

Abstract
Cyclic-di-GMP and cyclic-di-AMP are second messengers produced by bacteria and influence bacterial cell survival, differentiation, colonization, biofilm formation, virulence, and bacteria-host interactions. Here, we show that in both RAW264.7 macrophage cells and primary bone-marrow–derived macrophages (BMM) the production of IFNβ and IL-6, but not TNF, in response to cyclic-di-AMP and cyclic-di-GMP requires MPYS (also known as STING, MITA, and TMEM173). Furthermore, expression of MPYS was required for interferon response factor (IRF)-3 but not nuclear factor κB (NFκB) activation in response to these bacterial metabolites. We also confirm that MPYS is required for type I IFN production by cultured macrophages infected with the intracellular pathogens Listeria monocytogenes and Francisella tularensis. However, during systemic infection with either pathogen, MPYS deficiency did not impact bacterial burdens in infected spleens. Serum IFNβ and IL-6 concentrations in the infected control and MPYS−/− mice were also similar at 24 hpi, suggesting that these pathogens stimulate MPYS-independent cytokine production during in vivo infection. Our findings indicate that bifurcating MPYS-dependent and -independent pathways mediate sensing of cytosolic bacterial infections.
Introduction
Macrophages are amongst the first cells of the immune system to encounter invading pathogens. Hence, the ability of macrophages to sense pathogens and their products plays a crucial role in the initiation of inflammation and immune responses. A key mechanism by which macrophages mediate their function involves production of immune regulatory cytokines such as TNF, IL-6, and IFN-α/β. TNF and IL-6 play a largely protective role in the host response to bacterial infections (1–3). In contrast, the role of type I IFN in bacterial infection is more complex (4). type I IFNs were suggested to protect mice against Salmonella typhimurium and group B streptococcal infections (5, 6), and suppress intracellular replication of Legionella pheumophila (7, 8). In contrast, type I IFNs increase host susceptibility to Listeria monocytogenes (9–12), Mycobacterium tuberculosis (13–15), and Francisella tularensis (16). The mechanisms by which type I IFNs promote susceptibility during these infections are not presently clear. Nevertheless, it is important to understand how infection of host cells by intracellular bacteria elicits the production of type I IFNs.
A series of recent studies has established the critical role of the cytoplasmic DNA sensor AIM2 (absent in melanoma 2) in host defense against cytoplasmic bacteria, including Listeria monocytogenes and Francisella tularensis infections (17–23). Mice deficient in AIM2 are extremely susceptible to F. tularensis, suffering greater mortality and bacterial burden than wild-type mice. The increased susceptibility of AIM2 mutant mice is associated with defective caspase-1 activation and IL-1β secretion by infected macrophages (18, 20, 21). However, AIM2 is not required for type I IFN production (24), indicating that sensing of bacterial DNA or DNA-like molecules by AIM2 contributes to caspase 1 activation but not type I IFN production.
Interestingly, type I IFN production in response to intracellular DNA was more recently shown to involve a distinct cytoplasmic DNA sensor, IFI16/p204 (25). This study suggested that IFI16 recruits STING/MPYS to activate the TBK1-IRF3 signaling pathway. Transfection of L. monocytogenes DNA into the cytosol of host cells also activates the TBK1-IRF3 pathway leading to IFNβ production (26), but the extent to which intact bacterial DNA accesses the cytosol of host cells during infection is not clear. Since bacterial multidrug efflux pumps enhance induction of IFN-α/β during L. monocytogenes infection, small molecule substrates of these pumps also appear to elicit host cell production of type I IFN (27). Possible small molecule substrates of such pumps include cyclic dinucleotide monophosphates, such as c-di-AMP and c-di-GMP.
C-di-GMP influence bacterial cell survival, differentiation, colonization, biofilm formation and bacteria-host interactions (28–31). Diverse immune cell populations have been shown to respond to c-di-GMP treatments both in vivo and in vitro. For example, c-di-GMP induces dendritic cell (DC) maturation and triggers the production of IFNβ and a number of other cytokines and chemokines by DCs and macrophages (28, 29). It has also been shown that the amount of c-di-AMP secreted by L. monocytogenes strains correlates linearly with their IFNβ-inducing activity (32). Additionally, cytosolic delivery of c-di-AMP induces production of type I IFNs (32). IFNβ production in response to cytosolic c-di-AMP or c-di-GMP is dependent on TBK1 and IRF3 but independent of MyD88/Trif and MAVS (29, 32).
MPYS has been shown to play an essential role in the induction of IFNβ by intracellular dsDNA and by L. monocytogenes (33). However, it is not known whether MPYS acts as a general sensor of cytosolic bacterial infection in macrophages or contributes to IFNβ production in response to c-di-AMP or c-di-GMP. In this report, we address these questions and show that MPYS is essential for macrophage IL-6 and IFNβ production in response to cytosolic delivery of c-di-AMP and c-di-GMP as well as infections by the cytosolic bacterial pathogens Listeria monocytogenes and Francisella tularensis. These data reveal an important role for MPYS in the detection of cytosolic bacterial pathogens and their cyclic dinucleotide metabolites.
Materials and Methods
Knockdown of MPYS in RAW264.7 macrophages
RAW264.7 cells were transduced as previously described with retroviruses expressing either MPYS knockdown or luciferase knockdown constructs (34). Transduced cells were selected using medium containing 8 μg/ml puromycin. The efficiency of MPYS knockdown was confirmed by immunoblotting using anti-MPYS Ab (34).
Generation of MPYS-Knock Out mice
Linearized targeting vector, which covers ~10kb of the genomic region in MPYS locus on mouse chromosome 18, was transfected into JM8A3. N1 ES cells originated from C57BL/6J strain, followed by the selection for neomycin positive and diphtheria toxin (DTA) negative clones. Targeted clones were screened by PCR. From 52 clones, 6 positive clones were identified. Two of these ES clones were subjected to the generation of chimera mice by injection using C57BL/6J blastocysts as the host. The male chimeras (chimerism >95% determined by coat color) were mated with C57BL/6J female mice for germline transmission. Both ES clones had successful germline transmission. The heterozygous mice were interbred to obtain wild-type, heterozygous and homozygous littermates. The genotypes of the mice were determined by genomic PCR and intracellular MPYS staining in mouse peripheral blood. Animals were generated at the National Jewish Health Mouse Genetics Core Facility. Animal care and handling was performed as per IACUC Guidelines.
Intracellular MPYS staining
Mouse blood was collected by cheek bleeding. Red blood cells were lysed and white cells were harvested and washed in FACS buffer (PBS with 2% FBS, 0.05% sodium azide and 0.2μg/ml 2.4g2 Fc-receptor blocking Ab). Cells were then re-suspended in BD Cytofix/Cytoperm™ buffer (BD Bioscience) for 20min at RT. BD Perm/Wash buffer™ (BD Bioscience) was added into the cell suspension. Cells were collected and washed with BD Perm/wash buffer™ again. Cells were suspended in BD Perm/wash buffer™ containing rabbit anti-MPYS Ab for 20min at RT. Cells were collected and washed with BD Perm/wash buffer™ twice, and incubated with goat anti-rabbit Alexa-647 for 20min at RT. Cells were collected and washed with BD Perm/wash buffer™, then analyzed using a FACSCalibur. Data were analyzed by Flow-Jo software (Tree Star, Inc., San Carlos, CA).
Bone-marrow-derived macrophage culture
Bone-marrow-derived macrophages were generated as described (35). Briefly, bone marrow cells harvested from mouse femurs were cultured in DMEM (GIBCO) containing 20% FBS (Biosource), 10% L cell conditioned media as a source of CSF-1, 2mM L-glutamine (GIBCO), 1mM Sodium Pyruvate (GIBCO), 100units/ml penicillin+100μg/ml streptomycin (GIBCO) and 50μM 2-ME. The medium was exchanged after three days, and cells were stained for the macrophage marker F4/80 at day 7.
L. monocytogenes and F. tularensis infections
Macrophages were seeded in 12-well plates at 1×106 cells/well. The following day the medium was exchanged with fresh antibiotic-free medium. Listeria monocytogenes (strain 10403S) were grown to log phase and added to the cell cultures at a multiplicity of 10 infectious bacterial cells per macrophage cells (MOI). After 30 min, gentamicin (GIBCO, 50μg/ml) was added to the medium to kill all extracellular bacteria. At 1 hr post infection, the medium was exchanged. Francisella tularensis (strain LVS) were grown to log phase and added to cell cultures at an MOI of 100. Two hours later, gentamicin (50μg/ml) was added to the medium to kill all extracellular bacteria and one hour later the medium was exchanged. At time points indicated, supernatants were collected and analyzed for cytokines by ELISA. Cells were harvested, washed in PBS, and then lysed in .02% NP40 buffer. Serial dilutions of lysate were plated onto TSB agar plates.
Mouse infections
Mice between 6 to 8 wk of age were used for all in vivo experiments. Mice were infected (tail vein) with 0.5–2×104 cfu of log-phase L. monocytogenes (10403S) or 1–5 × 104 cfu F. tularensis (strain LVS). After 48–72h, spleens were harvested. Bacterial CFUs in were determined by dilution plating as previously described (12).
Cyclic-di-GMP and Cyclic-di-AMP activation
Macrophages were seeded in 12-well plate at 1×106 cells/well overnight before medium was exchanged. Cells were treated with Lipofectamine according to the manufacturer’s instructions. Briefly, in a tube designated A, we added 50μl DMEM (GIBCO) and increasing concentrations of c-di-GMP (2, 10, and 50 μM final concentration; Biolog) or c-di-AMP (1, 3, and 9μM final concentration; Biolog); To a tube designated B, we added 50μl DMEM and 2μl of lipofectamine™ 2000 (Invitrogen). Each tube was then vortexed for 1 sec and incubated at RT for 5min, before the solution from tube A was transferred to tube B, followed by vortexing for 2 sec and incubation at RT for 20min. The mixture was then added dropwise into the macrophage culture followed by incubation at 37°C. At designated time points supernatants were collected for cytokine ELISAs, and cells were washed in PBS and harvested for total RNA.
Cytokine ELISAs
Supernatants from treated and control macrophages were collected at indicated time points to measure concentrations of IL-6 and TNF (BD Pharmingen), or IFNβ (InterferonSource) using commercial ELISAs. Supernatants were stored at −20°C before assay.
RT-PCR
Macrophages were cultured and treated as described. Total RNA was isolated from triplicate wells of macrophages at the indicated times using the Qiagen RNeasy Plus mini kit (Qiagen) and reverse-transcribed using oligodeoxythymidylate primers and the IMPROM II FT system (Promega) as previously described (12). cDNAs were used as template for 29–32 cycles of PCR with Taq polymerase (Invitrogen). Primers and probes used quantitative PCR (TaqMan method) were: IFNβ sense (CTCATCATTCGAAGACTTACCAGAAAC); IFNβ antisense (CAGAGTCCGCCTCTGATGCT); IFNβ probe (ATGCCTCAGAATGAGTGGTGGTTGCA); GAPDH sense (GGGAAGCCCATCACCATCTT); GAPDH antisense (ACATACTCAGCACCGGCCTC); GAPDH probe (AGCGAGACCCCACTAACATCAAATGGG). ifnb transcript abundance was normalized to gapdh.
Results
MPYS knockdown selectively impairs IL-6 and IFNβ production in response to Lm and Ft infections in RAW264.7 cells
To investigate the requirement for MPYS in macrophage sensing of cytosolic bacterial pathogens, we used RAW 264.7 macrophage cell line that stably express short hairpin RNAs (shRNAs) targeting either luciferase (lucKD) or exon 5 of MPYS (MPYSKD) that have been reported before (36). Immunoblotting of cell lysates with anti-MPYS Ab confirmed the efficiency of MPYS knockdown in the MPYSKD, but not lucKD cells (Fig 1A). We next investigated the effects of MPYS knockdown on cytokine production following infection of the lucKD and MPYSKD cells with Lm or Ft. Both cells responded to LPS treatment with robust secretion of IFNβ protein (Fig. 1B). However, only the control cells efficiently secreted IFNβ when infected with Lm (MOI of 10) or Ft (MOI of 100). Likewise, the ability of macrophages to respond to infection by secretion of IL-6, but not TNF, required MPYS (Fig. 1C, D). These effects were not due to any discernable effects of MPYS expression on the ability of bacteria to infect the macrophages (not shown); the doubling times of Lm in lucKD and MPYSKD cells were 52.6 and 50 min. Thus, our data reveal the MPYS is essential for production of IL-6 and IFNβ, but not TNF, in response to cytosolic bacterial infection by both Gram-positive (L. monocytogenes) and Gram-negative (F. tularensis) bacteria.
Figure 1. MPYS is required for IFNβ production in response to infection with Listeria monocytogenes and Francisella tularensis.
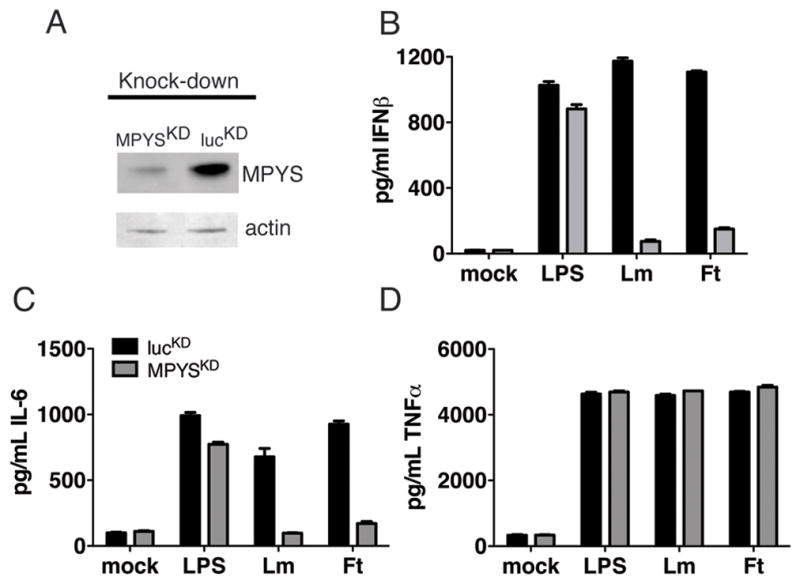
A Whole Cell Lysate (WCL) from RAW264.7 cells expressing luciferase knockdown (lucKD) or MPYS-knockdown construct (MPYSKD) were fractionated by SDS- Polyacrylamide Gel Electrophoresis (PAGE) (10% NuPAGE™), transferred to nitrocellulose and stained with anti-MPYS or anti-Actin Ab respectively. B–D. lucKD or MPYSKD macrophages were infected with Listeria monocytogenes (MOI 10) or Francisella tularensis LVS (MOI 100). Supernatants were collected at 12 hpi and ELISAs were performed for (B.) IFNβ, (C.) IL-6, and (D.) TNF.
MPYS knoc kdown impairs the recognition of cyclic dinucleotide monophosphates in RAW264.7 cells
It has been shown that secreted levels of the second messenger c-di-AMP correlate with the degree of type I IFN production in response to L. monocytogenes infection (32). Cytosolic delivery of the bacterial metabolites c-di-AMP or c-di-GMP to macrophage elicits IFNβ production (29, 32). To evaluate whether MPYS participates in sensing of these bacterial metabolites, we compared ifnb transcription and IFNβ secretion by lucKD and MPYSKD cells with increasing concentrations of cytosolic synthetic c-di-AMP and c-di-GMP (Fig. 2). The results clearly showed that the MPYSKD macrophages failed to produce IFNβ in response to either cyclic dinucleotide monophosphate.
Figure 2. MPYS is required for IFN β production in response to treatment with cyclic dinucleotides.

A RAW264.7 lucKD or MPYSKD macrophages were treated with 9 μM c-di-AMP or 50 μM c-di-GMP and harvested for RNA at 12 hr post treatment. Quantitative PCR (TaqMan method) was performed for IFNβ gene expression and results were normalized to those of GAPDH. B. RAW264.7 lucKD or MPYSKD cells were treated with increasing concentrations of c-di-AMP or c-di-GMP as before. At 12 hr post treatment, supernatants were collected and analyzed by ELISA for IFNβ production.
MPYS is required for IRF3 activation in response to cyclic dinucleotide monophosphates in RAW264.7 cells
Expression of TBK1 and IRF3 are required for production of IFN-α/β by mouse bone marrow derived macrophages (BMM) infected with Lm or cytosolic delivery of c-di-AMP or c-di-GMP (29, 32). We further observed that introduction of either cyclic dinucleotide monophosphate caused a prolonged increase in phosphorylation of IRF3 at Ser96 (p-IRF3) in the BMM (Fig 3A). Likewise, this prolonged increase in absolute and relative p-IRF3 was seen in control RAW264.7 lucKD cells receiving either c-di-GMP or c-di-AMP (Figs. 3B–E). Unlike control cells, MPYSKD macrophages failed to accumulate p-IRF3 in response to either c-di-AMP or c-di-GMP (Figs. 3B–E). Yet, MPYS was not required for the induced degradation of IκBα (Figs. 3B–E). These data indicate that MPYS lies upstream of IRF3 activation in the pathway leading from cyclic dinucleotide monophosphate sensing to ifnb transcription, but this requirement for MPYS does not extend to NFκB activation.
Figure 3. MPYS is selectively required for IRF3 activation in response to treatment with cyclic dinucleotides.

A Bone-marrow-derived macrophages (BMM) were treated with 20μM synthetic c-di-GMP or c-di-AMP as described in materials and methods. Whole cell lysates (WCL) were separated by SDS- PAGE (10% NuPAGE™), transferred to PVDF, and probed with Abs against p-IRF3 (Cell Signaling) and Actin (Santa Cruz). B. RAW264.7 lucKD or MPYSKD macrophages were treated with c-di-GMP (20μM) as before and harvested at the indicated times. WCL were separated and blotted as in A, then probed with Abs to p-IRF3, IκBα (Cell Signaling), actin or MPYS (34). C. The intensities of p-IRF3 (left panel) and IκBα (right panel) bands in B. were quantified using Odyssey 2.1 software and normalized to actin. The relative intensities of p-IRF3 and IκBα are plotted against time. D. RAW264.7 lucKD cells and MPYSKD cells were treated with c-di-AMP (20μM) as before. Activation of IRF3 and degradation of IκBα were evaluated as in B. E. The relative intensities of p-IRF3 and IκBα were determined as in C. Each experiment in this Figure has been done for at least three times.
We also evaluated IRF3 activation and IκBα degradation following Lm infection. In both BMM and RAW264.7 lucKD cells, we observed MPYS-dependent p-IRF3 at 3 h after infection, and MPYS-independent IκBα degradation within 4 h of infection (Supplemental Fig. 1).
MPYS-deficient BMM cells do not make IFNβ in response to cyclic dinucleotide monophosphates
To extend our observation that MPYS is required for sensing intracellular cyclic dinucleotide monophosphates to the primary cells, we used BMM cells generated from MPYS-KO mice recently developed in Dr. Cambier’s lab (Jin, L and Cambier JC, full characterization of the KO mice will be described elsewhere). The KO mice were generated through homologous recombination (Fig 4A) and the homologous integration event was confirmed by genomic PCR and sequencing (Supplemental Fig 2A, 2B, 2C). The endogenous MPYS gene was replaced by the targeting construct that has a neo gene inserted into the intron 5 of MPYS gene (Fig 4A). Western blot in splenocytes (Fig 4B) and intracellular staining in peripheral blood (Supplemental Fig 2D) confirmed the lack of MPYS protein expression. RT-PCR in cells from peripheral blood, using primers that amply exons 2, 3 and 4 of MPYS gene, also confirmed an absence of MPYS transcripts (Fig 4C). We next confirmed the requirement for MPYS expression in the response to c-di-GMP using BMM cultured from wt, MPYS+/−, and MPYS−/− mice (Fig. 5). Cytosolic delivery of c-di-GMP resulted in significant IFNβ secretion from the BMM only when MPYS was expressed. These results indicated that MPYS is required for the IFNβ response of BMM to cyclic di-nucleotide monophosphate.
Figure 4. Generation of MPYS-knockout mice by homologous recombination.

A Strategy to generate MPYS-knock out mice. The genomic sequence of mouse MPYS gene is derived from a BAC clone RP24-490M12 (~140kb). MPYS gene consists of 8 exons and spans from 65182 to 72058bp in the BAC. Protein translation starts from exon 3. A hypothetic gene, 1700066B19Rik, is ~2.8kb downstream of MPYS and there is no protein-coding gene in the ~30kb region upstream of MPYS. The targeting construct covers ~10kb genomic region in the MPYS locus (64515~74175). The targeting construct has a neo gene inserted in the intron 5 and a diphtheria toxin gene at the 3 end of the MPYS gene. The neo gene is flanked by Frt elements and one LoxP site. Another LoxP site is inserted in the intron 2. Thus, using tissue specific Flp or Cre transgenic mice, we can also generate conditional KO or conditional WT MPYS mice. B. Western blot analysis of MPYS from WT, KO and heterozygous MPYS littermates. Splenocytes were lysed in RIPA buffer containing 0.1% SDS and run on a reducing SDS-PAGE gel. The blot was probed with rabbit α-MPYS Ab. N.S. non-specific. This experiment has been repeated more than three times. C. RT-PCR analysis of MPYS transcript from peripheral blood. RT-PCR was done using cDNA from peripheral blood cells of WT, KO and heterozygous MPYS littermates with primers for exon 2, 3, and 4 of the MPYS transcript. This experiment has been repeated twice.
Figure 5. MPYS-deficient BMM cells are defective for IFNβ production in response to treatment with cyclic dinucleotide monophosphates.
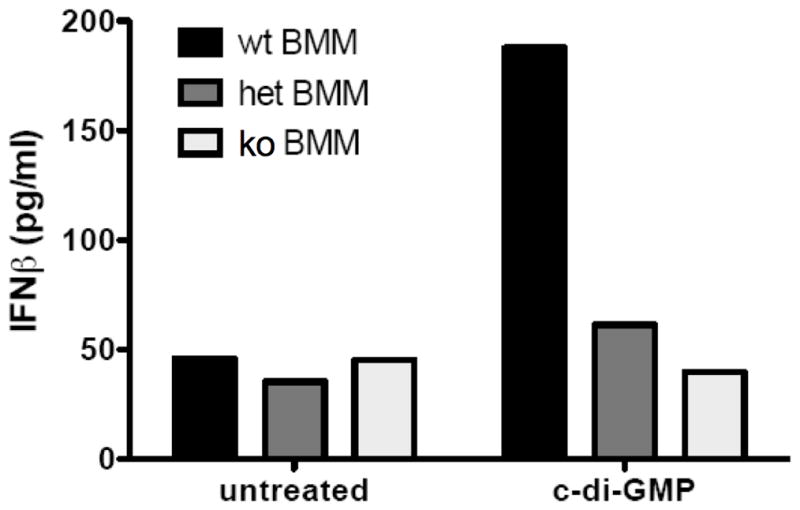
Bone-marrow-derived macrophages (BMM) from indicated mice were treated with 50μM synthetic c-di-GMP as before. At 12 hr post treatment, supernatants were collected and analyzed by ELISA for IFNβ production. Experiments were repeated twice with similar results.
Cyclic dinucleotide monophosphates fail to activate IRF3 in MPYS-deficient BMM
We next evaluated c-di-NMP induced IRF3 and NFκB activation in BMM from MPYS−/− and WT mice. Consistent with our observations in RAW264.7 MPYSKD cells (Fig. 3), MPYS−/− BMM cells did not have detectable phosphorylation of IRF3 following intracellular introduction of c-di-GMP and c-di-AMP, though the degradation of IκBα was still observed (Fig 6A and 6B). Thus MPYS is required for c-di-NMP induced IRF3 activation, but not NFκB activation, in RAW264.7 macrophage cell line as well as primary BMM.
Figure 6. MPYS-deficient BMM cells fail to activate IRF3 in response to treatment with cyclic dinucleotide monophosphates.

A–B Bone-marrow-derived macrophages (BMM) from indicated mice were treated with 20μM synthetic c-di-GMP (A.) or c-di-AMP (B.) for indicated times as before. WCL were separated by SDS-PAGE and probed with indicated Abs as in Fig 3. These experiments have been repeated twice.
The apparent MW of MPYS increases in response to L. monocytogenes and c-di-GMP activation
To further confirm the involvement of MPYS in L. monocytogenes and c-di-NMP induced type I IFN response, we investigate whether MPYS is activated by these stimuli. Previous studies indicated that MPYS forms transient homodimer in response to Sendai virus infection (36, 37) and intracellular dsDNA in 293T cells (38). We found that a high molecular weight MPYS form corresponding to the homodimer size was also elicited in response to L. monocytogenes infection of 293T cells (Fig. 7A). Furthermore, cytosolic c-di-GMP induced transient formation of these putative MPYS homodimers in BMM (Fig. 7B). These data suggest that homodimer formation is a hallmark of MPYS activation in response to diverse infectious and microbial stimuli.
Figure 7. MPYS forms homodimers in response to L. monocytogenes infection and cytosolic c-di-GMP activation.

A. HEK-293T cells were infected with L. monocytogenes as in Fig. 1 for the indicated time. Cells were then lysed in RIPA buffer containing 0.1% SDS and fractionated using non-reducing SDS-PAGE gels. Blots were probed with the anti-MPYS Ab. B. BMM cells were treated with c-di-GMP (20μM) as in Fig. 3. Cells were then lysed and fractionated on a non-reducing gel. Blots were probed with indicated Abs. High molecular weight MPYS that previous studies have reported to be homodimers are indicated. These experiments have been repeated more than three times.
MPYS deficiency does not dramatically impact bacterial burdens during L. monocytogenes and F. tularensis infection
As mentioned above, mice deficient for expression of the IFNAR demonstrate increased resistance to L. monocytogenes and F. tularensis. To determine whether MPYS deficiency might likewise reduce host susceptibility, we compared bacterial burdens from control B6, MPYS+/−, and MPYS−/− mice infected with L. monocytogenes. We found that bacterial burdens in spleens were comparable in infected WT and MPYS−/− mice at 72 hpi (Fig. 8A). This was a surprising observation since under similar conditions IFNAR1−/− mice have significantly reduced burdens of L. monocytogenes ((12); and not shown). We also found that F. tularensis burdens were similar at 48 hpi in WT, MPYS+/−, and MPYS−/− mice (Fig. 8B). Consistent with their similar susceptibilities, we found that MPYS deficiency significantly reduced production of IFNβ and IL-6 at early times (8 h) after L. monocytogenes and F. tularensis infections (Fig 8C and 8D, Supplemental Fig. 3), but not at later (24 h) times (Fig 8E and 8F). These data suggest MPYS is required only for the initial IFNβ and IL-6 production in response to infections by these cytosolic bacterial pathogens.
Figure 8. MPYS is required for early but not late IFN β or IL-6 productionin vivo.

A–B. MPYS−/−, MPYS+/− and their WT littermates (B6) were infected with 10,000 cfu of L. monocytogenes (i.v) A. or 5,000 cfu F. tularensis (i.p.) B. Spleens were harvested, homogenized and dilution plated to determine bacterial burdens 72h (A.) or 48h (B.) later. Each point indicates an individual mouse. Bars indicate the mean values. C-F. MPYS−/−, MPYS+/− and their WT littermates (B6) (n=3) were infected as above with L. monocytogenes (C. & E.) or F. tularensis (D. & F.). Sera were collected at indicated times. IFNβ and IL-6 concentrations were measured by ELISA. Experiments were done twice.
Discussion
In this report we established that the multi-transmembrane protein MPYS, plays an essential role in the sensing of cytosolic cyclic dinucleotide monophosphate in macrophages. MPYS mediated the activation of IRF3, but not NFκB, in response to the bacterial metabolites c-di-AMP and c-di-GMP. Thus, our findings are consistent with the notion that macrophages sense both Gram-positive and Gram-negative cytosolic bacteria via ligation of a receptor or receptors for cyclic dinucleotide metabolites that are released from these bacteria. While this manuscript was under review, Dr. Vance’s group also published evidence that implicates MPYS/STING in type I IFN production in response to c-di-NMPs (39).
Bacterial second messengers c-di-AMP and c-di-GMP are ubiquitously expressed in bacterial species but absent in higher eukaryotes (39). It was previously suggested that c-di-GMP acts as a danger signal when sensed eukaryotic cells (40). Indeed, several studies have shown that c-di-GMP has strong immunostimulatory properties (28, 41). More recent studies demonstrated that introduction of c-di-GMP or c-di-AMP into macrophages also activated type I IFN production independent of TLRs and intracellular RNA sensing pathways (29). Our studies thus provide mechanistic insight into the host response to these ubiquitously expressed bacterial second messengers. Specifically, by demonstrating an essential role of MPYS in the activation of IRF3 (but not NFκB) and the production of type I IFNs by macrophages treated with c-di-AMP, c-di-GMP, or infections with L. monocytogenes and F. tularensis. These studies do not necessarily indicate that MPYS is itself a receptor for c-di-AMP or c-di-GMP. Rather, the differences in kinetics of IRF3 activation in response to c-di-GMP and c-di-AMP and the lack of a requirement for MPYS in the activation of NFκB by these ligands, suggest that a proximal receptor or receptors is responsible for binding these cyclic dinucleotides prior to independent activation of both the MPYS-TBK1-IRF3 and the NFκB pathways.
MPYS is a potent type I IFN stimulator (33). It mediates type I IFN responses to intracellular dsDNA of bacteria or mammalian origin (42). This intracellular dsDNA response is motif-independent but length-dependent (26). Recent studies identified the proteins AIM2 and IFI16 as sensors of intact cytosolic DNA. AIM2 activates caspase-1 and subsequent release of IL1β and IL-18 during Lm and Ft infections (17–22, 43). It is not known whether AIM2 also senses the presence of bacterial cyclic dinucleotides, however, AIM2 does not mediate the activation of IRF3 and thus does not lead to production of type I IFNs (24, 25). IFI16 interacts with MPYS and activates IRF3 in response to dsDNA of > 25 nt (~13kDa) (25). Thus, while IFI16 may activate MPYS-IRF3 in response to large dsDNA fragments released from dying bacteria, we speculate that another receptor may sense the small molecule cyclic dinucleotide monophosphates (~600Da). Regardless of the receptors involved in cyclic dinucleotide sensing by phagocytes, it will be important to better understand how MPYS interacts with such receptors and activates the TBK1-IRF3 pathway.
Our data here have confirmed previous reports that MPYS is important for type I IFN production in response to in vitro infection of macrophages by L. monocytogenes and F. tularensis (18, 33), but indicate that the situation is more complex during in vivo infections. Consistent with our observation that wt and MPYS−/− mice have comparable bacterial loads during systemic L. monocytogenes and F. tularensis infections, MPYS was required only for the early (8hpi) but not late (24hpi) IFNβ and IL-6 production in response to these bacteria. These data are reminiscent of results from Dr. Barber’s group showing that, in vivo, MPYS was required only for early (6hpi) but not late (12hpi) IFNα and β production in response to vesicular stomatitis virus (VSV) infection. Likewise, Sauer et al published that mutations in MPYS did not impact splenic L. monocytogenes burdens at 24hpi (39). At this early time, bacterial burdens in tissues of wt and IFNAR1−/− mice are similar, but they diverge by 72 hpi with L. monocytogenes. We thus evaluated L. monocytogenes burdens at this later time (Fig. 7) and found that MPYS−/− mice also harbored similar bacterial loads in the spleens at this time. Together, these data demonstrate that alternate, MPYS-independent, mechanisms exist to elicit production of type I IFNs and the ensuing suppression of host resistance during in vivo L. monocytogenes and F. tularensis infections.
Although MPYS was clearly required for production of type I IFN in response to L. monocytogenes infection of BMM and GM-DC (Supplemental FigS4; (42)), our data from infected mice revealed additional MPYS-independent production of type I IFN in vivo. A subpopulation of macrophages or TNF and iNOS-producing DC (Tip-DCs) are thought to be the main source for in vivo production of IFNβ during in vivo L. monocytogenes infection (44–46). The type I IFN production by these cells, or perhaps other cell types that are infected in vivo, is presumably independent of MPYS and may instead utilize other stimulators. It will be of interest to investigate the nature of this MPYS-independent pathway or pathways.
The detection of bacterial components in the cytosol of macrophages appears to be the major mechanism responsible for type I IFN production by cultured phagocytes. It was originally thought that such detection enabled the host to stimulate an appropriate response to the presence of intracellular bacteria (47). Yet, many of the intracellular bacteria that elicit production of type I IFNs appear to replicate better in mice that are capable of responding to these cytokines. It is unclear why the host produces type I IFNs during bacterial infections in which the production of such IFNs is deleterious to host resistance. It is also unclear why type I IFN production (often counter-protective) and IL-6 production (often protective) are linked. We and others have speculated that IFN production may be a consequence of the strong evolutionary pressure to respond against aggressive viral infections, where type I IFNs are protective. Alternatively, type I IFN production may limit damage from unchecked inflammatory responses (48, 49). Alternatively, or additionally, certain pathogens may have evolved strategies to avoid the antibacterial consequences of type I IFNs while benefiting from the ability of these cytokines to down-regulate inflammatory responses.
In summary, the results of this study revealed that MPYS is required for type I IFN production by cultured macrophages and GM-DCs in response to the bacterial second messengers c-di-AMP and c-di-NMP. However, studies with MPYS−/− mice reveal that additional mechanisms for type I IFN production during in vivo bacterial infections. Further study is needed to characterize the nature of these mechanisms and their contributions to host defense as well as host susceptibility to infectious and other diseases.
Supplementary Material
Footnotes
Supported in part by NIH grants R01AI065638 and R01AI055701 to L.L.L. and R01AI062739-05S1, R01AI062739-05S2, R01AI062739-05, and P01AI0222950-22 to J.C.C. J.C.C. is an Ida and Cecil Green Professor of Immunology.
References
- 1.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 2.Dinarello CA. Anti-cytokine therapeutics and infections. Vaccine. 2003;21(Suppl 2):S24–34. doi: 10.1016/s0264-410x(03)00196-8. [DOI] [PubMed] [Google Scholar]
- 3.Raychaudhuri SP, Nguyen CT, Raychaudhuri SK, Gershwin ME. Incidence and nature of infectious disease in patients treated with anti-TNF agents. Autoimmun Rev. 2009;9:67–81. doi: 10.1016/j.autrev.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. 2005;5:675–687. doi: 10.1038/nri1684. [DOI] [PubMed] [Google Scholar]
- 5.Freudenberg MA, Merlin T, Kalis C, Chvatchko Y, Stubig H, Galanos C. Cutting edge: a murine, IL-12-independent pathway of IFN-gamma induction by gram-negative bacteria based on STAT4 activation by Type I IFN and IL-18 signaling. J Immunol. 2002;169:1665–1668. doi: 10.4049/jimmunol.169.4.1665. [DOI] [PubMed] [Google Scholar]
- 6.Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, Tomasello F, Gambuzza M, Macri G, Ruggeri A, Leanderson T, Teti G. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol. 2007;178:3126–3133. doi: 10.4049/jimmunol.178.5.3126. [DOI] [PubMed] [Google Scholar]
- 7.Opitz B, Vinzing M, van Laak V, Schmeck B, Heine G, Gunther S, Preissner R, Slevogt H, N’Guessan PD, Eitel J, Goldmann T, Flieger A, Suttorp N, Hippenstiel S. Legionella pneumophila induces IFNbeta in lung epithelial cells via IPS-1 and IRF3, which also control bacterial replication. J Biol Chem. 2006;281:36173–36179. doi: 10.1074/jbc.M604638200. [DOI] [PubMed] [Google Scholar]
- 8.Coers J, Vance RE, Fontana MF, Dietrich WF. Restriction of Legionella pneumophila growth in macrophages requires the concerted action of cytokine and Naip5/Ipaf signalling pathways. Cell Microbiol. 2007;9:2344–2357. doi: 10.1111/j.1462-5822.2007.00963.x. [DOI] [PubMed] [Google Scholar]
- 9.O’Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, Perry AK, Nguyen BO, Lane TF, Taniguchi T, Miller JF, Cheng G. Type I Interferon Production Enhances Susceptibility to Listeria monocytogenes Infection. J Exp Med. 2004;200:437–445. doi: 10.1084/jem.20040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to listeria infection. J Exp Med. 2004;200:535–540. doi: 10.1084/jem.20040769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Auerbuch V, Brockstedt DG, Meyer-Morse N, O’Riordan M, Portnoy DA. Mice Lacking the Type I Interferon Receptor Are Resistant to Listeria monocytogenes. J Exp Med. 2004;200:527–533. doi: 10.1084/jem.20040976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J Exp Med. 2010;207:327–337. doi: 10.1084/jem.20091746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, Barry CE, 3rd, Freedman VH, Kaplan G. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc Natl Acad Sci U S A. 2001;98:5752–5757. doi: 10.1073/pnas.091096998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stanley SA, Johndrow JE, Manzanillo P, Cox JS. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol. 2007;178:3143–3152. doi: 10.4049/jimmunol.178.5.3143. [DOI] [PubMed] [Google Scholar]
- 15.Antonelli LR, Gigliotti Rothfuchs A, Goncalves R, Roffe E, Cheever AW, Bafica A, Salazar AM, Feng CG, Sher A. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J Clin Invest. 2010;120:1674–1682. doi: 10.1172/JCI40817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henry T, Kirimanjeswara GS, Ruby T, Jones JW, Peng K, Perret M, Ho L, Sauer JD, Iwakura Y, Metzger DW, Monack DM. Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. J Immunol. 2010;184:3755–3767. doi: 10.4049/jimmunol.0902065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsuchiya K, Hara H, Kawamura I, Nomura T, Yamamoto T, Daim S, Dewamitta SR, Shen Y, Fang R, Mitsuyama M. Involvement of absent in melanoma 2 in inflammasome activation in macrophages infected with Listeria monocytogenes. J Immunol. 2010;185:1186–1195. doi: 10.4049/jimmunol.1001058. [DOI] [PubMed] [Google Scholar]
- 18.Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, Dixit VM, Monack DM. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci U S A. 2010;107:9771–9776. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sauer JD, Witte CE, Zemansky J, Hanson B, Lauer P, Portnoy DA. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe. 2010;7:412–419. doi: 10.1016/j.chom.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, Eisenlohr L, Landel CP, Alnemri ES. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol. 2010;11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim S, Bauernfeind F, Ablasser A, Hartmann G, Fitzgerald KA, Latz E, Hornung V. Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur J Immunol. 2010;40:1545–1551. doi: 10.1002/eji.201040425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu J, Fernandes-Alnemri T, Alnemri ES. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J Clin Immunol. 2010;30:693–702. doi: 10.1007/s10875-010-9425-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hornung V, Latz E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur J Immunol. 2010;40:620–623. doi: 10.1002/eji.200940185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Crimmins GT, Herskovits AA, Rehder K, Sivick KE, Lauer P, Dubensky TW, Jr, Portnoy DA. Listeria monocytogenes multidrug resistance transporters activate a cytosolic surveillance pathway of innate immunity. Proc Natl Acad Sci U S A. 2008;105:10191–10196. doi: 10.1073/pnas.0804170105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karaolis DK, Means TK, Yang D, Takahashi M, Yoshimura T, Muraille E, Philpott D, Schroeder JT, Hyodo M, Hayakawa Y, Talbot BG, Brouillette E, Malouin F. Bacterial c-di-GMP is an immunostimulatory molecule. J Immunol. 2007;178:2171–2181. doi: 10.4049/jimmunol.178.4.2171. [DOI] [PubMed] [Google Scholar]
- 29.McWhirter SM, Barbalat R, Monroe KM, Fontana MF, Hyodo M, Joncker NT, Ishii KJ, Akira S, Colonna M, Chen ZJ, Fitzgerald KA, Hayakawa Y, Vance RE. A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J Exp Med. 2009;206:1899–1911. doi: 10.1084/jem.20082874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romling U, Gomelsky M, Galperin MY. C-di-GMP: the dawning of a novel bacterial signalling system. Mol Microbiol. 2005;57:629–639. doi: 10.1111/j.1365-2958.2005.04697.x. [DOI] [PubMed] [Google Scholar]
- 31.Tamayo R, Pratt JT, Camilli A. Roles of cyclic diguanylate in the regulation of bacterial pathogenesis. Annu Rev Microbiol. 2007;61:131–148. doi: 10.1146/annurev.micro.61.080706.093426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woodward JJ, Iavarone AT, Portnoy DA. c-di-AMP Secreted by Intracellular Listeria monocytogenes Activates a Host Type I Interferon Response. Science. 2010;328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin L, Waterman PM, Jonscher KR, Short CM, Reisdorph NA, Cambier JC. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol Cell Biol. 2008;28:5014–5026. doi: 10.1128/MCB.00640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Humann J, Bjordahl R, Andreasen K, Lenz LL. Expression of the p60 autolysin enhances NK cell activation and is required for Listeria monocytogenes expansion in IFN-{gamma}-responsive mice. J Immunol. 2007;178:2407–2414. doi: 10.4049/jimmunol.178.4.2407. [DOI] [PubMed] [Google Scholar]
- 36.Jin L, Lenz LL, Cambier JC. Cellular reactive oxygen species inhibit MPYS induction of IFNbeta. PLoS One. 2010;5:e15142. doi: 10.1371/journal.pone.0015142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, Jiang Z. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci U S A. 2009;106:8653–8658. doi: 10.1073/pnas.0900850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, Kawai T, Akira S. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity. 2010;33:765–776. doi: 10.1016/j.immuni.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 39.Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, Vance RE. The ENU-induced Goldenticket (Gt) mouse mutant reveals an essential function of Sting (Tmem173, Mita, Mpys, Eris) in the in vivo interferon response to Listeria monocytogenes and cyclic-di-nucleotides. Infect Immun. 2010;79:688–694. doi: 10.1128/IAI.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karaolis DK, Cheng K, Lipsky M, Elnabawi A, Catalano J, Hyodo M, Hayakawa Y, Raufman JP. 3′,5′-Cyclic diguanylic acid (c-di-GMP) inhibits basal and growth factor-stimulated human colon cancer cell proliferation. Biochem Biophys Res Commun. 2005;329:40–45. doi: 10.1016/j.bbrc.2005.01.093. [DOI] [PubMed] [Google Scholar]
- 41.Ogunniyi AD, Paton JC, Kirby AC, McCullers JA, Cook J, Hyodo M, Hayakawa Y, Karaolis DK. c-di-GMP is an effective immunomodulator and vaccine adjuvant against pneumococcal infection. Vaccine. 2008;26:4676–4685. doi: 10.1016/j.vaccine.2008.06.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warren SE, Armstrong A, Hamilton MK, Mao DP, Leaf IA, Miao EA, Aderem A. Cutting edge: Cytosolic bacterial DNA activates the inflammasome via Aim2. J Immunol. 2010;185:818–821. doi: 10.4049/jimmunol.1000724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stockinger S, Kastner R, Kernbauer E, Pilz A, Westermayer S, Reutterer B, Soulat D, Stengl G, Vogl C, Frenz T, Waibler Z, Taniguchi T, Rulicke T, Kalinke U, Muller M, Decker T. Characterization of the interferon-producing cell in mice infected with Listeria monocytogenes. PLoS Pathog. 2009;5:e1000355. doi: 10.1371/journal.ppat.1000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Solodova E, Jablonska J, Weiss S, Lienenklaus S. Production of IFN-beta during Listeria monocytogenes Infection Is Restricted to Monocyte/Macrophage Lineage. PLoS One. 2011;6:e18543. doi: 10.1371/journal.pone.0018543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dresing P, Borkens S, Kocur M, Kropp S, Scheu S. A fluorescence reporter model defines “Tip-DCs” as the cellular source of interferon beta in murine listeriosis. PLoS One. 2010;5:e15567. doi: 10.1371/journal.pone.0015567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Riordan M, Yi CH, Gonzales R, Lee KD, Porntoy DA. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc Natl Acad Sci U S A. 2002;99:13861–13866. doi: 10.1073/pnas.202476699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rayamajhi M, Humann J, Kearney S, Hill KK, Lenz LL. Antagonistic crosstalk between type I and II interferons and increased host susceptibility to bacterial infections. Virulence. 2010;1:1–5. doi: 10.4161/viru.1.5.12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trinchieri G. Type I interferon: friend or foe? J Exp Med. 2010;207:2053–2063. doi: 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.