

Abstract
Human hematopoietic stem/progenitor cells are usually obtained from bone marrow, cord blood, or peripheral blood and are used to study hematopoiesis and leukemogenesis. They have the capacity to differentiate into lymphoid and myeloid lineages. The colony forming cell (CFC) assay is used to study the proliferation and differentiation pattern of hematopoietic progenitors by their ability to form colonies in a semisolid medium. The number and the morphology of the colonies formed by a fixed number of input cells provide preliminary information about the ability of progenitors to differentiate and proliferate. Cells can be harvested from individual colonies or from the whole plate to further assess their numbers and differentiation states using flow cytometry and morphologic evaluation of Giemsa-stained slides. This assay is useful for assessing myeloid but not lymphoid differentiation. The term myeloid in this context is used in its wider sense to encompass granulocytic, monocytic, erythroid, and megakaryocytic lineages.
We have used this assay to assess the effects of oncogenes on the differentiation of primary human CD34+ cells derived from peripheral blood. For this purpose cells are transduced with either control retroviral construct or a construct expressing the oncogene of interest, in this case NUP98-HOXA9. We employ a commonly used retroviral vector, MSCV-IRES-GFP, that expresses a bicistronic mRNA that produces the gene of interest and a GFP marker. Cells are pre-activated by growing in the presence of cytokines for two days prior to retroviral transduction. After another two days, GFP+ cells are isolated by fluorescence-activated cell sorting (FACS) and mixed with a methylcellulose-containing semisolid medium supplemented with cytokines and incubated till colonies appear on the surface, typically 14 days. The number and morphology of the colonies are documented. Cells are then removed from the plates, washed, counted, and subjected to flow cytometry and morphologic examination. Flow cytometry with antibodies specific to the cell surface markers expressed during hematopoiesis provides information about lineage and maturation stage. Morphological studies of individual cells under a microscope after Wright- Giemsa staining provide further information with regard to lineage and maturation. Comparison of cells transduced with control empty vector to those transduced with an oncogene reveals the effects of the oncogene on hematopoietic differentiation.
Protocol
1. PREPARATION OF REAGENTS
Prepare sterile 1000x stock solutions of Fms-related tyrosine kinase 3 (FLT-3) ligand, granulocyte/macrophage colony-stimulating factor (GM-CSF), stem cell factor (SCF), thrombopoietin (TPO), interleukin (IL)-3, and IL-6 according to manufacturer's instructions. Prepare sterile Retronectin stock solution (1mg/ml) also according to manufacturer's instructions. Divide these stock solutions into small aliquots and keep at -20 °C to avoid repeated freeze-thaw.
Prepare 2% FBS in IMDM.
Prepare complete IMDM medium with final concentrations of 20% FBS, 2 mM Glutamine, 100 units/ml Penicillin/Streptomycin (PS). Add the following cytokines just before use: 100 ng/ml FLT-3 ligand, 20 ng/ml GM-CSF, 100 ng/ml SCF, 100 ng/ml TPO, 50 ng/ml IL-3, and 100 ng/ml IL-6.
Prepare 1% BSA in HBSS (Ca & Mg-free, without phenol red) and 2% BSA in D-PBS (Ca & Mg-free). Sterilize the solutions by passing through 0.22 μm syringe filters and store at 4 °C.
Prepare 0.02% EDTA in HBSS and store at 4°C.
Prepare GaLV-pseudotyped retrovirus encoding the DNA of interest and store in aliquots at -80 °C. This procedure is outside the scope of this article, but is described elsewhere (1-3).
For Giemsa Staining
Just before use, filter the Wright/Giemsa stain solution twice through 1 μm filter papers into a clean glass bottle.
Immediately before use, prepare Wright/Giemsa buffer pH 6.4, by mixing 50 ml of the filtered Wright/Giemsa stain solution and 200 ml of phosphate buffer solution, pH 6.4 - Giordano formula.
Prepare phosphate buffer solution, pH 6.0 by dissolving 27.3 g of KH2PO4 and 4.62 g of Na2HPO4 in 3.5 L H2O.
Prepare 50% methanol in H2O.
2. PROCEDURE
Thawing and pre-activation of CD34+ cells
Thaw a vial of frozen CD34+ cells quickly at 37 °C by gently shaking until a last small ice crystal is left and transfer the cell suspension (1 ml) to a 50 ml conical tube. Gently rinse off the remaining cells from the vial with 1 ml of room temperature 2% FBS/IMDM and add it drop-wise to the 50 ml tube while swirling gently. Wait for 3 minutes. Next, slowly add 2 ml of 2% FBS/IMDM, while mixing gently, and wait for 3 minutes. Repeat the procedure by adding 2% FBS/IMDM that is the same volume as the diluted cell suspension at 3 min intervals, swirling gently in between additions, until the final volume reaches 32 ml.
Centrifuge at 250 x g for 10 minutes at room temperature and remove the supernatant leaving behind around 0.5 ml.
Wash the pellet by suspending in 20 ml 2% FBS/IMDM and centrifuging at 200 x g for 8 minutes at room temperature.
Remove the supernatant and suspend the cells in complete IMDM medium at approximately 0.5 x 106 cells/ml. Take 10 μl of the cell suspension into a microtube, mix with the same volume of Trypan blue solution (0.4%) and count using a hemocytometer.
Dilute the cells to 0.1-0.15 X 106 cells/ml by adding complete IMDM medium supplemented with cytokines (see 1.3)
Culture the cells in a humidified 37°C incubator with 5% CO2 for 2 days.
Retroviral transduction by virus pre-loading
On the day of transduction, count cells before starting virus preloading to determine the number of wells to be prepared.
Dilute the Retronectin stock solution to 25 μg/ml and add 400 μl to each well of a 24-well non-treated tissue culture plate. Incubate for 2 hours at room temperature in the laminar-flow biosafety hood to pre-coat.
Remove the Retronectin solution and add 400 μl of 2% BSA in D-PBS to each well. Incubate for 30 minutes at room temperature.
Thaw virus stock solutions and keep on ice. Remove the BSA solution, add 0.5 ml of virus preparation to each well, and centrifuge at 2200 x g at 4°C for 15 minutes.
Remove the virus solution from wells and repeat the virus loading described in 2.10 three more times.
Rinse each well with cold 0.5 ml HBSS (+ Ca & Mg) or IMDM.
Collect the pre-activated CD34+ cells by centrifuging at 200 x g for 8 minutes at room temperature.
Resuspend the cells at 0.1-0.15 x 106 cells per 1.5 ml in complete IMDM medium with cytokines.
Add 1.5 ml cell suspension to each well and incubate in a humidified 37°C incubator with 5% CO2 for 2 days.
Cell sorting and collection of transduced cells
Gently but thoroughly suspend the cells in the above virus-loaded plates and filter through a 50 μm CellTrics cell filter into a conical tube. Take 10 μl of the cell suspension into a microtube, mix with an equal volume of Trypan blue solution and count using a hemocytometer. Rinse each well with 0.8 ml of cold 0.02% EDTA/HBSS (Ca & Mg-free without phenol red) and add to the same collection tube through a 50 μm CellTrics cell filter.
Centrifuge the cells at 200 x g for 8 minutes at 4°C, and wash the pellet with cold HBSS (Ca & Mg-free without phenol red) by centrifuging at 200 x g for 8 minutes at 4°C.
Resuspend the pellet in 1% BSA/HBSS (Ca & Mg-free without phenol red) to a concentration of 15 X 106/ml or a minimum volume of 0.250 ml for cell sorting. Keep the cells on ice in the dark. Prepare a tube containing cold 20% FBS/IMDM/Glutamine/PS medium for collecting the sorted cells. Sorting is performed at the High Speed Cell Sorter Core of the Alvin J. Siteman Cancer Center at Washington University School of Medicine using a MoFlo high-speed sorter (Dako, Glostrup, Denmark).
CFC assay
Thaw the required number of 3 ml aliquots of CFC assay media. Vortex vigorously to mix and let the tubes stand at least 5 min to let any bubbles present to rise to the surface before adding cells.
To obtain the cell concentration for each sorted sample, divide the cell number provided by the sorting facility by the approximate cell suspension volume. Take 3,000 virus-transduced, sorted cells into a sterile microtube containing cold 2% FBS/IMDM. Pre-adjust the volume of 2% FBS/IMDM so that the final suspension volume will be approximately 0.3 ml.
Suspend the cells and transfer the entire 0.3 ml cell suspension to a 3 ml aliquot of Methocult GF+ H4435, which consists of 1% methylcellulose, 30% FBS, 1% BSA, 10-4 M 2-mercaptoethanol, 2 mM L-glutamine, 50 ng/ml SCF, 20 ng/ml GM-CSF, 20 ng/ml IL-3, 20 ng/ml IL-6, 20 ng/ml G- CSF, and 3 units/ml erythropoietin in IMDM. Human Methylcellulose Enriched Media (R & D Systems) which consists of 1.3% methylcellulose, 25% FBS, 1% BSA, 5 x 10-5 M 2-mercaptoethanol, 2 mM L-glutamine, 50 ng/ml SCF, 20 ng/ml GM-CSF, 20 ng/ml IL-3, 20 ng/ml IL-6, 20 ng/ml G- CSF, and 3 units/ml erythropoietin in IMDM can be also used.
Vortex vigorously to make the mixture rise fully and fall 3-4 times. Let the tube stand still for 3 min.
Attach a 16 gauge blunt-end needle to a 3 ml syringe and draw up 2.2 ml. Do not draw up large bubbles; expel them at the beginning by pushing out a couple of times. Push out 1.1 ml each into two 30 mm non-treated dish and spread out the mixture evenly by rotating.
Place duplicate plates in a 100 mm plate together with a water dish containing 3 ml sterile water. Culture for 14 - 17 days.
Characterize and score the colonies according to their morphology with an inverted microscope at 40x magnification in a culture dish marked with a scoring grid. For the purposes of our assay, the colonies are classified into 3 categories: pure erythroid, myelomonocytic, and mixed. See manufacturer's instructions for further colony subclassification.
The entire CFC assay plate may be scanned without magnification using a regular scanner at 600 dpi, and low-power (40x) photomicrographs of colonies may be taken using an inverted microscope equipped with a color camera.
For further analysis of differentiation and proliferation, cells from the entire CFC assay plate are recovered by suspending in several volumes of room temperature 2% FBS/IMDM. After centrifugation at 300 x g for 10 min at 4°C, the cells are resuspended in 2% FBS/IMDM, counted, and either stained with antibodies for flow cytometry or transferred to slides using a Cytospin centrifuge for Giemsa staining.
Antibody staining and flow cytometry
Centrifuge cells at 200 x g for 8 min at 4°C and resuspend in cold 50% normal mouse serum in HBSS (Ca & Mg-free without phenol red).
Place approximately 5 x 105 cells in 80 μl per tube (12 x 75 mm round-bottom tubes) and keep on ice. Add antibody mixture (approximately 20 μl mixture or negative control solution for no-antibody controls) and mix by lightly tapping the tubes. Prepare calibration control tubes with no antibody or with an anti-CD45 antibody for each fluorochrome. Place tubes on ice in the dark and incubate for 20 minutes. Mix by lightly tapping tubes after the first 10 min. After the 20 min incubation, fill the tubes with cold HBSS and centrifuge at 200 x g for 8 min at 4°C. Resuspend the pellet in cold 0.5% paraformaldehyde/HBSS. The following mixtures of antibodies are typically used for our analysis. For myeloid differentiation: CD11b (phycoerythrin-conjugated clone ICRF44)/CD33 (allophycocyanin-conjugated clone WM53)/CD45 (phycoerythrin-Cy7-congjugated clone HI30). For erythroid differentiation: CD71 (phycoerythrin-conjugated clone M-A712)/CD235a (allophycocyanin-conjugated clone GA-R2)/CD45 (phycoerythrin-Cy7-congjugated clone HI30).
Flow cytometry is performed at the High Speed Cell Sorter Core of the Alvin J. Siteman Cancer Center at Washington University School of Medicine on a FACScan flow cytometer upgraded to 5 colors and two lasers (BD Biosciences) and analyzed using CellQuest (BD Biosciences) or FlowJO v7.2.4 (Tree Star, Inc., Ashland, OR, USA) software.
Giemsa staining
Approximately 30,000 cells recovered from CFC assay plates described above are suspended in 0.3 ml 2% FBS/IMDM and transferred to a slide using a Cytospin centrifuge at 1,000 rpm for 10 min at room temperature.
- Set up nine staining vessels containing solutions in the following order.
- Absolute Methanol
- Wright/Giemsa stock solution
- Wright/Giemsa buffer, pH 6.4
- 50% Methanol in H2O
- H2O
- H2O
- Phosphate buffer, pH 6.0
- H2O
- H2O
Place the slides into a slide carrier and dip in absolute methanol (#1) for 2 min and blot off excess methanol.
Immediately, dip the slide carrier in Wright/Giemsa stock solution (#2) for 5 min.
Transfer the carrier into Wright/Giemsa buffer, pH 6.4 (#3) and incubate for 10 min.
Dip the carrier twice in 50% methanol (#4), 10 times in H2O (#5), another 10 times in H2O (#6), and then 5 times in Phosphate buffer, pH 6.0 (#7).
Transfer the carrier into H2O (#8) and incubate for 2 min. Repeat the wash in H2O (#9).
Allow the slides to air dry completely in the carrier. Remove each slide and wipe the back of the slide with methanol-soaked Kimwipes to remove stains. Place a drop of Cytoseal 60 and a cover glass to seal.
A 500-cell differential count is performed for each Giemsa-stained slide using an Olympus BX51 microscope. Cells are divided into five categories: primitive cells include blasts and promyelocytes; intermediate myeloid cells include myelocytes and metamyelocytes; mature myeloid cells include bands, segmented neutrophils, monocytes, and macrophages; intermediate erythroid cells include cells with intermediate hemoglobinization; and mature erythroid cells include cells with full hemoglobinization. Photomicrographs are taken with an Olympus DP71 camera with a 60x oil objective.
3.0) REPRESENTATIVE RESULTS:
CFC assay: Some of the input cell population will proliferate, differentiate, and form colonies on the semi-solid medium during the 14-17-day incubation period. In this experiment, it is evident that expression of the NUP98-HOXA9 oncogene caused the formation of prominent red (erythroid colonies) (Figure 1) (3).
Antibody Staining and Flow Cytometry: Flow cytometric immunophenotyping of cells collected from CFC assay plates provides information on the differentiation state of the cell population. By combined use of antibodies against certain differentiation markers, the lineage and degree of maturation of cells can be assessed. In this case CD235a was used to identify erythroid cells. As shown in Figure 2A, NUP98-HOXA9 caused an increase in the percentage of CD235a+ erythroid cells, but the brightness of CD235a expression by these cells was diminished compared to controls, indicating inhibition of erythroid maturation. In Figure 2B, myeloid cells are identified by virtue of their CD33 positivity. NUP98-HOXA9 caused a decrease in the numbers of CD33+ cells, with a decrease in the brightness of CD11b expression, consistent with inhibition of myeloid maturation (3). Thus the flow cytometry results show that NUP98-HOXA9 caused an erythroid hyperplasia with inhibition of both myeloid and erythroid maturation.
Giemsa Staining: Morphological examination of cells collected from CFC assay plates provides information on the lineage and degree of maturation of the cell population. The conclusions obtained morphologically may differ from those obtained by flow cytometric studies, and thus these methods are complementary to each other (3). The experiment is repeated at least 3 times, and each time a 500-cell differential count is performed to distinguish 5 categories of cells as described above. Examples of these cells are pointed out in Figure 3. The results are tabulated and analyzed in order to determine whether there are statistically significant differences between the oncogene-transduced sample and the control. In this case, the results showed that NUP98-HOXA9 caused an overall increase in the numbers of cells, with erythroid hyperplasia and inhibition of both erythroid and myeloid maturation (3) (not shown).
FIGURE LEGENDS
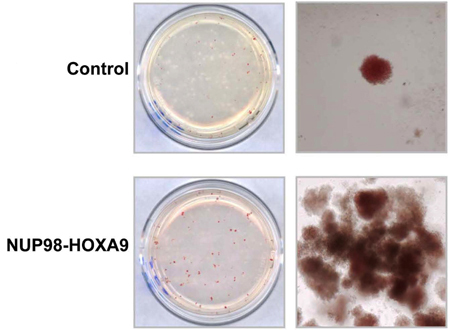 Figure 1: The effects of NUP98-HOXA9 on human CFC morphology.
Primary human CD34+ cells were retrovirally transduced with either control MSCV-IRES-GFP vector or vector expressing NUP98-HOXA9, and cells were sorted for GFP positivity. One thousand cells were seeded into each of two duplicate plates for CFC assay and the experiment was repeated 3 4 independent times. Representative plates without magnification (left) and low power photomicrographs of representative erythroid colonies (right) are shown (3).
Figure 1: The effects of NUP98-HOXA9 on human CFC morphology.
Primary human CD34+ cells were retrovirally transduced with either control MSCV-IRES-GFP vector or vector expressing NUP98-HOXA9, and cells were sorted for GFP positivity. One thousand cells were seeded into each of two duplicate plates for CFC assay and the experiment was repeated 3 4 independent times. Representative plates without magnification (left) and low power photomicrographs of representative erythroid colonies (right) are shown (3).
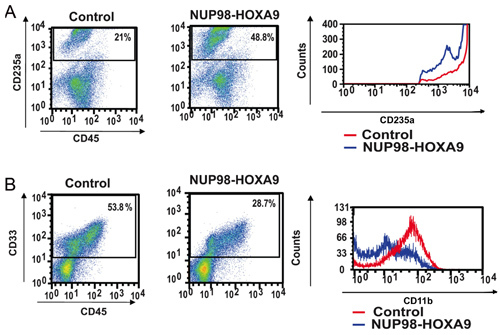 Figure 2: Flow cytometry shows disruption of human primary CD34+ cell differentiation by NUP98-HOXA9.
(A) Flow cytometry for erythroid differentiation: Cells from the CFC plates (see Figure 1) were harvested and stained with antibodies to CD45 and CD235a. The CD235a+ gate was plotted on a histogram (right panel) to show the expression of CD235a relative to control cells (B) Flow cytometry for myeloid differentiation: Cells from the CFC plates were harvested and stained with CD45 and CD33. The CD33+ gate was plotted on a histogram to show CD11b expression compared to control (right panel). The percentages of cells falling within each gate are shown (3).
Figure 2: Flow cytometry shows disruption of human primary CD34+ cell differentiation by NUP98-HOXA9.
(A) Flow cytometry for erythroid differentiation: Cells from the CFC plates (see Figure 1) were harvested and stained with antibodies to CD45 and CD235a. The CD235a+ gate was plotted on a histogram (right panel) to show the expression of CD235a relative to control cells (B) Flow cytometry for myeloid differentiation: Cells from the CFC plates were harvested and stained with CD45 and CD33. The CD33+ gate was plotted on a histogram to show CD11b expression compared to control (right panel). The percentages of cells falling within each gate are shown (3).
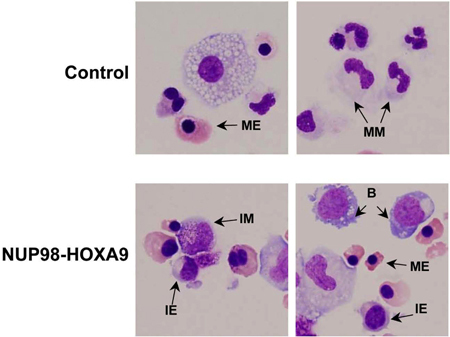 Figure 3: Cell morphology shows disruption of differentiation by NUP98-HOXA9 compared to vector control.
Cytospin smears were prepared from CFC plates and stained with Giemsa. Photomicrographs were taken from representative fields with a 60x oil objective. B: blast; MM: mature myeloid, IM: intermediate myeloid, ME: mature erythroid, IE: intermediate erythroid (3).
Figure 3: Cell morphology shows disruption of differentiation by NUP98-HOXA9 compared to vector control.
Cytospin smears were prepared from CFC plates and stained with Giemsa. Photomicrographs were taken from representative fields with a 60x oil objective. B: blast; MM: mature myeloid, IM: intermediate myeloid, ME: mature erythroid, IE: intermediate erythroid (3).
Discussion
The CFC assay has been used extensively to determine the proliferation and differentiation patterns of hematopoietic progenitors and to study the effects of oncogenes (4, 5). It has the advantage over liquid cultures of being a clonal assay, such that the colonies represent the progeny of a single progenitor and can be individually removed for further analysis. The limitation of the CFC assay is that it is not adequate for the detection of more immature progenitors or hematopoietic stem cells; such cells are detected using the long-term culture-initiating cell (LTC-IC) assay (6, 7). The experiments described here demonstrate that the AML-associated oncogene NUP98-HOXA9 inhibits myeloid and erythroid maturation and causes erythroid hyperplasia (1, 3).
Since the methylcellulose-based culture is relatively long-term and the CFC medium does not contain antibiotics, it is of paramount importance to prevent contamination of the cell cultures, particularly during and after sorting. The slightest bacterial or fungal contamination will overwhelm the culture. Interpretation of flow cytometry and cell morphology requires experience; consultation with trained personnel is recommended. Only FBS that has been pre-screened for suitability to use with primary human myeloid progenitors should be used; the product that we routinely use is listed in the reagents table. Methylcellulose-based CFC media are very viscous and special care should be taken while handling them to mix evenly and eliminate bubbles before pouring into plates. Inclusion of a water dish with the CFC plates in a larger plate is important to prevent drying of the CFC medium during the prolonged incubation.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank the Alvin J. Siteman Cancer Center at Washington University School of Medicine and Barnes-Jewish Hospital in St Louis, MO, for the use of the High Speed Cell Sorter Core, which provided flow cytometry equipment and sorting services. This work was supported by National Institutes of Health Grants R01 HL082549 and K02 HL084179 (NRY). The Siteman Cancer Center is supported in part by an NCI Cancer Center Support Grant P30 CA91842.
References
- Takeda A, Goolsby C, Yaseen NR. NUP98-HOXA9 induces long-term proliferation and blocks differentiation of primary human CD34+ hematopoietic cells. Cancer Res. 2006:66–6628. doi: 10.1158/0008-5472.CAN-06-0458. [DOI] [PubMed] [Google Scholar]
- Yassin ER, Abdul-Nabi AM, Takeda A, Yaseen NR. Effects of the NUP98-DDX10 oncogene on primary human CD34+ cells: role of a conserved helicase motif. Leukemia. 2010 doi: 10.1038/leu.2010.42. Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassin ER, Sarma NJ, Abdul-Nabi AM, Dombrowski J, Han Y, Takeda A, Yaseen NR. Dissection of the transformation of primary human hematopoietic cells by the oncogene NUP98-HOXA9. PLoS One. 2009;4:e6719–e6719. doi: 10.1371/journal.pone.0006719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen-Druey C, Tichelli A, Meyer-Monard S. Human hematopoietic colonies in health and disease. Acta Haematol. 2005;113:5–96. doi: 10.1159/000081987. [DOI] [PubMed] [Google Scholar]
- Pereira C, Clarke E, Damen J. Hematopoietic colony-forming cell assays. Methods Mol Biol. 2007;407:177–208. doi: 10.1007/978-1-59745-536-7_14. [DOI] [PubMed] [Google Scholar]
- Coulombel L. Identification of hematopoietic stem/progenitor cells: strength and drawbacks of functional assays. Oncogene. 2004;23:7210–7222. doi: 10.1038/sj.onc.1207941. [DOI] [PubMed] [Google Scholar]
- Hogge DE, Lansdorp PM, Reid D, Gerhard B, Eaves CJ. Enhanced detection, maintenance, and differentiation of primitive human hematopoietic cells in cultures containing murine fibroblasts engineered to produce human steel factor, interleukin-3, and granulocyte colony-stimulating factor. Blood. 1996;88:3765–3773. [PubMed] [Google Scholar]