

Abstract
Medaka is a small egg-laying freshwater fish that allows both genetic and embryological analyses and is one of the three vertebrate model organisms in which genome-wide phenotype-driven mutant screens were carried out 1. Divergence of functional overlap of related genes between medaka and zebrafish allows identification of novel phenotypes that are unidentifiable in a single species 2, thus medaka and zebrafish are complementary for genetic dissection of the vertebrate genome functions. Manipulation of medaka embryos, such as dechorionation, mounting embryos for imaging and cell transplantation, are key procedures to work on both medaka and zebrafish in a laboratory. Cell transplantation examines cell autonomy of medaka mutations. Chimeras are generated by transplanting labeled cells from donor embryos into unlabeled recipient embryos. Donor cells can be transplanted to specific areas of the recipient embryos based on the fate maps 3 so that clones from transplanted cells can be integrated in the tissue of interest during development. Due to the hard chorion and soft embryos, manipulation of medaka embryos is more involved than in zebrafish. In this video, we show detailed procedures to manipulate medaka embryos.
Protocol
1. Development of the embryos
When they are laid, eggs are clustered because of attachment filaments on the chorion. To let embryos develop normally, it is necessary to separate eggs. Tangle and cut attachment filaments by holding the attachment filaments with two forceps.
After unclustering, eggs are separated from feces and algae and transferred to fresh embryo medium at a maximum density of 40 eggs per 6cm Petri dish.
Medaka embryos develop slightly slower than zebrafish at 27°C. The timing of hatching is different between the two species; medaka embryos hatch from the chorion in 7 days and immediately start to swim and eat, whereas zebrafish embryos hatch in 2 days but start to swim and eat in 5-6 days. Development of medaka is staged according to Iwamatsu's staging4.
The development of medaka embryos can be conveniently adjusted to experimental plans by selecting the appropriate temperature. Development of medaka embryos can be stopped at 4°C in early development. After stage 24 when heart beating starts, development can be slowed using a minimum temperature of 18°C.
The timing of appearance of organs/tissues is slightly different in medaka compared with zebrafish, i.e. in medaka somitogenesis occurs after the onset of brain development whereas in zebrafish somitegenesis precedes brain development.
2. Removing the chorion
The chorion of medaka consists of two protective layers with a hard inner layer and a soft outer surface. Thus, a two-step protease treatment employing pronase and hatching enzyme is necessary to remove this chorion.
Once dechorionated, embryos should be kept in 1X BSS. Semi-sterile conditions will enhance the successful culture of dechorionated embryos, especially when longer periods of observation are required. These include using sterile solutions (e.g. sterilized 1X BSS with antibiotics) and tools sterilized with 70% ethanol followed by rinsing with 1X BSS.
As dechorionated medaka embryos are softer and more fragile than dechorionated zebrafish embryos, extra care must be taken to ensure they do not contact air or bubbles in the pipette, as this will cause immediate collapse. To ensure minimal damage to embryos, a wide-mouthed heat polished glass pipette with pipette pump should be used for transferring embryos and a hair loop should be utilized to orientate embryos for observation. Non-adherent Petri dishes should be used to prevent embryos from attaching to surfaces.
Prior to dechorionation it should be checked that eggs have been sufficiently separated and cleaned-up.
Since both pronase and hatching enzymes are proteinases, they should be kept on ice and minimize exposure of embryos to these enzymes.
Transfer eggs to sandpaper (p2000 grit size, waterproof) placed in the lid of a 9cm Petri dish. Remove excess medium but ensure a sufficient volume remains as to prevent drying of embryos.
Gently roll embryos for around 45-60 seconds to remove some of the outer surface hairs and lightly score the surface of the chorion (Figure 1). Transfer embryos back to original Petri dish and examine. When rolling embryos on sandpaper use the forefinger, applying minimal pressure and keeping finger parallel to surface of sand paper. Do not roll more than 5-7 embryos at once. This precaution will minimize the risk of crushing embryos beneath each other.
Replace egg medium in dish with 20mg/ml pronase and incubate embryos for 40-60 minutes at 27°C. Ensure embryos are sufficiently covered by pronase and a lid is present on the dish. Unused pronase should be kept on ice during this step to minimize self-digestion and can be reused until activity is lost (approximately 1-2 weeks).
Recover pronase for reuse and wash embryos 5X in embryo medium to remove traces of pronase as this will inactivate the hatching enzyme to be added.
Remove embryo medium and cover embryos with hatching enzyme, ensuring embryos sit as a monolayer in the dish. If eggs sit on top of one another in the dish, those at the bottom will be crushed as the chorion dissolves. Keep unused hatching enzyme on ice.
Incubate embryos at 27°C and after 15 minutes periodically check the progress of hatching using a stereomicroscope. It will be seen that a number of lunar crater-like holes begin to appear in the inner layer of the chorion, which soon dissolves following this leaving the soft outer layer of the chorion easily removed manually. The entire process of hatching may take 15-60 minutes.
As soon as embryos come out from the chorion, transfer embryos to a Petri dish containing 1X BSS. Ensure not to transfer hatching enzyme to the 1X BSS by touching the tip of the pipette onto the surface of the BSS allowing the embryos to gently roll out.
Once all embryos are transferred from hatching enzyme, make a final transfer to another fresh dish of 1X BSS. This ensures embryos are not exposed to any remnants of hatching enzyme, as this will damage exposed embryos. Once in this final dish any embryos still possessing the outer layer of the chorion can be manually liberated using sterilized micro-forceps under the stereomicroscope. If embryos need to be developed following dechorionation, penicillin/streptomycin should be added to the BSS to prevent bacterial growth.
 Figure 1. Comparison of rolled and unrolled embryos during dechorionation. Observe how the rolled embryos in panel B lack the hairs seen on the unrolled embryos in panel A.
Figure 1. Comparison of rolled and unrolled embryos during dechorionation. Observe how the rolled embryos in panel B lack the hairs seen on the unrolled embryos in panel A.
3. Mounting dechorionated embryos
Agarose embedding is useful for longer periods of imaging (e.g. time-lapse imaging) for live embryos as well as for detailed observations of fixed embryos. During gastrulation and early organogenesis (stage 14 to stage 28), medaka embryos exhibit waves of rhythmic contractile movements across the periderm, a tissue layer covering both the developing embryo and the yolk 5. Embryos are treated with 3.5mM 1-heptanol to stop contractile movements 6.
To stop movement of embryos after stage 28 (64hpf), embryos are anaesthetized by adding drops of Tricaine mesilate (TMS) before embedding. TMS is also added to the agarose (add only a minimal amount, this dose needs to be optimized, approximately a 1:25 dilution).
Thaw a small amount of 3% low gelling temperature agarose (in 1X BSS) by heating to 37°C and maintain at this temperature.
The following steps need to be carried out swiftly to ensure that the agarose does not solidify before orientating the embryo. Using a wide-mouthed glass pipette transfer enough molten agarose to fill the cap depression of an Eppendorf tube.
Transfer one dechorionated embryo to the cap depression minimizing carrying over BSS with the embryo. Immediately uptake the agarose and embryo from the cap depression and transfer to a Petri dish culture chamber. A 3.5cm Petri dish with a glass window at the bottom is used. For imaging using an inverted microscope, embryos are orientated body facedown and placed close to the cover glass. For imaging using an upright microscope, the thickness of agarose is minimized.
Transfer the 3.5cm Petri dish into a 14cm diameter Petri dish containing ice and water (water filled to roughly 1/3 total depth of large dish). Whilst holding the chamber down firmly on the bottom of the 14cm Petri dish, use a hair loop to gently orientate the embryo as desired in the molten agarose and hold the embryo until the agarose solidifies by gently raising and lowering the smaller dish to its original position in the ice-cold water.
4. Cell transplantation in medaka embryos
The goal of this procedure is to determine whether the gene of interest acts cell-autonomously (within a cell) or non cell-autonomously (between cells).
Donor cells can be labeled with a tracer dye such as rhodamine-dextran prior to transplantation (as outlined in the accompanying JoVE protocol 'Microinjection Of Medaka Embryos') or a transgenic strain with GFP expression may be utilized, allowing transplantation assessment. A combination of both labeling techniques is often useful to overcome auto-fluorescence that can be encountered. For time-lapse studies following transplantation, GFP expression is particularly useful.
Use a wide-mouthed glass pipette with pipette pump throughout this procedure and sterilize all tools (including slides) beforehand with 70% EtOH followed by thorough rinsing with sterile 1X BSS. Recipient embryos are usually developed to around stage 12 as this allows discrimination of the ventral and dorsal poles when carrying out transplantation.
Commence dechorionating embryos 1.5 hours prior to transplantation and setup injection apparatus during enzyme incubations.
Place a cavity microscope slide into a 9cm diameter Petri dish.
Add a small amount of 3% methylcellulose to the centre of the cavity slide using a sterile pipette tip and spread thinly.
Dry methylcellulose for approximately 1-2 minutes.
Add 350μl of sterile 1X BSS to fill slide depression.
Transfer one donor embryo and up to four recipient embryos to the slide using a glass pipette.
Orientate embryos using a hair loop so the blastoderm of the embryo is facing upwards. Embryos can be leant against each other to further increase stability.
Gently insert the micro-needle into donor blastoderm and slowly uptake 10-20 cells.
Gently insert needle into required area of recipient embryo blastoderm and expel the cells slowly. Great care should be taken when inserting cells into the recipient blastoderm so as not to disrupt the cell-yolk sac boundary as this will result in death of the embryo.
Pour sterilized 1X BSS into the Petri dish. Carefully pour the BSS into dish as close to the side of the dish as possible so as not to disrupt the embryos. Never pour the BSS directly onto the embryos and add sufficient BSS such that the slide and embryos are immersed.
Add 100μl of penicillin/streptomycin to the dish and cover. Carefully transfer dish to a 27°C incubator to allow normal development.
Embryos can be periodically observed as desired. When using transplantation to carry out gain-of-function and/or phenotype rescue experiments, some morphological changes observed might be due to effects of transplantation. Thus multiple transplantations are necessary. After 2-3 days, melanophores should be present on the yolk sac, head, eyes and trunk. If present on transplanted embryos then a successful chimera has most likely been produced (Figure 3).
If necessary, genotype donor embryo(s) by transferring to PCR tube(s) containing 25μl of 20mg/ml proteinase K. Incubate at 55°C for 4 hours followed by 10 minutes at 94°C and carry out PCR.
5. Representative results
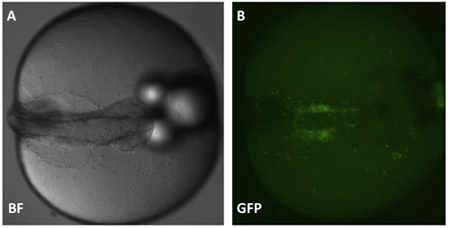 Figure 2. Example position of an agarose-embedded embryo. Anterior is shown to the right and view is dorsal. Panel B: endothelial cells can be seen on either side of the embryo body and are labeled using GFP driven by the fli promoter.
Figure 2. Example position of an agarose-embedded embryo. Anterior is shown to the right and view is dorsal. Panel B: endothelial cells can be seen on either side of the embryo body and are labeled using GFP driven by the fli promoter.
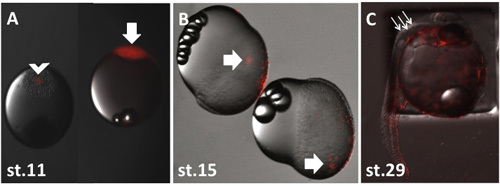 Figure 3. Images of post-transplantation embryos. Image A shows a donor and recipient embryo immediately post-transplantation. The donor embryo is shown on the right with a completely red blastoderm (arrow). The recipient embryo is shown on the left and is easily identified by the small mass of red transplanted cells visible in the blastoderm (arrowhead). Image B shows two recipient embryos approximately 7 hours post-transplantation (incubated at 27°C). Notice the transplanted cells have migrated from the site of transplantation and are now dispersed across the blastoderm (arrows). Image C shows a recipient embryo at st.29 (approximately 74hpf). Note the pigmentation in the eye and the presence of melanophores in the trunk and brain region (arrows). The rhodamine-labelled cells can be seen to be colonising many of the embryonic structures indicating the successful production of a chimera.
Figure 3. Images of post-transplantation embryos. Image A shows a donor and recipient embryo immediately post-transplantation. The donor embryo is shown on the right with a completely red blastoderm (arrow). The recipient embryo is shown on the left and is easily identified by the small mass of red transplanted cells visible in the blastoderm (arrowhead). Image B shows two recipient embryos approximately 7 hours post-transplantation (incubated at 27°C). Notice the transplanted cells have migrated from the site of transplantation and are now dispersed across the blastoderm (arrows). Image C shows a recipient embryo at st.29 (approximately 74hpf). Note the pigmentation in the eye and the presence of melanophores in the trunk and brain region (arrows). The rhodamine-labelled cells can be seen to be colonising many of the embryonic structures indicating the successful production of a chimera.
Discussion
In this video we demonstrate how to dechorionate and carry out cell transplanataion on medaka embryos. This is a powerful technique to produce chimeric embryos for the study of gene/protein function during development, as well as for elucidating the autonomy of the gene/protein in question. Medaka are a useful vertebrate model system for this technique due to well established fate maps and their transparency lending them well to in vivo real-time imaging. Transplantation can be combined with microinjection (as outlined in the accompanying JoVE protocol 'Microinjection Of Medaka Embryos') so that the behaviour of transplanted cells can be easily observed.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work is supported by a grant from the MRC to M. F-S.
References
- Furutani-Seiki M, Sasado T, Morinaga C. A systematic genome-wide screen for mutations affecting organogenesis in Medaka, Oryzias latipes. Mech Dev. 2004;121:647–658. doi: 10.1016/j.mod.2004.04.016. [DOI] [PubMed] [Google Scholar]
- Furutani-Seiki M, Wittbrodt J. Medaka and zebrafish, an evolutionary twin study. Mech Dev. 2004;121:629–6237. doi: 10.1016/j.mod.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Hirose Y, Varga ZM, Kondoh H, Furutani-Seiki M. Single cell lineage and regionalization of cell populations during Medaka neurulation. Development. 2004;131:2553–2563. doi: 10.1242/dev.01140. [DOI] [PubMed] [Google Scholar]
- Iwamatsu T. Stages of normal development in the medaka Oryzias latipes. Zool Sci. 1994;11:825–839. doi: 10.1016/j.mod.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Cope J, Fluck R, Nicklas L, Plumhoff LA, Sincock S. The stellate layer and rhythmic contractions of the Oryzias latipes embryo. J Exp Zool. 1990;254:270–275. doi: 10.1002/jez.1402540305. [DOI] [PubMed] [Google Scholar]
- Brend T, Holley SA. Zebrafish Whole Mount High-Resolution Double Fluorescent In Situ Hybridization. J Vis Exp. 2009 doi: 10.3791/1229. [DOI] [PMC free article] [PubMed] [Google Scholar]