

Abstract
Immunoprecipitation detected by flow cytometry (IP-FCM) is an efficient method for detecting and quantifying protein-protein interactions. The basic principle extends that of sandwich ELISA, wherein the captured primary analyte can be detected together with other molecules physically associated within multiprotein complexes. The procedure involves covalent coupling of polystyrene latex microbeads with immunoprecipitating monoclonal antibodies (mAb) specific for a protein of interest, incubating these beads with cell lysates, probing captured protein complexes with fluorochrome-conjugated probes, and analyzing bead-associated fluorescence by flow cytometry. IP-FCM is extremely sensitive, allows analysis of proteins in their native (non-denatured) state, and is amenable to either semi-quantitative or quantitative analysis. As additional advantages, IP-FCM requires no genetic engineering or specialized equipment, other than a flow cytometer, and it can be readily adapted for high-throughput applications.
Protocol
**This video protocol is based on an associated publication 1: High-sensitivity detection and quantitative analysis of native protein-protein interactions and multiprotein complexes by flow cytometry. Adam G. Schrum, Diana Gil, Elaine P. Dopfer, David L. Wiest, Laurence A. Turka, Wolfgang W. A. Schamel, Ed Palmer. Science's STKE 2007 (389): pl2, June 5, 2007, [DOI: 10.1126/stke.3892007pl2]. Please click here to see this publication.
Prior to Starting, Prepare the Following Aqueous Stock Solutions:
| MES Coupling Buffer: | Store at 25°C |
| MES, pH 6.0 | 50 mM |
| EDTA | 1 mM |
| Quenching/Blocking/Storage (QBS) Buffer: | Store at 4°C |
| BSA | 1% |
| Sodium azide | 0.02% |
| PBS, pH 7.4 | 1x |
| FCM Buffer: | Store at 4°C |
| Tris, pH 7.4 | 50 mM |
| NaCl | 100 mM |
| Sodium azide | 0.02% |
| FBS | 5% |
| PBS, pH 7.4 | 1x |
Prepare Immediately Prior to Use:
| EDAC-MES: | Dissolve 50 mg/mL EDAC powder in MES Coupling Buffer |
| Digitonin solution (2% w/v): | Dissolve digitonin powder in dH2O by heating to 95°C for 5min then cooling on ice |
| Lysis Buffer: | |
| Tris, pH 7.4 | 50 mM |
| NaCl | 150 Mm |
| Digitonin solution | 1% |
| Protease Inhibitors | 1x |
| Keep on ice |
1. Coupling of mAb to Beads
Determine the concentration of beads from the purchased stock by diluting in PBS and counting with a hemacytometer. Start with a 1:10000 dilution of beads for counting.
Pipette 18 x 106 beads into a 1.5-mL microcentrifuge tube.
Wash the beads 2 to 3 times in 0.5 to 1.0 mL MES Coupling Buffer, centrifuging at 20,000g for 3 minutes at 25°C after each wash.
Resuspend the beads in 50 μL MES Coupling Buffer.
Activate the carboxyl groups on the beads by adding 20 μL of freshly prepared EDAC-MES.
Mix gently for 15 min at 25°C by manually pipetting up and down.
Wash the activated beads 2 to 3 times in 0.5 to 1.0 mL PBS, centrifuging at 20,000g for 3 minutes at 25°C after each wash.
Resuspend the activated beads in 50 μL PBS.
Add 50 μL of the IP mAb (at 0.2-1.0 mg/ mL stock concentration) to the activated bead solution.
Mix for 3 to 4 hours at 25°C by placing the tube on a vibrating shaker. Shake sufficiently to prevent settling of the beads on the bottom of the tube.
Wash mAb-coupled beads 2 to 3 times in 0.5 to 1.0 mL PBS, centrifuging at 20,000g for 3 minutes at 25°C after each wash.
Resuspend the beads in 100 μL QBS Buffer. These can be stored at 4°C.
Suspending the beads well, dilute 1:200-1:10,000 in PBS and count with a hemacytometer. The concentration must be measured for each preparative batch, so that the number of beads used in each IP or pull-down experiment can be precisely controlled.
2. Post-nuclear Lysate Preparation and Ip
The lysis method and optimal lysis conditions will depend on the cell type and protein-protein interactions being examined. A number of protocols exist and can be modified for your application.
Lyse 20 x 106 'small' cells, such as lymphocytes, or 5 x 106 'large' cells, such as macrophages or tumor cells, in 100 μL fresh Lysis buffer in a 1.5-mL microcentrifuge tube for 20min on ice. Scale the lysis volume as needed.
To remove nuclei and insoluble cellular debris, centrifuge the lysate at 20,000g for 10 min at 4°C. Keep the supernatant and discard the pellet.
Add 0.5 x 105 to 2.5 x 105 mAb-coupled beads to the clarified lysate, pipetting gently to mix. The minimum volume we use to perform a single IP is 5 μL.
Place on a vertical rotating wheel 4 hours to overnight in a cold room. Set to sufficient velocity to prevent the beads from settling. It is acceptable for the liquid of low-volume IPs to remain in the bottom of the tube during rotation.
3. Probing of Bead-captured Protein with Fluorochrome-conjugated Antibodies
Wash the IP beads two times in 0.2 to 1.0 mL ice-cold FCM Buffer, centrifuging at 20,000g at 4°C for 3 min after each wash.
Resuspend the beads in 100 μL FCM Buffer and aliquot 20 μL into each of five or more 1.5-mL microcentrifuge tubes or wells of a chimney-bottom plate.
Add fluorochrome-conjugated mAbs to the samples. Perform the FCM stain according to the vendor's instructions or according to empirically determined concentrations. As a starting point, add 0.2 to 1 μL of stock antibody solution (at 0.2-1.0 mg/ mL) per tube or well, and incubate for 40 min on ice.
Wash probed beads in 1.5-ml tubes two times in 1.0 mL ice-cold FCM Buffer, centrifuging at 20,000g at 4°C for 3 min after each wash. Gently remove the supernatant after each wash, taking care not to disturb the beads. For a chimney-bottom plate, wash two times with 0.2 mL ice-cold FCM Buffer, centrifuging at 1000g for 5 min at 4°C after each wash. A multichannel pipettor is useful for this step. To remove supernatant from beads in a chimney-bottom plate, 'flick' the plate once, then, while still upside-down, blot any drips from the edges of the wells. The residual volume in each well, containing the beads, will be about 20 μL.
Resuspend the beads in 200 μL FCM Buffer per sample. Transfer to labeled FACS tubes. Samples are now ready for FCM.
4. FCM Acquisition
The CML beads described in this protocol are 3 to 5 μm in diameter, approximately half the diameter of a quiescent mouse lymphocyte. Therefore, it may be necessary to manually increase the Forward Scatter (FSC) amp gain and the Side Scatter (SSC) voltage in order for the population of bead events to register on scale. Individual IP beads should form a single tightly clustered population. The settings and gate should be adjusted to exclude bead doublets and debris.
Prior to running beads or bead standards, remove the droplet containment sleeve that surrounds the sample inlet on some cytometers. Allow the sheath fluid to drip between samples, as this will clear the inlet and prevent beads from being carried over from one sample to another.
Use unlabeled beads, a negative control of fluorescence, and Rainbow Calibration Particles (RCPs), a positive control of fluorescence, to adjust settings so that both the negative and positive fluorescence extremes appear simultaneously on the log-scale x-axis. This ensures that the fluorescence from the experimental samples will also be on-scale. Note that RCPs are smaller than the CML beads, so FSC and SSC parameters, as well as gate position, may need to be temporarily altered for that sample.
Return to unlabeled-bead settings once calibration with RCPs is complete. If only one fluorescent probe per sample is used, no fluorescence compensation is necessary. In general, we probe multiprotein complexes with a single fluorochrome-conjugated mAb per FCM sample, and we examine multiple interaction partners by staining parallel bead samples with mAbs specific for the various subunits.
Acquire and save fluorescence data files. Analysis can then be performed using flow cytometry software such as CellQuest, FlowJo, or CFlow.
5. Representative Results
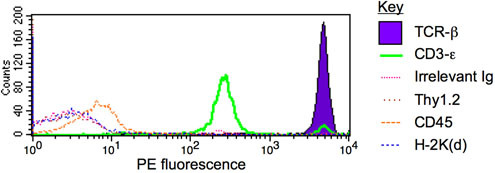 Figure 1. IP-FCM for TCR/CD3 multiprotein complex. T cells from the mouse strain BALB/c were lysed in 1% digitonin, and the lysate was subjected to IP-FCM using anti-CD3ε beads. The captured complexes contained significant quantities of TCR-β (purple region) and co-associated CD3-ε (green), but close to background levels (defined by the irrelevant immunoglobulin probe, pink trace) of other proteins such as Thy1.2, CD45, or H-2K(d) (brown, orange, and blue traces, respectively). See similar previously published results1-6.
Figure 1. IP-FCM for TCR/CD3 multiprotein complex. T cells from the mouse strain BALB/c were lysed in 1% digitonin, and the lysate was subjected to IP-FCM using anti-CD3ε beads. The captured complexes contained significant quantities of TCR-β (purple region) and co-associated CD3-ε (green), but close to background levels (defined by the irrelevant immunoglobulin probe, pink trace) of other proteins such as Thy1.2, CD45, or H-2K(d) (brown, orange, and blue traces, respectively). See similar previously published results1-6.
Discussion
Information about protein-protein interactions is highly relevant to the analysis of many cellular processes such as signal transduction, lineage maturation, cell-cycle progression, and apoptosis cascades. IP-FCM provides a quick, quantitative, and sensitive way to examine the interaction of proteins and define members of multiprotein complexes in their native conformation. Beads can be coupled and incubated with cell lysates in one day and can be probed and analyzed the next day. A 96-well plate format allows for large numbers of samples to be analyzed at one time to provide efficient data collection for statistical or screening purposes. Using fluorescent bead standards, the number of proteins captured by each bead can be estimated. Very little source material is needed for capture and detection, so limited samples and scarce analytes can still be analyzed for multiple interactions. Although IP-FCM does not require genetic engineering, epitope-tagging, denaturation, or in-vitro mixing of proteins in a non-physiological environment, it can be coupled with these and other techniques, making it a valuable and accessible tool with applicability to many biological systems.
Troubleshooting:
Many IP-FCM experiments generate useful protein interaction data the first time they are attempted. However, optimization of IP-FCM can improve antibody conjugation to beads, protein complex capture, and fluorescent probe binding.
The efficiency of IP antibody conjugation can be determined by probing coupled beads directly with an anti-immunoglobulin antibody. If this efficiency is low, increasing the concentration of antibody during the coupling reaction can allow more IP antibodies to attach to each bead. This can increase the binding capacity of the IP bead batch, resulting in enhanced capture and detection of analytes. Other primary-amine containing molecules (e.g. Tris, bovine serum albumin) should not be present during the coupling reaction, as these can compete with the mAb for bead attachment and result in low mAb coupling. If conjugation of mAb to beads proves problematic for other reasons, beads can instead be coupled to avidin/streptavidin, and biotinylated antibodies can subsequently be non-covalently bound and used for immunprecipitation.
Despite good antibody conjugation, initial detection of bead-associated fluorescence may be low. First, complex capture itself may be low. Because IP-FCM depends on the concentration of analytes, increasing the number of cells lysed per unit lysis volume can increase analyte capture and detection. Additionally, capture may be enhanced by decreasing the number of IP beads incubated with the lysate, which distributes captured complexes across fewer beads, and results in increased average fluorescence per bead when probed. Second, it is possible that access of the probe antibody to the captured complexes is sterically hindered by the immunoprecipitating antibody. In this case, the problem can be addressed by using different antibodies to capture and/or probe.
Acknowledgments
This work was supported by the Eagles Innovation Award (Fraternal Order of Eagles) and by the Mayo Foundation.
References
- Schrum AG. High-sensitivity detection and quantitative analysis of native protein-protein interactions and multiprotein complexes by flow cytometry. Sci STKE. 2007:pl2–pl2. doi: 10.1126/stke.3892007pl2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrum AG. Visualization of multiprotein complexes by flow cytometry. Curr Protoc Immunol. 2009;Chapter 5:9–9. doi: 10.1002/0471142735.im0509s87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeiro E. T cell division and death are segregated by mutation of TCRbeta chain constant domains. Immunity. 2004;21:515–526. doi: 10.1016/j.immuni.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Teixeiro E. Different T Cell receptor signals determine CD8+ memory versus effector development. Science. 2009;323:502–505. doi: 10.1126/science.1163612. [DOI] [PubMed] [Google Scholar]
- Treves S. Junctate is a key element in calcium entry induced by activation of InsP3 receptors and/or calcium store depletion. J Cell Biol. 2004;166:537–548. doi: 10.1083/jcb.200404079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil D, Schrum AG, Alarcon B, Palmer E. T cell receptor engagement by peptide-MHC ligands induces a conformational change in the CD3 complex of thymocytes. J Exp Med. 2005;201:517–522. doi: 10.1084/jem.20042036. [DOI] [PMC free article] [PubMed] [Google Scholar]