

Abstract
The first series of peptidyl aldehyde inhibitors that incorporate in their structure a glutamine surrogate has been designed and synthesized based on the known substrate specificity of Norwalk virus 3C protease. The inhibitory activity of the compounds with the protease and with a norovirus cell-based replicon system was investigated. Members of this class of compounds exhibited noteworthy activity both in vitro and in a cell-based replicon system.
Keywords: norovirus 3C protease, transition state inhibitors
Noroviruses are a leading cause of food-borne and water-borne non-bacterial acute gastroenteritis.1 Norovirus infections constitute an important health problem with an estimated 23 million cases of gastroenteritis occurring annually in the U.S., causing 50,000 hospitalizations and 300 deaths.2 There are currently no effective vaccines or antiviral therapeutics for the treatment of norovirus infection.
Noroviruses are small enveloped viruses of the Caliciviridae family.3 The genome of the Norwalk virus, a prototype of noroviruses, is comprised of a single-stranded, positive sense RNA molecule of ~7.7 Kilo bases that consists of three open reading frames (ORFs) that encode a 200 kDa polyprotein (ORF1), a major capsid protein VP1 (ORF2), and a small basic protein VP2 (ORF3). The mature polyprotein is co- and post-translationally processed by a virus-encoded protease to generate mature non-structural proteins.4 Processing of the mature polyprotein is mediated by this 3C protease, a (chymo)trypsin-like cysteine protease having a Cys-His-Glu catalytic triad and an extended binding site. The substrate specificity of norovirus 3C protease has been determined using in-vitro transcription/translation studies, and peptidyl chromogenic and fluorogenic substrates.5-7 The protease shows a strong preference for a –D/E-F/Y-X-L-Q-G-P- sequence (where X is H, E or Q) corresponding to the subsites S5-S4-S3-S2-S1-S1’-S2’-. Cleavage is at the P1-P1’ (Q-G) scissile bond. X-ray crystal structures of norovirus 3C protease alone8-9 or covalently-bound to an inhibitor, a peptidyl Michael acceptor, have been reported.7
Norovirus 3C protease plays an essential role in virus replication, consequently, orally-bioavailable drug-like agents that inhibit the 3C protease are of value as potential antiviral therapeutics. We describe herein the results of preliminary studies related to the inhibition of Norwalk virus 3C protease by a series of peptidyl aldehyde inhibitors (Figure 1).
Figure 1.

General representation of the interaction between a cysteine protease and a transition state inhibitor.
Initial design considerations included the use of a glutamine surrogate10 for optimal synthetic tractability and design flexibility (vide infra). Furthermore, our overarching goal was to identify a suitably-functionalized di-peptide or tri-peptide inhibitor that could be further transformed into a molecule possessing molecular properties that are important for oral bioavailability and favorable ADME/Tox characteristics.11-13 The design of the inhibitors was further augmented by insights gained via the use of computer graphics and modeling and the X-ray crystal structure of the enzyme.7 The synthesis of inhibitors 1-10 was carried out as shown in Scheme 1.14 The glutamine surrogate starting material was synthesized using literature procedures.15
Scheme 1.

Synthesis of inhibitors 1-10
Deblocking with TFA, followed by coupling with an appropriate Cbz-protected amino acid ester, yielded a product which was subsequently reduced to the alcohol with lithium borohydride. Dess-Martin oxidation yielded the desired aldehydes. Alpha-ketoamide 10 was synthesized by reacting the corresponding peptidyl aldehyde with isopropyl isonitrile in the presence of acetic acid, followed by mild hydrolysis of the diastereomeric acetate ester to yield the α-hydroxyamide, and then Dess-Martin oxidation.16 The interaction of compounds 1-10 with Norwalk virus 3C protease17 was investigated and the results are summarized in Table 1.
Table 1.
Inhibitory activity of compounds (1-10)
| Compound | Structure | IC50 (μM) |
|---|---|---|
| 1 |
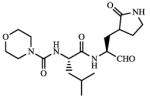
|
7.2 |
| 2 |

|
Inactivea |
| 3 |
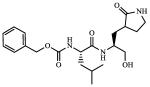
|
Inactivea |
| 4 |
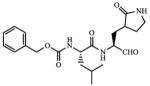
|
1.82 |
| 5 |
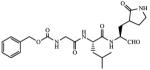
|
1.45 |
| 6 |
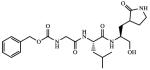
|
Inactivea |
| 7 |
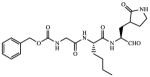
|
0.87 |
| 8 |
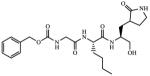
|
Inactivea |
| 9 |
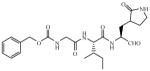
|
7.5 |
| 10 |
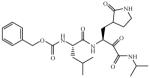
|
Inactivea |
Compounds were designated as inactive if the percent inhibition was < 25 when incubated with the enzyme for 30 minutes at an [ I ] / [ S ] ratio of 25.
Incubation of compound 4 with Norwalk virus 3C protease lead to dose-dependent inhibition of the enzyme (Figure 2). It is evident from Table 1 that the presence of the aldehyde warhead is essential for inhibitory activity since the precursor alcohols were either inactive or had minimal activity (compare, for example, compounds 3 and 4, 5 and 6, 7 and 8, Table 1). Furthermore, the nature of the cap is of paramount importance (compare, for example, compounds 1 and 4, Table 1). In order to gain a better insight and understanding into the binding of Inhibitor 4 to the active site of the enzyme, computer modeling was used to demonstrate that 4 is capable of adopting a low energy conformation that closely resembles the conformer of the co-crystallized peptide (Figure 3).7,18 Thus, in addition to covalent bond formation between the active site cysteine residue (Cys139) and the inhibitor aldehyde carbonyl (see general illustration in Figure 1), inhibitor 4 engages in multiple favorable binding interactions with the enzyme, including lipophilic interactions involving the –CH2-CH2-segment of the ligand lactam with the –CH2-CH2- segment of Pro136, the leucine side chain in the inhibitor with His30, Ile109 and Val 114, and interactions of the phenyl ring in the Cbz cap – partially occupying the S4 pocket – with Ile109. In addition, a network of hydrogen bonds involving Thr134 (backbone carbonyl), Ala158 (backbone carbonyl), Gln110 (side chain carbonyl), and Ala160 (backbone amide proton) is clearly evident. Extending the inhibitor by an additional amino acid (as in compound 5) improved potency, albeit not dramatically (compare compounds 4 and 5, Table 1). Modeling studies suggested that replacement of Leu by other hydrophobic amino acids might result in an optimal fit of the amino acid side chain in the S2 pocket, improving potency. Indeed, compound 7 with a P2 Nle was found to be a sub-micromolar inhibitor of the enzyme, however, replacement of Leu with Ile (compound 9, Table 1) was detrimental to inhibitory activity. α-Ketoamide 10 was devoid of inhibitory activity, suggesting that steric congestion in the vicinity of the S1’ subsite is severe.
Figure 2.

Log dose-response curve for the inhibition of NV 3C protease by inhibitor 4.
Figure 3.

Predicted covalently-bound conformer21 for NV 3C protease inhibitor 4 (stick structure with green carbon atoms and CPK-colored N and O atoms) contrasted with the peptidic inhibitor acetyl-Glu-Phe-Gln-Leu-Gln-CH=CH-COO− (stick structure with gray carbon atoms and CPK-colored N and O atoms) resolved in the 1IPH crystal structure.7 The NV 3C protease binding site is shown as a Connolly surface colored as follows: yellow = non-polar groups, white = partially polar C, H atoms, red = polar O, blue = polar N, cyan = polar H. Key pharmacophore residues are labeled according to the positions on the receptor surface from which they interact with the ligand (except for Q110 whose approximate position is marked but whose surface is not shown because the residue is above the plane of the molecule).
The activity of inhibitors 4-5 against the Norwalk norovirus was investigated using a cell-based replicon system.19-23 Compounds 4 and 5 were found to be active against the virus with effective doses that inhibit 50% of norovirus replication, ED50s, of 2.1 and 7.8 μM, respectively. The median toxic dose, TD50, for both 4 and 5 was found to be >320 μM. Compounds 4 and 5 also inhibit the replication of murine norovirus (MNV) in RAW267.4 cells with ED50s of 5.5 and 20.3 μM, respectively.24 The TD50s for both 4 and 5 with RAW267.4 were found to be >320 μM.24 The results of an ongoing hit-to-lead optimization campaign will be reported in due course.
In conclusion, the first series of transition state inhibitors of norovirus protease has been reported. Members of this series of compounds exhibited noteworthy activity in a cell-based replicon system of norovirus infection.
Acknowledgements
The generous financial support of this work by the National Institutes of Health (U01AI081891) is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.(a) Glass RI, Parasha UD, Estes MK. New Engl. J. Med. 2009;361:1776. doi: 10.1056/NEJMra0804575. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tan M, Jiang X. Curr. Opin. Investig. Drugs. 2008;9:146. [PubMed] [Google Scholar]
- 2.Mead PS, Slutsker L, Dietz V, McCaig LF, Breese JS, Shapiro C, Griffin PM, Tauxe RV. Emerg. Infect. Dis. 1999;5:607. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Green YK. In: Fields Virology. Knipe DM, Howley PM, editors. Vol. 1. Lippincott, Williams & Wilkins; Philadelphia: 2007. pp. 949–979. [Google Scholar]
- 4.Blakeney SJ, Cahill A, Reilly PA. Virology. 2003;308:216. doi: 10.1016/s0042-6822(03)00004-7. [DOI] [PubMed] [Google Scholar]
- 5.Hardy ME, Crone TJ, Brower JE, Ettayebi K. Virus Res. 2002;89:29. doi: 10.1016/s0168-1702(02)00114-4. [DOI] [PubMed] [Google Scholar]
- 6.Someya Y, Takeda N, Miyamura T. Virus Res. 2005;110:91. doi: 10.1016/j.virusres.2005.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hussey RJ, Coates L, Gill RS, Erskine PT, Coker S-F, Mitchell E, Cooper JB, Wood S, Broadbridge R, Clarke IN, Lambden PR, Shoolingin-Jordan PM. Biochemistry. 2011;50:240. doi: 10.1021/bi1008497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeitle CE, Estes MK, Venkatarman B. V. Prasad. J. Virol. 2006;80:5050. doi: 10.1128/JVI.80.10.5050-5058.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamura K, Someya Y, Kumasaka T, Ueno G, Yamamoto M, Sato T, Takeda N, Miyamura T, Tanaka N. J. Virol. 2005;79:13685. doi: 10.1128/JVI.79.21.13685-13693.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Webber SE, Okano K, Little TL, Reich SH, Xin Y, Fuhrman SA, Matthews DA, Love RA, Hendrickson TF, Patick AK, Meador JW, Ferre RA, Brown EL, Ford CE, Binford SL, Worland ST. J. Med. Chem. 1998;41:2786. doi: 10.1021/jm980071x. [DOI] [PubMed] [Google Scholar]; (b) Dragovich PS, Prins TJ, Zhou R, Webber SE, Marakovits JT, Fuhrman SA, Patick AK, Matthews DA, Lee CA, Ford CE, Burke BJ, Rejto PA, Hendrickson TF, Tuntland T, Brown EL, Meador JW, Ferre RA, Harr JE, Kosa MB, Worland ST. J. Med. Chem. 1999;42:1213. doi: 10.1021/jm9805384. [DOI] [PubMed] [Google Scholar]
- 11.Lipinski CA. J. Pharmacol. Toxicol. Meth. 2000;44:235. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 12.Veber DF. J. Med. Chem. 2002;45:2615. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 13.Ritchie TJ, Ertl P, Lewis R. Drug Discov. Today. 2011;16:65. doi: 10.1016/j.drudis.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 14.All compounds were characterized by 1H NMR and HRMS and had a >95% purity.
- 15.Mou K, Xu B, Ma C, Yang X, Zou X, Lu Y, Xu P. Bioorg. Med. Chem. Lett. 2008;18:2198. doi: 10.1016/j.bmcl.2007.12.077. [DOI] [PubMed] [Google Scholar]
- 16.Bogen SL, Arasappan A, Velazquez F, Blackman M, Huelgas R, Pan W, Siegel E, Nair LG, Venkatraman S, Guo Z, Dolle R, Shi N-Y, Njoroge FG. Bioorg. Med. Chem. 2010;18:1854. doi: 10.1016/j.bmc.2010.01.044. [DOI] [PubMed] [Google Scholar]
- 17.Recombinant NV protease was assayed as follows: 10 μL of 50 μM NV protease was added to a thermostatted cuvette at 30 °C containing 180 uL of 50 mM NaH2PO4 buffer, pH 8.0, containing 120 mM NaCl and 6 mM DTT, and 5 uL DMSO. Five μL of 12 mM Edans-EPDFHLQGPEDLAK-Dabcyl substrate in DMSO was then added and the increase in fluorescence was monitored for 30 minutes at an excitation and emission wavelength of 360 and 460 nm, respectively, using a HORIBA FluoroMax 4 spectrofluorometer. Hydrolysis curves were linear. The final enzyme and substrate concentrations were 2.5 μM and 300 nM, respectively. In a typical inhibition run, 10 μL of 50 μM NV protease was added to a thermostatted cuvette at 30 °C containing 180 μL of 50 mM NaH2PO4 buffer, pH 8.0, containing 120 mM NaCl and 6 mM DTT, and 5 μL of inhibitor in DMSO. After a 30 minute incubation period, 5 μL of 12 μM Edans-EPDFHLQGPEDLAK-Dabcyl substrate in DMSO was then added and the increase in fluorescence was monitored for 30 minutes at an excitation and emission wavelength of 360 and 460 nm, respectively.
- 18.A prospective bound conformer for NV 3C protease inhibitor 4 was determined via a genetic algorithms conformational optimization using the SYBYL program (SYBYL 8.0, The Tripos Associates, St. Louis, MO, 2008) The covalently-bound ligand-receptor complex was prepared from the PDB 1IPH crystal structure by deleting the co-crystallized ligand and adding the new ligand (one atom at a time) in an analogous conformation via the “Add Atom” utility so as to have conformational control during construction of the ligand and ensure automatically specification of low energy bond lengths and bond angles. Hydrogens were added to the entire complex according to the automatic SYBYL algorithm (assuming cationic Lys and Arg residues, and anionic Asp and Glu) and were positionally optimized via molecular mechanics with all heavy atoms held rigid and default convergence criteria using the Tripos Molecular Force Field24 and Gasteiger-Marsili charges.25 The resulting complex was then subjected to a genetics algorithm conformational search implemented in SYBYL, requesting identification of the top twenty most favorable conformations. The search yielded only one plausible low-energy conformation (depicted in Figure 3).
- 19.The effects of compounds 4 and 5 were examined in NV replicon-harboring cells (HG23 cells).20 The detailed procedures for studying the antiviral effects using HG23 cells have been reported elsewhere. 20-22 Briefly, 1-day old, 80-90% confluent HG23 cells were treated with varying concentrations of compound 4 or 5 (0 [mock-DMSO]-320 μM) to examine its effects on the replication of NV. At 24 or 48 hrs of treatment, the NV protein or genome were analyzed with Western blot analysis, or qRT-PCR, respectively. The ED50s of compounds 4 or 5 for NV genome levels were determined at 24 hr post-treatment. The cytotoxic effects of compounds 4 or 5 on HG23 cells were determined using a cell cytotoxicity assay kit (Promega, Madison, WI) to calculate the median toxic dose (TD50) at 48 hr of treatment.
- 20.Chang KO, Sosnovtsev SV, Belliot G, Green KY. Virology. 2006;2:463. doi: 10.1016/j.virol.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 21.Chang KO, George DW. J Virol. 2007;22:12111. doi: 10.1128/JVI.00560-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang KO. J Virol. 2009;83:8587. doi: 10.1128/JVI.00005-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The effects of compounds 4 and 5 were also examined in murine norovirus-1 (MNV-1). MNV-1 can be cultured in the murine macrophage-like cell line RAW267.4, thus MNV-1 can be a surrogate system to examine the effects of antiviral compounds on norovirus replication in cells. Confluent RAW267.4 cells in 6-well plates were inoculated with MNV-1 at a multiplicity of infection (MOI) of 2 with varying concentrations (0-320 uM) of compounds 4 and 5. Virus infected cells were then incubated for an additional 12 and 24 hr. After freezing and thawing plates 3 times, the replication of MNV-1 in the presence of the compound was measured by the 50% tissue culture infective dose (TCID50) assay. The nonspecific cytotoxic effects in RAW267.4 cells by ribavirin were monitored by the method described above.
- 24.Clark M, Cramer RD, III, Van Opdenbosch N. J. Comput. Chem. 1989;10:982. [Google Scholar]
- 25.Gasteiger J, Marsili M. Tetrahedron Lett. 1978:3181. [Google Scholar]