfuse trypsin to detach cellular adhesions from the gel, and take an image of the fluorescent beads in the same imaging field where the cell adhered. Comparison of strained and unstrained bead positions allows for the quantification of gel substrate displacement under contraction.
Representative Results:
The above protocol describes the experimental procedure for preparing compliant PAA gels for studying cell contractility and is illustrated in Figure 1. The gel surface obtained with this protocol is relatively flat and smooth, with fluorescent beads embedded evenly throughout (Figure 2A).
If measuring gel contraction at the location of focal adhesions, imaging of the cell (Figure 3A) and gel surface (Figure 3B) should be done at the confocal optical plane of focal adhesions. The contraction of a gel can be visualized by displacement of embedded fluorescent beads (Figure 3B) at the gel surface when cells are adherent (strained) versus detached (unstrained). The use of computational algorithms can yield traction stresses associated with bead displacement and corresponding elastic modulus of the gel (Figure 3C and 3D) (Sabass et al., 2008). If imaging takes place deeper within the gel, then bead displacements will be smaller and not representative of traction forces exerted at focal adhesions.
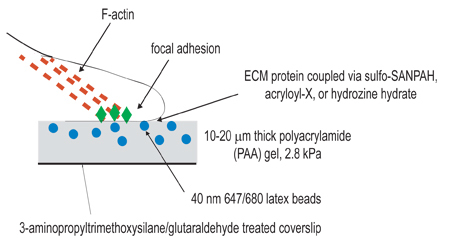 Figure 1. Schematic illustration of experimental setup. The overall goal of this procedure is to create compliant matrices for the purpose of studying cellular contraction. The first step of the experimental procedure is to activate coverslips by amino-silane/glutaraldehyde treatment for the purpose of anchoring polymerized gels. The second step is to polymerize a polyacrylamide gel, containing fluorescent beads, onto the activated coverslip. The third step involves the chemical cross-linking of extracellular ligand to the surface of the polyacrylamide gel, using one of the three coupling techniques listed in step 3. Cells are then plated onto the gel and allowed to adhere and spread. Under active cellular contraction, beads embedded in the gel displace.
Figure 1. Schematic illustration of experimental setup. The overall goal of this procedure is to create compliant matrices for the purpose of studying cellular contraction. The first step of the experimental procedure is to activate coverslips by amino-silane/glutaraldehyde treatment for the purpose of anchoring polymerized gels. The second step is to polymerize a polyacrylamide gel, containing fluorescent beads, onto the activated coverslip. The third step involves the chemical cross-linking of extracellular ligand to the surface of the polyacrylamide gel, using one of the three coupling techniques listed in step 3. Cells are then plated onto the gel and allowed to adhere and spread. Under active cellular contraction, beads embedded in the gel displace.
 Figure 2. Optical confocal slice of top surface of PAA gel, as visualized by (A.) fluorescent 40nm beads embedded within gel and (B.) fibronectin immunofluorescence.
Figure 2. Optical confocal slice of top surface of PAA gel, as visualized by (A.) fluorescent 40nm beads embedded within gel and (B.) fibronectin immunofluorescence.
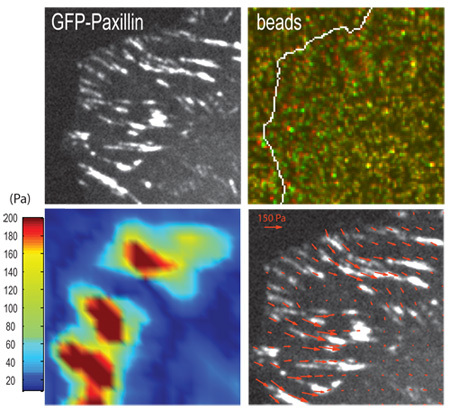 Figure 3. Representative result for a traction force experiment. (A.) Focal adhesions in a human osteosarcoma U2OS cell are marked by GFP-paxillin and (B.) positions of fluorescent beads embedded in the PAA gel underlying focal adhesions in the strained (green) and unstrained (red) states. Arrows indicate examples of bead displacement. (C.) Traction stress vectors and (D.) corresponding heat-scale map of traction stresses derived from the contraction of the gel, using computational algorithms (Sabass et al., 2008). Scale bar = 5 μm.
Figure 3. Representative result for a traction force experiment. (A.) Focal adhesions in a human osteosarcoma U2OS cell are marked by GFP-paxillin and (B.) positions of fluorescent beads embedded in the PAA gel underlying focal adhesions in the strained (green) and unstrained (red) states. Arrows indicate examples of bead displacement. (C.) Traction stress vectors and (D.) corresponding heat-scale map of traction stresses derived from the contraction of the gel, using computational algorithms (Sabass et al., 2008). Scale bar = 5 μm.
Table 1:
Example Stock and Working PAA Solutions (Data in table 1 was first obtained from Yeung et. al. and independently confirmed in our laboratory.)
| Stock PAA Solution | ||||
| Shear Modulus of PAA Gel (Pa) | 230 | 2833 | 8640 | 16344 |
| 40% Acrylamide (mL) | 1.25 | 3.12 | 2.34 | 2.50 |
| 2% Bis-Acrylamide (mL) | 0.50 | 0.83 | 1.88 | 0.60 |
| Water (mL) | 3.25 | 1.04 | 0.78 | 1. 90 |
| Total Volume (mL): | 5 | 5 | 5 | 5 |
| Working PAA Solution | ||||
| Stock Solution Used (Pa) | 230 | 2833 | 8640 | 16344 |
| Stock Solution Volume (μL) | 150 | 150 | 200 | 300 |
| Water (μL) | 341.75 | 341.75 | 291.75 | 191.75 |
| Beads (μL) | 5 | 5 | 5 | 5 |
| TEMED (μL) | 0.75 | 0.75 | 0.75 | 0.75 |
| 10% APS (μL) | 2.5 | 2.5 | 2.5 | 2.5 |
| Total Volume (μL): | 500 | 500 | 500 | 500 |
| Final Acrylamide % | 3 | 7.5 | 7.5 | 12 |
| Final Bis-Acrylamide % | 0.06 | 0.1 | 0.3 | 0.15 |
Table 2:
Shear Modulus of PAA substrates of various final acrylamide and bis-acrylamide percentages
| 12% Acrylamide | 7.5% Acrylamide | |||
| % Bis-Acrylamide | Shear Modulus (Pa) | % Bis-Acrylamide | Shear Modulus (Pa) | |
| 0.145 | 16344 | 0.01 | 689 | |
| 0.28 | 30067 | 0.03 | 1535 | |
| 0.45 | 34263 | 0.05 | 2286 | |
| 0.55 | 42375 | 0.075 | 2833 | |
| 0.575 | 50873 | 0.1 | 4069 | |
| 0.6 | 55293 | 0.2 | 5356 | |
| 0.3 | 8640 | |||
| 5% Acrylamide | 3% Acrylamide | |||
| % Bis-Acrylamide | Shear Modulus (Pa) | % Bis-Acrylamide | Shear Modulus (Pa) | |
| 0.05 | 430 | 0.02 | 1.3 | |
| 0.075 | 600 | 0.04 | 54 | |
| 0.1 | 1431 |
Discussion
The procedure described here for the setup of a traction force microscopy (TFM) experiment, along with the implementation of computational tracking routines (Sabass et al., 2008), allows for the quantification of cellular forces with micron-scale spatial resolution. To optimize the experimental protocol, it is critical to form a pure and uniform gel substrate with uniform coating of ECM ligand. We discuss potential pitfalls below:
Non-uniform Gel Surface or Tears:
In our experience, there are several steps that appear critical to forming a nice uniform gel. If your gel surface appears to have dark holes in it, it is likely that excess Rain-X droplets were not completely removed from the glass slide, forming an uneven hydrophobic surface for your gel to polymerize against. Also, we have found that for extremely soft gels (<100 Pa), the gel surface becomes highly uneven due to constrained swelling of the adherent PAA gel on the coverslip surface; one way to minimize this effect is to replace water with an appropriate buffer such that osmotic pressure is maintained from gel polymerization to imaging.
Rips or tears in the gel surface can arise from several factors. If gel polymerization is not completed prior to disassembly of the coverslip-slide sandwich, this can lead to large ruptures or tears. Too much adhesion to the glass slide (i.e. elimination of the Rain-X coating) or too weak adhesion to the coverslip surface (i.e. problems with activation step of coverslip surface or keeping activated coverslips in a dusty environment) can also lead to large tears or rips in the surface. Finally, if the PAA gel becomes dehydrated while in the coverslip/slide sandwich, tears are more likely to occur and cracks can be visualized across the gel surface. These problems become more prevalent when working with gels with <1 kPa stiffness. Tears or Rips can often be identified by low magnification (10-20x) phase contrast imaging of the gel (i.e. on a tissue culture microscope) prior to ECM coupling.
Choice of ECM Coupling Method:
Bulk incorporation of ECM-conjugated to Acryloyl -X is, by far, the easiest method to execute (Reinhart-King et al., 2005). We have confirmed that incorporation ECM/Acryoyl-X at 10% volume fraction does not impact PAA gel stiffness. Two benefits of this technique are: (1) as described here, it uses the least amount of ECM protein so it is optimal for situations where the ECM protein is expensive and (2) the bulk conjugation also facilitates the fabrication of gels with very large surface area, which we have used to coat large glass slides with a PAA gel for Western Blotting or RT-PCR experiments. Two limitations with this technique are that the changes in total amount of surface-available ligand (we have found surface density of ligand to saturate at 10% volume fraction) and that some proteins, like collagen, do not incorporate into the gel in a homogeneous manner due to their tendency to spontaneously polymerize.
Coupling protein on the PAA gel surface using hydrazine hydrate provides the largest range in ligand density, but is also the most complicated to execute. We have found that the surface density of fibronectin can be altered significantly by changing the concentration of the oxidized fibronectin protein (from 1-50 μg/mL), such that ligand available can be precisely tuned. At sufficiently high concentrations of fibronectin and sodium meta-periodate in the oxidation reaction, the fibronectin can aggregate and strongly hinder the deposition of a uniform coat of protein. Careful testing as indicated in the next section is needed to ensure the deposition of the desired quantity and quality of protein. We have also used this chemistry in conjunction with micro-contact printing of ECM proteins to control the spatial distribution of ligands (Stricker et al., 2010). Another advantage of this coupling is that it requires significantly (100x) lower concentrations of ECM ligand in the coupling step to result in a similar surface density, so less ECM protein is required. Disadvantages of this technique are the time involved and the lack of appropriate carbohydrate groups necessary for oxidation of some proteins.
The method of coupling the PAA gel surface with sulfo-SANPAH is robust and we have used it to couple a numerous range of proteins to PAA gel surfaces. The most significant disadvantage is that a very high concentration of ECM protein is required during the conjugation step (1 mg/mL) and results in a moderate amount of surface-available ligand. Thus, this method can result in consumption of a very high amount of ECM protein. Another pitfall can be poor coupling due to loss of reactivity of sulfo-SANPAH due to poor storage or time delays during preparation. However, these inconsistencies are minimal with the method described here.
Confirmation and Quantification of Surface Available Ligand:
To confirm the density of protein ligand on the gel surface, we have used immunofluorescence against fibronectin (Figure 2B) or collagen and have compared the intensity of images of the gel surface against a range of calibrated standards (a range of fibronectin concentrations adsorbed on glass). More quantitative measures can be done by radioactive labeling (Rajagopalan et al., 2004). The ECM protocols here describe methods to fabricate gels that have approximately the same surface density of fibronectin as one would obtain by adsorbing 10 μg/mL fibronectin on an untreated glass coverslip for 1 hr at room temperature. This is sufficient for most cells to become well adhered and spread. This is the maximal surface density we have obtained using sulfo-SANPAH or AcryoylX methods; higher surface densities can be obtained with the hydrazine hydrate method.
Poor coupling could result from improper handling and subsequent loss of reactivity of sulfo-SANPAH and Acryloyl -X. Furthermore, it is important to maintain the pH of the extracellular matrix protein at around 7.0 when using sulfo-SANPAH or Acryloyl-X as the cross-linking agent to ensure proper cross-linking to the PAA gel. Also, in the oxidization step of the hydrazine hydrate protocol, we have found that higher concentrations of sodium metaperiodate than the amount recommended here yield large aggregates of protein that do not couple uniformly to the gel surface.
Please note that cells may take longer to spread and adhere onto soft gel surfaces than one may be used to working with tissue culture plastic or glass coverslips.
Reproducibility of Gel Stiffness:
The gel recipes described here yield gels with an extremely reproducible stiffness over the course of many years in several labs and multiple hands, which have independently confirmed gel stiffness using bulk rheology. Nevertheless, it is advisable to confirm gel stiffness by rheology methods. Note that the stiffness reported here is the shear modulus, not the Young's modulus. The two values are related by E = 2G(1+υ), where E is the Young's Modulus, G is the shear modulus, and υ is the Poisson's ratio of the material.
The presence of oxygen and contaminants within buffers or laboratory water, such as metals and non-buffer ions, can inhibit acrylamide polymerization, leading to inconsistent results. Furthermore, the 5 mL stock solution should be sufficient to facilitate the fabrication of thousands of gels over the course of the experiment and, thus, minimize inconsistencies. Nevertheless, it is advisable to confirm gel stiffness by rheology methods.
Impact on ECM coupling on gel stiffness:
We have confirmed that bulk conjugation of ECM proteins using Acroyl-X does not change the stiffness of PAA gels. Others have confirmed that conjugation with sulfo-SANPAH does not impact local stiffness (Engler et al., 2004).
Choice of bead size:
We have found 40 nm beads to be an optimal bead size to facilitate the use of high density of beads, as is shown in our representative image. These beads are bright enough to facilitate imaging with both wide field and confocal microscopy, but not too large to have any measurable effect on the stiffness of our polyacrylamide gel. To facilitate imaging, larger beads may be used.
Useful applications of traction force microscopy (TFM):
The applications of compliant hydrogels and TFM are wide-ranging. We, and others, have used this protocol to study many aspects of cellular differentiation, proliferation, migration and contraction, as well as focal adhesion assembly, maintenance and disassembly. Pharmacological agents that either inhibit or activate protein activity have been used in conjunction with TFM to investigate the role of specific proteins on cellular traction forces. In addition, the kinetic properties of proteins can be assessed with the use of fluorescent speckle microscopy (Gardel et al., 2008) or fluorescence recovery after photobleaching (FRAP) and correlated to traction forces. Furthermore, application of siRNA to probe for the affect of protein knockdown on traction force exertion is also a useful method to assess the role of specific proteins in altering cellular biophysical properties. Altogether, the combination of TFM with high-resolution fluorescence microscopy yields a powerful approach to correlate dynamic and mechanical properties of the cell.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank the lab of Ulrich Schwarz for computational tracking software used in quantification of cellular traction forces (Sabass et al., 2008). This work was supported by a Burroughs Wellcome Career Award and NIH Director's Pioneer Award (DP10D00354) to M.L. Gardel and Medical Scientist National Research Service Award (5 T32 GM07281) to S.P. Winter.
References
- Damljanovic V, Lajerholm BC, Jacobson K. Bulk and micropatterned conjugation of extracellular matrix proteins to characterized polyacrylamid substrates for cell mechanotransduction assays. Biotechniques. 2005;39(6):847–851. doi: 10.2144/000112026. [DOI] [PubMed] [Google Scholar]
- Engler A, Bacakova LNewman, Hategan C, Griffin A, M DDischer. Substrate compliance versus ligand in cell on gel responses. Biophys J. 2004;86((1 Pt 1)):617–628. doi: 10.1016/S0006-3495(04)74140-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardel ML, Sabass B, Ji L, Danuser G, Schwarz US, Waterman CM. Traction stress in focal adhesions correlates biphasically with actin retrograde flow speed. J Cell Biol. 2008;183:999–1005. doi: 10.1083/jcb.200810060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan P, Marganski WA, Brown XQ, Wong JY. Direct comparison of the spread area, contractility, and migration of balb/c 3T3 fibroblasts adhered to fibronectin- and RGD-modified substrata. Biophys J. 2004;87(4):2818–2827. doi: 10.1529/biophysj.103.037218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart-King CA, Dembo M, Hammer DA. The dynamics and mechanics of endothelial cell spreading. Biophys J. 2005;89:676–689. doi: 10.1529/biophysj.104.054320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker J, Sabass B, Schwarz US, Gardel ML. Optimization of traction force microscopy for micron-sized focal adhesions. J. Phys: Condensed Matter. 2010;22:194104–194114. doi: 10.1088/0953-8984/22/19/194104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabass B, Gardel ML, Waterman CM, Schwarz US. High resolution traction force microscopy based on experimental and computational advances. Biophys J. 2008;94:207–220. doi: 10.1529/biophysj.107.113670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung T. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton. 2005;60(1):24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer CM. Microtubule/organelle motility assays. Curr Protoc Cell Biol. 1998:13.1.1–13.1.21. [Google Scholar]