

Abstract
Viral RNA-dependent RNA polymerase (RdRP) enzymes are essential for the replication of positive-strand RNA viruses and established targets for the development of selective antiviral therapeutics. In this work we have carried out a high-throughput screen of 154,267 compounds to identify poliovirus polymerase inhibitors using a fluorescence based RNA elongation assay. Screening and subsequent validation experiments using kinetic methods and RNA product analysis resulted in the identification of seven inhibitors that affect the RNA binding, initiation, or elongation activity of the polymerase. X-ray crystallography data show clear density for five of the compounds in the active site of the poliovirus polymerase elongation complex. The inhibitors occupy the NTP binding site by stacking on the priming nucleotide and interacting with the templating base, yet competition studies show fairly weak IC50 values in the low μM range. A comparison with nucleotide bound structures suggests that weak binding is likely due to the lack of a triphosphate group on the inhibitors. Consequently, the inhibitors are primarily effective at blocking polymerase initiation and do not effectively compete with NTP binding during processive elongation. These findings are discussed in the context of the polymerase elongation complex structure and allosteric control of the viral RdRP catalytic cycle.
Keywords: RNA-dependent RNA polymerase, RdRP, inhibitor, poliovirus, HTS, RNA
1. Introduction
Positive-strand RNA viruses encompass a wide range of serious human and animal pathogens for which there are few antiviral pharmaceuticals. The group includes hepatitis C virus (HCV), flaviviruses such as dengue, yellow fever, and West Nile, alphaviruses such as eastern and western equine encephalitis, and the picornaviruses that include foot-and-mouth disease, coxsackie, and polio viruses. While the molecular details of viral replication pathways and associated proteins vary greatly among these viruses, they all rely on a virally encoded RNA-dependent RNA polymerase (RdRP) to replicate their genomes. As a group, RdRPs share a set of core fingers, palm, and thumb structural domains whose overall fold resembles a cupped right hand (Ferrer-Orta et al., 2006; Ng et al., 2008). The picornaviruses have the smallest RdRPs that contain these core domains within a ≈460 residue protein and the larger viral polymerases add other functional elements to this core. For example, the HCV and bovine viral diarrhea virus polymerases include C-terminal extensions and thumb insertions that provide a platform for primer-independent de novo initiation and flaviviral enzymes add an N-terminal domain with RNA capping activity.
The RdRP is essential for viral replication and does not have a host cell homolog, making it a target for antiviral pharmaceuticals (Das et al., 2010; Malet et al., 2008; Sampath and Padmanabhan, 2009; Sarisky, 2004). The viral RdRPs are generally considered low-fidelity enzymes, in large part because they lack proofreading abilities and consequently they can be targeted with broad-spectrum chain terminators or mutagenic nucleoside analogs such as ribavirin (Crotty et al., 2001; Crotty et al., 2000; Vignuzzi et al., 2005) or T-705 (Furuta et al., 2009; Mendenhall et al., 2011). However, nucleoside analogs are less than ideal antiviral drugs because they must be phosphorylated after cell entry, inherently leading to non-specific interactions because they mimic the natural nucleotides to some extent. As a result, non-nucleoside inhibitors targeting allosteric control sites on a single viral polymerase have the potential for much higher specificity and better potency as antiviral drugs. For example, several such inhibitors targeting the HCV RdRP are in late stages of development, including compounds that interfere with formation of an elongation complex by preventing RNA binding or interacting with the initiation/priming platform found on the HCV polymerase thumb domain (Kwong et al., 2008; Legrand-Abravanel et al., 2010).
Although the structure of more than a dozen viral RdRPs have been solved, the rational design of inhibitors targeting viral genome synthesis itself has historically been hindered by a lack of structural information about active polymerase-RNA elongation complexes. This is unfortunate because the elongation complex undergoes thousands of catalytic cycles during the processive synthesis of negative and positive sense RNA strands, providing ample opportunities for allosteric inhibitors to access their binding sites and interfere with replication. The recently solved structure of a poliovirus polymerase elongation complex improves the prospects for such drug design efforts by providing important insights into how the viral RdRPs select nucleotide triphosphates and subsequently close their active sites for catalysis (Gong and Peersen, 2010). In contrast to most other polymerases that use a pre-insertion site for initial NTP recognition and a swinging motion of the fingers domain to positioning the nascent base pair into a preformed active site, the viral RdRPs close their active site for catalysis with a novel conformational change in the palm domain. This new mode of active site closure is highly conserved among positive-strand RNA virus RdRPs, providing a unique structural transition that could be targeted by rational drug design approaches.
To identify viral polymerase inhibitors and assess how they interact with an elongation complex, we carried out a high-throughput screen using poliovirus polymerase and the fluorescence based Polymerase Elongation Template Element (PETE) assay previously developed in our laboratory (Mestas et al., 2007). The PETE assay relies on fluorescence polarization (FP) to detect elongation-dependent changes in the mobility of a fluorescein label at the very 5′ end of a RNA template strand as it is drawn into the active site of the polymerase . From the structure of the elongation complex there are direct contacts with the +2 and +3 downstream templating bases (Gong and Peersen, 2010) that give rise to the clear fluorescence changes observed in microplate format assays (Mestas et al., 2007), and by more sensitive stopped-flow fluorescence we can detect differences in fluorescence when the polymerase is five nucleotides from the end of the template (Gong et al., 2009). Using preassembled polymerase elongation complexes, the assay can detect both loss of RNA binding, resulting in a reduced FP signal due to increased RNA mobility, and inhibition of elongation activity, detected as a failure to increase the FP signal upon NTP addition. A screen of ≈155,000 compounds resulted in the identification of a dozen compounds showing significant polymerase inhibition and five of these are observed to bind the polymerase active site by X-ray crystallography of the poliovirus polymerase elongation complex. The identification of these compounds and their known binding geometry set the stage for the rational design of antiviral compounds that have the potential to be effective against a wide range of positive-strand RNA viruses.
2. Experimental Procedures
2.1 PETE assay stability
The stability and reproducibility of the PETE assay in a 384-well microplate format was initially examined using a Perkin-Elmer Victor 3 platereader. For these experiments, 5 ml of 1 μM polymerase was incubated with 20 nM fluorescently labeled 8-6 PETE RNA (Fig. 1A) and 200 nM ATP on ice for 30 minutes prior to aliquoting the solution into a 12×8 array of 45 ul samples in a room temperature 384-well microplate. Eight different elongation experiments were then carried out by adding remaining NTPs to a final concentration of 200 nM after pre-incubation times that ranged from 0:00 to 5:15 hours. A total of 12 identical reactions were initiated at each of the eight pre-incubation times and FP data from the entire 12×8 array was obtained every 45 minutes up to a final timepoint at 8 hours after the mixture was added to the microplates. The reading of 12 replicate samples for each reaction allowed us to obtain statistics regarding the reproducibility of the experiment and were adequate to calculate a preliminary Z’ value (i.e. Z-factor) for the assay as Z’ = 1 − 3(σp + σn)/∣μp − μn∣, where μ and σ are the averages and standard deviations of the pre- and post-NTP addition FP signals.
Fig. 1.

PETE polymerase RNA binding and elongation assay used for high throughput screening. (A) Sequence of the 5′ fluorescein labeled hairpin “8-6 PETE” RNA primer-template used as a substrate. The 8-6 nomenclature refers to a 8 base duplex region followed by a 6 base single stranded templating region. (B,C) Stability of the PETE assay over time as demonstrated by incubation of polymerase-RNA complexes for various times before and after the addition of NTPs that trigger elongation to the end of the template. Pre-incubation with the first nucleotide to be incorporated (ATP) results in formation of elongation complexes that are stable (≈275 mP) for up to 8 hours and addition of remaining NTPs increases signal to ≈400 mP. (C) In the absence of the first nucleotide there is a steady decrease in the FP signal from the pre-initiation complexes (≈275 to 225 mP), but the signal does increase to ≈400 mP upon NTP addition that triggers elongation to the end of the template. (D) HTS control data showing reproducibility of signals from pre-formed elongation complexes (EC) and the effects of polymerase-RNA dissociation by NaCl, partial elongation with 2′-deoxy UTP, and full elongation with all NTPs. The Z’-factors between the initial stalled elongation complex (EC) and the three other states are shown in the far right column. The microplate representation shows raw FP data color-coded according to intensity by the Wallac software.
2.2 High-throughput screening
All screening experiments were carried out at the ICCB-Longwood Screening Facility at Harvard Medical School (Boston, MA). The stalled elongation complex used as a substrate in the HTS assay was generated by pre-incubating 600 nM 3Dpol with 12 nM 5′ fluorescein labeled 8-6 PETE RNA and 240 nM ATP for 30 minutes at room temperature in 50 mM HEPES, pH 7.0, 40 mM NaCl, 1.5 mM magnesium acetate, 60 μM ZnCl2, 4 mM DTT, and 0.1% Igepal detergent. This solution was then dispensed into 384-well microplates at a volume of 25 uL/well and the small molecule compounds were added by a robotic 100 nL pin drop. Small molecule compounds were solubilized in DMSO at a standard concentration of 5 mg/ml, resulting in a final concentration of 20 μg/ml (corresponding to ≈57 μM based on a 350 Da typical compound mass). After an incubation time of ≈60 minutes, the total fluorescence and fluorescence polarization signals from the labeled RNA were measured with Perkin Elmer EnVision microplatereaders. This first reading provides data indicating whether or not compounds interfered with RNA binding to the stalled elongation complex. Polymerase elongation activity was then tested by adding 5 uL of a solution containing GTP, CTP, and UTP at 1.2 μM each, resulting in a final concentration of 200 nM for each of the four NTPs and 17 μg/ml (≈47 μM ) for the compounds. This allows for elongation to the end of the RNA template and assay data were obtained with a second reading done after an incubation period of ≈60 minutes, which is four times longer than the ≈15 minutes needed for elongation under non-inhibited conditions (Mestas et al., 2007).
Each microplate in the screening library also contained 24 empty control wells, of which 12 were used as negative controls, where added NTPs would result in complete elongation (i.e. lack of inhibition) and the remaining 12 were used as positive controls by not adding any NTP solution (i.e. complete inhibition). In addition, the behavior of every batch of pre-assembled elongation complex was tested using a 384-well control plate that contained four distinct control sections with 96 replicate wells each (Fig. 1D). One section contained only buffer to give FP signals from the initial stalled elongation complex, the second section contained all four NTPs to obtain signals reflecting complete elongation, the third section contained chain terminating 3′ deoxy-UTP to obtain a signal reflecting partial elongation by only one nucleotide, and the fourth section contained 300 mM NaCl to prevent RNA from binding to the polymerase and forming the elongation complex.
2.3 Inhibitor selection
The HTS data were analyzed and potential 3Dpol inhibitors were identified using a correlation plot combining standard Z-scores with a 3Dpol elongation efficiency measurement (Fig. 2A). The Z-scores for each compound were calculated by the normal method of Z = (FPcompound − FPplate_mean) ÷ StdDevplate_mean. We then calculated an elongation efficiency value as E = (FPcompound − FP+) ÷ (FP− − FP+), which reflects how the FP signal obtained in the presence of a compound compares to those of the unelongated positive controls (FP+) and fully elongated negative controls (FP−). Compounds that result in a FP signal greater than the starting material but less than the fully elongated controls will thus have E values between 0% and 100%, while compounds that cause the RNA to dissociate from 3Dpol will have negative E values due to the FP value associated with free RNA being lower than the unelongated FP+ value. Finally, the elongation reaction also results in an increase in the total fluorescence intensity due to deprotonation of the fluorescein as it approaches the positively charged polymerase surface (Gong et al., 2009), providing an additional assessment of elongation. Compounds that resulted in total fluorescence readings higher than that of the fully elongated control samples were rejected from the analysis.
Fig. 2.

High throughput screening results. (A) Correlation plot of fluorescence polarization (FP) vs. total fluorescence (TF) signals observed for a screen of 13,023 natural products extracts (blue circles) that show a number of potential elongation inhibitors resulting in signals intermediate between those of the ±NTP controls (red dots). (B) Summary of final set of 1025 compounds considered as hits after HTS. The graph show the elongation efficiency relative to ±NTP controls (E-value) plotted against the Z-score that reflects standard deviations from the mean signal observed for all samples on the same microplate. HTS hits were classified as strong, medium, or weak inhibitors using the indicated cutoff values.
2.4 Microplate based kinetic validation assays
To further validate the potential inhibitors from the HTS experiments and obtain initial dose response data we used a microplate based kinetic assay in which the rate of the NTP-dependent FP increase in the presence of compounds was observed after addition of NTPs. These experiments were done in 384-well plates on a Victor 3 plate reader where sample injectors we used to add 25 ul of 0.4 μM (200 nM final) each GTP, UTP, and CTP solution to 25 uL of pre-assembled elongation complex (1 μM 3Dpol, 15 nM RNA, 0.5 μM ATP), with both solutions in a buffer composed of 50 mM HEPES, pH 7.0, 40 mM NaCl, 1.5 mM magnesium acetate, 60 μM ZnCl2, 4 mM DTT, and 0.1% Igepal detergent. The high polymerase concentration is used to drive the weak binding equilibrium for the RNA (Kd ≈2 μM ), maximizing the amount of bound RNA that will provide an observable signal change upon elongation (Gong et al., 2009). Three sequential experiments were done to test the inhibitory effects of each compound at concentrations of 10, 20, and 40 μg/ml (≈29, 57, and 114 μM based on a 350 Da typical compound molecular weight). For each experiment we collected 35 data points over ≈1 minute prior to NTP injection to obtain a baseline FP value and then 150 points over ≈4 minutes after NTP injection to follow the time course of the elongation induced FP signal change (Fig. 3). When mathematically fitting the resulting data to single exponential curves, we omitted the initial ≈15 seconds of data due to mixing artifacts from the microplate reader based injection system.
Fig. 3.

HTS hit validation experiment using PETE assay in a kinetic format. (A) Data from the kinetic assay showing time course of FP signals before and after the sample injector based addition of 25 uL NTP solution to 25 uL of preformed [8-6 RNA+3Dpol+ATP] elongation complex. Replacing UTP with chain terminating 3′-deoxy UTP prevents full elongation and eliminates the NTP dependent signal increase. The rate and amplitude of the elongation phase data were determined by curve fitting the latter part of the data (black dots) to a floating time-offset single exponential function whose starting amplitude was fixed at the pre-NTP addition value. (B) An example of dose dependent inhibition by compound 10 at three different concentrations. Rates and amplitudes resulting from curve fitting of the highest inhibitor concentrations are listed in Tables 1 and S1 and pre-NTP addition baseline data are omitted for clarity.
2.5 Elongation product analysis
Polymerase initiation and elongation activity in the presence of small molecule compounds was examined using a gel based analysis of RNA products from two different hairpin RNAs (Fig. 4A,B). We first formed elongation complexes on RNA1 in the absence of compounds, then added compounds to test if they destabilized the preformed RNA1 complexes, then added RNA2 to see if the compounds affected initiation on this RNA, and finally added NTPs to drive the complete elongation of both RNAs. We have previously used these two RNAs to examine poliovirus polymerase initiation rates and elongation complex stability in a study where they were designated 9+2_24 (RNA1) and 10+1_12 (RNA2) (Hobdey et al., 2010).
Fig. 4.

Gel based assay examining inhibition of both initiation and elongation activity. (A) Experimental workflow allowing for 1: initiation on RNA1 in absence of compounds, 2: addition and incubation with ≈114 μM compound, 3: time course of initiation on RNA2 in the presence of ≈72 μM compound, and 4: time course of subsequent elongation of both RNAs in the presence of ≈36 μM compound. Compound concentrations are based on 350 Da molecular weight. (B) Schematic of the two RNAs used in the assay to test for initiation and elongation inhibition. The starting RNA hairpin sequence is in black and the one or two nucleotides that are added during the initiation step with ATP and GTP are shown in color and boxed. The modified deoxythimidine used for IRdye labeling is shown in bold. (C) Electrophoretic analysis of reaction products showing inhibition by compounds 11 and 14. Left most panel shows the migration of RNA1 and its slower migrating +2 product upon incorporation of G and A during step 1. The next panel shows initiation on RNA2 to generate a +1 product during step 3 (4 timepoints) and then elongation of both RNA1 and RNA2 to longer +20 and +7 products when ATP, GTP and UTP are added in step 4 (3 timepoints). The two right panels show effects of compounds 11 and 14 on initiation and elongation during steps 3 and 4. Compound 11 abolishes initiation on RNA2 and destabilized elongation complexes that were pre-formed on RNA1, resulting in significant amounts of RNA1 +2 product being left behind during step 4. Compound 14 predominantly acts to slow step 3 initiation on RNA2, with only slight destabilization of the pre-formed RNA1 complex. Neither compound has a significant effect on elongation rate based on this quenched reaction product time course analysis.
Experimentally, the reactions were assembled in a stepwise manner that involved continual dilution of the potential inhibitory compounds as components were added to the reaction (Fig. 4A). When both RNAs were present, they were always at equal concentrations, although differences in IRdye labeling efficiency and degradation during the experiments affect final band intensities. The 15-fold molar excess of 3Dpol is used to drive the weak RNA binding equilibrium which has a Kd of ≈4 μM. The experiments were done in a buffer composed of 75 mM NaCl, 4 mM magnesium acetate, 50 mM HEPES, pH 6.5, and 2 mM TCEP (tris(2-carboxyethyl)phosphine). The sequential reaction assembly protocol was as follows: First, step 1 allowed for initiation on RNA1 in the absence of compounds via the incorporation of two nucleotides to yield a RNA1 +2 product by incubating 1 μM RNA1 with 15 μM 3Dpol and 40 μM GTP and ATP for 15 minutes at room temperature. In step 2 the small molecule compounds were added while diluting the solution 5-fold, resulting in concentrations of ≈144 μM (50 μg/ml) compound, 0.2 μM RNA1, 3 μM polymerase and 8 μM each ATP and GTP. This was incubated for 15 minutes at room temperature to allow compounds to bind to the pre-formed RNA1 elongation complex or to the excess polymerase. Step 3 then allowed for initiation on the second RNA in the presence of compound by adding an equal volume of 0.2 μM RNA2, resulting in final concentration of ≈72 μM (25 μg/ml) compound, 0.1 μM each RNA, 1.5 μM 3Dpol, and 4 μM ATP and GTP. Initiation on RNA2 was evaluated by the formation of its +1 product in EDTA-quenched samples taken after 0, 2, 7 and 15 minutes. Finally, step 4 allowed for elongation to +20 (RNA1) or +7 (RNA2) products of both RNAs by adding ATP, GTP, and UTP to a final concentration of 40uM in another 2-fold dilution step. The final compound concentration was then ≈36 μM (12.5 μg/ml) and quenched samples were taken after 1, 2 and 5 minutes of elongation. The reaction products were separated on a denaturing 7 M urea polyacrylamide gel and quantified using an IRdye 800RS (Li-Cor Biosciences) label attached to a deoxy-thymidine in the tetraloop of both RNAs as previously described (Hobdey et al., 2010).
2.6 Stopped-flow kinetics
The ability of the compounds to inhibit the processive elongation of pre-formed elongation complexes was tested using the PETE assay in a stopped-flow format to obtain an elongation rate in nucleotides per second by measuring the time needed for 3Dpol to replicate to the end of an 18 nucleotide long single stranded template. Experiments were carried out at as previously described (Gong et al., 2009) with compounds being added at final concentrations of 5 and 25 μM after mixing in the stopped-flow instrument.
2.7 IC50 Determination
To assess the inhibitory strength of the final set of compounds we carried out titrations to measure the effects of compound concentrations on elongation complex formation and subsequent elongation to the end of the template. Compounds were mixed with 1 μM polymerase, 15 nM RNA, and 200 nM ATP and incubated for 60 minutes prior to reading FP and total fluorescence signals to determine at what concentrations the compounds affected polymerase–RNA complex formation or stability. The remaining NTPs were then added to a final concentration of 200 nM each to drive elongation and FP signals were measured after another 30 minute incubation period. Experiments were done in buffer containing 50 mM HEPES, pH 7.0, 40 mM NaCl, 4 mM magnesium acetate, and 2 mM TCEP. IC50 values for inhibition of the formation and/or elongation steps were determined from the midpoints of the resulting inhibition curves.
2.8 X-ray crystallography
Stalled poliovirus 3Dpol elongation complexes were generated, purified, and crystallized as previously described (Gong and Peersen, 2010). Crystals were then harvested, with buffer gradually exchanged into cryostabilizer solution, and finally transferred to cryo-solution containing the small molecule compounds. Compounds were present at 5 mM concentration made from 100 mM stocks in DMSO, except for compound 11 that was only soluble to 34 mM in DMSO, resulting in a final concentration of 1.7 mM in the cryo-solution. Crystals were soaked for 15 to 20 hours prior to being frozen in liquid nitrogen and shipped to the Advanced Light source (Berkeley, CA) for remote data collection at the Molecular Biology Consortium beamline 4.2.2. Resulting diffraction data were processed using d*Trek and structures were refined using Phenix, as previously described (Gong and Peersen, 2010). The complete refinement of the compound-bound structures will be reported elsewhere.
3. Results
3.1 Stability of PETE Assay
HTS experiments require that the fluorescence polarization signals associated with the stalled and elongated 3Dpol complexes remain stable for several hours at room temperature so as to allow time for the dispensing of small molecule compounds and subsequent reading of the FP data. These steps result in a typical minimum incubation time of about one hour each. In addition, the HTS format PETE assay includes a second reaction whereby NTPs are added after the initial data reading to test for polymerase elongation activity in the presence of the compounds. To verify that the signals associated with the complexes were sufficiently stable over such timeframes, we assembled stalled elongation complexes and monitored their FP signals as a function of incubation time at room temperature both before and after elongation by the addition of NTPs. The initial complex generated by mixing 3Dpol with “8-6” RNA (Fig. 1A) and ATP, the first nucleotide to be incorporated, resulted in a FP reading of ≈270 mP that remains stable for more than five hours (Fig. 1B). Addition of the remaining NTPs to enable elongation to the end of the template, which immobilizes the 5′ fluorescein label, resulted in a significant and stable increase to ≈400 mP. The Z’ factor (Zhang et al., 1999) for the NTP-dependent elongation event was 0.87±0.02 as calculated using the mean±sd FP values from 12 replicates of the time points 45 minutes before and 45 minutes after each NTP addition, indicating that the assay is highly robust over the needed timeframe. The microplate based data also show that full elongation of the RNA leads to an increase in both the total fluorescence and FP signals, which is due to deprotonation and immobilization of the fluorescein reporter group as the 5′ end of the template is pulled into the polymerase active site (Gong et al., 2009).
The assay can also be configured to identify compounds that interfere with pre-initiation 3Dpol-RNA complex formation in the absence of ATP. Such complexes displayed an initial FP signal of 270±3 mP as compared to ≈170 mP for free RNA, but they were unstable and the signal exhibited a gradual decrease to 220±5 mP over a five hour pre-reaction incubation time (Fig. 1C). The slow reduction in the signal from the RNA-polymerase complex means that the Z’ for the RNA binding reaction is constantly decreasing; it starts at a value of 0.78 and decreases to 0.66 after three hours of incubation and then to 0.27 after five hours of incubation. This signal drop significantly hampers the interpretability of the data, and, consequently, the RNA binding assay format was not used for HTS.
3.2 High-throughput screening
A total of 154,267 compounds from the available ICCB-Longwood Screening Facility libraries were tested using the PETE assay with two data readings done per plate to monitor both dissociation of RNA from pre-formed elongation complexes and the extent of elongation upon addition of NTPs. The Z’ value of the assay was calculated based on the 24 control wells on each microplate, resulting in an overall average Z’-score of 0.91±0.02 from all library plates used in the screen. In addition, every batch of pre-assembled 3Dpol elongation complex was assayed using the specially designed 4-section control plate that also controls for RNA dissociation and partial elongation, which had Z’-scores of 0.26 and 0.58, respectively (Fig. 1D). Dissociation of the RNA from the polymerase results in a reduction in the FP signal that is consistent with more rapid tumbling of the RNA in solution. It should be noted that while each of the Z’ scores found at the screening facility are consistent with the Z’-scores observed on our home plate reader, the absolute fluorescence and FP values differ significantly between plate readers and background correction methods used. Igepal detergent was used in the assay to minimize sample sticking to the microplate wells and to eliminate surface tension effects that cause scatter in the fluorescence data. Note that this may have affected our ability to detect inhibition by particularly hydrophobic compounds that could be trapped in detergent micelles.
The correlation observed between total fluorescence and FP values is demonstrated by results from a library screen containing natural extract samples. This chemically diverse library resulted in an unusually wide range of partial elongation effects and total fluorescence values as compared to the results from more standard small molecule libraries (Fig. 2A). These data also show the tight clustering of the positive and negative control sample signals and the detection of abnormally high total fluorescence signals encountered with fluorogenic compounds. We calculated Z-scores and elongation efficiency (E) values for all the compounds based on comparisons with controls on the same microplate. The first step in the selection process for potential polymerase inhibitors was to generate scatter plots comparing the FP and total fluorescence signals arising from all the compounds in a single screening run and then reject any compounds whose total fluorescence signal was higher than that observed for the fully elongated controls (Fig. 2A). The Z-scores and E-values of all the remaining inhibitory compounds from the full ≈155,000 compounds screen were then combined on a single scatter plot, yielding 1025 compounds that showed detectable levels of 3Dpol inhibition (Fig. 2B). Of these, 492 were classified as medium to strong inhibitors based on Z-score absolute values (2–5 for medium, >5 for strong) and elongation efficiency E-values (<75% for medium, <50% for strong, <0% indicates RNA dissociation). This set of 492 compounds reflects a reasonable HTS hit rate of 0.32% to select compounds for further study using more involved kinetic assays.
3.3 Validation Assays
3.3.1 Microplate based kinetic validation assays
The effects of these 492 compounds on 3Dpol elongation activity were then tested in a kinetic realtime elongation assay at three different compound concentrations of 10, 20, and 40 μg/ml (≈29, 57, and 114 μM based on an average 350 Da compound weight). Microplate reader sample injectors were used to add 25 uL of NTP solution to 25 uL of pre-assembled elongation complex with compounds the resulting elongation-dependent increase in the FP signal was observed over a 4 minute time window (Fig. 3). The compounds generally fell into one of three different categories: a) those that reduced the starting FP signal and/or the amplitude of the FP change, but not the rate of the FP signal change, b) those that reduced the rate of the FP signal change, and c) those with no discernible effects, i.e. the initial HTS results could not be replicated. As the starting FP value is indicative of how much RNA is bound to 3Dpol (Mestas et al., 2007) and the amplitude of the FP change is indicative of how much of the RNA is being elongated (Gong et al., 2009; Hobdey et al., 2010), the category a data is indicative of compounds that interfere with RNA binding and/or elongation competence and the category b data is indicative of compounds that simply slow the 3Dpol elongation rate. To verify that extended incubation times in the microplate reader did not affect the integrity of the assay we carried out DMSO control reactions both immediately before and immediately after testing each set of three different compound concentrations. Replacing UTP in the elongation step with a chain terminating 3′-deoxy-UTP eliminated the NTP dependent increase in the FP signal (Fig. 3A), further showing that the signal change was due to elongation at the end of the template strand. Using this kinetic assay, we narrowed the list of polymerase inhibitors from HTS down to the 56 compounds that are listed in Appendix A.
3.3.2 Elongation product analysis
To further examine which aspects of 3Dpol activity were being affected by the remaining 56 compounds we used two different RNA molecules to test for inhibition of initiation, inhibition of elongation, or destabilization of the elongation complex, all in the same reaction mixture and at inhibitor concentrations ranging from 50 to 12.5 μg/ml (≈140 to 36 μM) (Fig. 4A,B). This analysis revealed that the predominant mode of inhibition was at the level of initiation and destabilization of pre-formed elongation complexes. As shown by the gels in Fig. 4C, most of the effective compounds abolished initiation on the second RNA (RNA2) template but had little or no effect on the elongation of complexes that were pre-formed with RNA1 in the absence of the compound. We did not observe any intermediate length products nor significant slowing of the elongation step to form +20 or +7 products for any of the compounds, indicating that processive elongation by active elongation complexes is not efficiently inhibited by 12.5ug/ml (≈36 μM) compound concentrations. However, we did observe that several compounds appeared to destabilize the pre-formed elongation complex, resulting in some of the preformed +2 product of RNA1 being left behind in the elongation step. The compounds that resulted in this effect also significantly slowed initiation on the second RNA, leading us to speculate that the compounds may be interfering with nucleic acid binding.
Based on these results, compound availability and pricing, and the desire to compare similar compounds with minor structural changes, seven compounds were selected for further examination of their mode of action with more detailed kinetic and X-ray crystallography studies.
3.3.3 Stopped-flow Kinetics
We used the PETE RNA elongation assay in a stopped-flow rapid kinetics format (Gong et al., 2009) to examine if the compounds affected the Vmax and Kmapp for NTP driven elongation of pre-formed 3Dpol elongation complexes. However, no significant inhibition of processive elongation rates were observed in the presence of the 5–50 μM NTP concentrations needed for efficient elongation. This included testing compound concentrations as high as 25 μM with NTP concentrations as low as 5 μM (data not shown). These data indicate that the compounds cannot efficiently compete with biologically relevant micromolar NTP concentrations, as compared to the 200 nM nucleotide concentration used during HTS.
3.3.4 IC50 Determinations
We tested the concentration dependence of these seven compounds for inhibition of both initiation and elongation using the microplate format PETE assay to determine an IC50 midpoint of the inhibition curves. The data show that some of the compounds caused dissociation of the 3Dpol-RNA complex prior to NTP addition, while others prevented elongation without significant effects on RNA binding (Fig. 5). However, all the compounds tested were fairly weak inhibitors with IC50 values of ≈50 μM at best (Table 1).
Fig. 5.

IC50 determination for RNA binding and elongation inhibition. Total fluorescence signal arising from preformed elongation complexes [3Dpol + 8-6 RNA + ATP] in the presence of various compound concentrations were examined before (○) and after (●) the addition of 200nM NTPs to trigger full elongation, as shown by the kinetic data in Fig. 3A. IC50 values were determined from the midpoints of the resulting elongation inhibition curves.
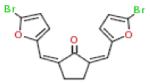
Table 1.
Structures and inhibitor data for the seven compounds selected for structural and IC50 studies. See Supplementary Content for a ranked list of the best 56 inhibitors.
| Compound Number |
Structure | HTS Z-score1 |
HTS E-score2 |
Validation Type3 |
IC50 (μM) |
|---|---|---|---|---|---|
| 6 |

|
−13 | 56 | AR | 458±21 |
| 7 |

|
−8 | 58 | KAR+X | 147±14 |
| 10 |

|
−12 | 47 | AR+X | 82±11 |
| 11 |
|
−8 | 43 | A+X | ND4 |
| 15 |

|
−3 | 66 | KAR+X | 91±7 |
| 25 |

|
−8 | 22 | AR | 55±3 |
| 34 |

|
−11 | 24 | AR+X | 56±2 |
Z-score based on fluorescence polarization = (FPcompound − FPplate_mean) ÷ StdDevplate_mean
Elongation efficiency as compared to ±NTP controls: E = (FPcompound − FP+) ÷ (FP− − FP+)
IC50 for compound 11 could not be determined due to fluorescence at high concentrations.
3.4 X-ray Crystallography
To examine how the compounds interacted with the polymerase-RNA complex, crystals of the elongation complex were soaked in 5 mM compound for 15-20 hours prior to being frozen in liquid nitrogen and shipped to the Advanced Light Source for synchrotron data collection. Data were collected and processed and the structures were solved and refined as previously described (Gong and Peersen, 2010). Clear positive difference density reflecting the added small molecule was observed for five of the compounds tested (Fig. 6A, Table 1) and these compounds were all found to bind in the incoming NTP binding site. Stacking interactions between the priming nucleotide and the planar rings on the compounds appear to be the major energetic determinant of binding. While we could clearly model some of the compounds based on the strong initial difference electron density maps, the density was generally rather weak and suggestive of low affinity binding. For comparison, we also soaked cytosine and cytidine into the crystals in order to compare their densities to that of the complete cytidine triphosphate that is the natural substrate given the templating guanosine in this crystal form. The resulting electron density maps showed that significantly weaker density is observed with just the nucleobase or nucleoside forms of cytosine as compared to the complete CTP (Fig. 6B), indicating that the triphosphate moiety plays a major role in NTP binding to the poliovirus polymerase elongation complex. The density observed for the five inhibitory compounds was comparable to the weak density observed for cytosine and cytidine. The complete refinement of the compound-bound structures will be reported elsewhere.
Figure 6.

Electron density for inhibitors bound in active site of polymerase elongation complex. (A) Fo-Fc (green/red) and 2Fo-Fc (cyan) maps showing clear positive difference density (green) for three compounds in the incoming NTP binding site where they stack on the priming nucleotide and appear to interact with the hydrogen bonding face of the templating base. (B) Results from analogous soaking experiments with the cognate nucleotide showing density for only the cytosine nucleobase, the cytidine nucleoside, and the complete cytidine triphosphate. The inhibitory compounds show density levels that are comparable to those of cytosine and cytidine, but significantly weaker that the difference density observed for the triphosphorylated nucleoside.
Discussion
The polymerase template elongation element (PETE) assay utilizes an elongation dependent change in the FP signal of a fluorescein molecule located at the very 5′ end of a RNA template to detect polymerase activity in either microplate or stopped-flow kinetics formats (Gong et al., 2009; Mestas et al., 2007). Here we have used this assay in a high-throughput screen to look for inhibitors of the poliovirus RNA-dependent RNA polymerase. After screening ≈155,000 compounds and performing follow-up studies, we identified a pool of seven inhibitory compounds and five of these were found to bind the active site of the polymerase elongation complex by X-ray crystallographic analysis. However, the IC50 values of the compounds are quite high and they do not efficiently compete with NTPs for binding. The structural analysis indicates that this is likely due to a lack of triphosphate-like charge interactions with the inhibitors in their current form.
The PETE assay performed well in a HTS format, yielding Z’ scores of 0.91±0.02 and a hit rate of ≈0.32% for the identification of inhibitors based on end-point analysis of an elongation reaction in the presence of 200 nM NTPs. This allowed us to select 492 potential inhibitors for follow-up studies using the assay in a microplate based kinetic format where we monitored both the amount and rate of RNA elongation by fluorescence polarization. The analysis further reduced the pool to 56 compounds that retained significant levels of inhibition and warranted further investigation via direct analysis of RNA elongation products in a gel based assay and effects on elongation rates in a stopped-flow format assay. The gel based assay showed that several compounds inhibited the initiation of RNA elongation, some of them destabilized preformed elongation complexes, but none of them showed significant ability to slow the polymerase elongation rate in the presence of micromolar NTP concentrations.
The compound titration data from the IC50 determinations also showed that some compounds inhibit activity by destabilizing the elongation complex, providing a mode of inhibition via promoting the dissociation of the RNA from the polymerase. For example, the compound 7 titration shows a decrease of RNA binding in the absence of NTPs with a midpoint concentration of ≈300 μM, and then a ≈5-fold lower IC50 for the elongation phase when NTPs are added to the reaction (Fig. 5). These compounds may act by interfering with RNA binding to the template entry channel in a manner similar to that of recently identified flaviviral polymerase inhibitors (Niyomrattanakit et al., 2010). In contrast, compound 25 is only effective as an inhibitor during elongation and does not affect RNA binding. The molecular mechanism by which such compounds destabilize the elongation complex are not clear, in large part because the interactions that hold the processive complex together are also not fully understood. Biochemical data have shown that incorporation of the first nucleotide is preceded by a conformational change that takes on the order of a minute and that the addition of this nucleotide increases the lifetime of the polymerase-RNA elongation complex from minutes to hours (Arnold and Cameron, 2000). However, no major structural changes are observed in the polymerase upon formation of the elongation complex (Gong and Peersen, 2010), suggesting this conformational change step may simply involve the template-primer junction arriving at the proper binding geometry for catalysis in the active site.
Crystallographic analysis showed clear difference electron density for five compounds that bind directly into the polymerase active site as competitive inhibitors of NTP binding (Fig. 4, Table 1). The inhibitors interact with the elongation complex via stacking and hydrogen bonding interactions that mimic those observed for an incoming nucleotide. This includes compound 11 for we could not determine an IC50 value due to fluorogenic properties and low solubility, as have been noted in a previous study, where it was identified as a botulinum neurotoxin endopeptidase inhibitor (Cai et al., 2010). The density observed for the five compounds is comparable to that seen with cytosine and cytidine, but much lower than that seen for cytidine triphosphate. Although a crystal soaking is not an ideal binding experiment, these data nonetheless suggest that the compounds are interacting rather weakly with the 3Dpol active site. When the compounds are present at high concentrations, such as the ≈50 μM used during HTS, they can effectively compete with 200 nM NTP concentrations and thus be identified as inhibitors. However, when tested at lower compound concentrations and in the presence of higher ≈20 μM NTP concentrations that reflect the nucleotide Km for 3Dpol elongation (Arnold and Cameron, 2004; Gong et al., 2009), the compounds no longer function as effective competitive inhibitors. Thus, although the assay was originally designed to look for elongation phase inhibitors, it has predominantly identified weak inhibitors that are instead only effective at the polymerase initiation phase where the nucleotide incorporation rate is 4–5-fold slower than during processive elongation (Arnold and Cameron, 2004).
The crystal structures show that the compounds are held in the polymerase active site by stacking interactions with the priming base that will always be present and by sequence specific hydrogen bonding interactions with the templating base. The latter interaction underscores that it may be worth designing future screening assays in a multiplexed format that samples multiple templating nucleotides because they will provide different sets of interactions in the active site. However, even such an assay is unlikely to overcome the aforementioned problem of weak interactions and identifying compounds that cannot compete with the binding of charged NTPs to an active elongation complex. Alternatively, the PETE elongation assay could be made more stringent by using higher NTP concentrations, i.e. in the low micromolar range consistent with biological in vivo concentrations (Traut, 1994), but then it is unlikely we would have identified any strong inhibitors in this 150,000 compound screen. Alternatively, a more efficient screening workflow could increase sensitivity by detecting signals shortly (i.e. minutes) after NTP addition, as opposed to the equipment-dictated ≈1 hour minimum wait time between compound delivery and data collection in our assay. Last, the PETE assay can also be configured to look for initiation phase inhibitors, but this is at the expense of Z’-scores (Fig. 1B) and based on our experience there will also be problems with RNA degradation (data not shown) because the nucleic acid substrate is no longer protected from trace RNAses by pre-binding to the polymerase.
It is important to note that while none of the compounds identified appear to be particularly strong elongation phase inhibitors based on in vitro biochemical assays, they are nonetheless potential lead compounds whose potency and interactions in the active site may be optimized through medicinal chemistry efforts. As such, the compounds and structure do outline sets of interactions that are important and sufficient for polymerase active site binding. The greater issue regarding the discovery by activity-based screening for RdRP inhibitors, however, may be the limited structural transitions involved in active site closure and subsequent catalysis in these enzymes. The positive-strand RNA virus RdRPs close their active site for catalysis with a subtle conformational change of Motif A in the palm domain, rather than the large swinging motion of the fingers domain typically observed in other classes of single subunit polymerases (Gong and Peersen, 2010). This will make it difficult to identify binding sites for allosteric inhibitors that can function while bound far away from the active site, which probably explains the preponderance of initiation inhibitors identified in our screen of over 150,000 compounds. In fact, RdRP active site inhibitor development may best be done using rational design methods bootstrapped with homology modeling based on the poliovirus 3Dpol elongation complex structure (and others as they become available) or using fragment screening approaches where weak binding sites for compound substructure fragments are first identified crystallographically and then assembled synthetically into larger and more potent inhibitors (Murray and Blundell, 2010).
The lack of efficient elongation phase inhibitors from the screen is also consistent with the development of HCV polymerase antivirals over the past decade that has focused on either nucleotide analogs or on compounds that prevent initiation (Dhanak et al., 2002; Gu et al., 2003; Reich et al., 2010; Tomei et al., 2003; Wang et al., 2003). Similarly, two recently identified inhibitors target the template RNA binding channel on the fingers domain of denguevirus (Niyomrattanakit et al., 2010) and bovine viral diarrhea virus (Castro et al., 2011) polymerases, and mutational studies suggest there are inhibitor binding pockets on the dengue polymerase thumb domain that can interfere with initiation of genomic RNA synthesis (Zou et al., 2011). Unlike the small picornaviral polymerases that carry out primer-dependent initiation, these larger RdRPs are capable of de novo initiation and undergo large conformational changes in their thumb domain during the transition to an elongation complex. Non-nucleoside inhibitors can interfere with this transition, but because the drug binding sites are not directly involved in the catalytic site they are unfortunately prone to the emergence of resistance mutations (Legrand-Abravanel et al., 2010).
In conclusion, our high-throughput screen for poliovirus RNA-dependent RNA polymerase inhibitors has identified several compounds that show moderate inhibitory potential and crystallographic analysis shows that they all bind in the enzyme active site. The compounds occupy the nucleotide triphosphate binding site and are held there by stacking interactions with upstream product RNA and interactions with the base-pairing face of the templating nucleotide. The compounds are most effective at preventing initiation by the polymerase, likely because the lack of a triphosphate group means that they cannot efficiently compete with micromolar concentrations of the natural nucleotides present during the polymerase elongation phase. These compounds and our structural data set the stage for further optimization of interactions with the active site to increase the potency by which the compounds can interfere with RdRP initiation or the stability of the polymerase elongation complex.
Supplementary Material
Acknowledgements
We would like to thank Su Chiang and David Wrobel for insights on assay configuration and data analysis, and Jay Nix for synchrotron data collection support. This work was supported by National Institutes of Health Grants R01-AI059130 (to OBP), U54-AI065357 (to OBP via Rocky Mountain Regional Center of Excellence) and U54-AI057159 (to New England Regional Center of Excellence).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arnold JJ, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3D(pol)). Assembly of stable, elongation-competent complexes by using a symmetrical primer-template substrate (sym/sub) J. Biol. Chem. 2000;275:5329–5336. doi: 10.1074/jbc.275.8.5329. [DOI] [PubMed] [Google Scholar]
- Arnold JJ, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3Dpol): pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mg2+ Biochemistry. 2004;43:5126–5137. doi: 10.1021/bi035212y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai S, Lindo P, Park JB, Vasa K, Singh BR. The identification and biochemical characterization of drug-like compounds that inhibit botulinum neurotoxin serotype A endopeptidase activity. Toxicon. 2010;55:818–826. doi: 10.1016/j.toxicon.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro EF, Fabian LE, Caputto ME, Gagey D, Finkielsztein LM, Moltrasio GY, Moglioni AG, Campos RH, Cavallaro LV. Bovine viral diarrhea virus RNA synthesis inhibition by the thiosemicarbazone derived from 5, 6-dimethoxy-1-indanone. J. Virol. 2011;85:5436–5445. doi: 10.1128/JVI.00859-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S, Cameron CE, Andino R. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. USA. 2001;98:6895–6900. doi: 10.1073/pnas.111085598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S, Maag D, Arnold JJ, Zhong W, Lau JY, Hong Z, Andino R, Cameron CE. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000;6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- Das K, Aramini JM, Ma LC, Krug RM, Arnold E. Structures of influenza A proteins and insights into antiviral drug targets. Nat. Struct. Mol. Biol. 2010;17:530–538. doi: 10.1038/nsmb.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanak D, Duffy KJ, Johnston VK, Lin-Goerke J, Darcy M, Shaw AN, Gu B, Silverman C, Gates AT, Nonnemacher MR, Earnshaw DL, Casper DJ, Kaura A, Baker A, Greenwood C, Gutshall LL, Maley D, DelVecchio A, Macarron R, Hofmann GA, Alnoah Z, Cheng HY, Chan G, Khandekar S, Keenan RM, Sarisky RT. Identification and biological characterization of heterocyclic inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Biol. Chem. 2002;277:38322–38327. doi: 10.1074/jbc.M205566200. [DOI] [PubMed] [Google Scholar]
- Ferrer-Orta C, Arias A, Escarmis C, Verdaguer N. A comparison of viral RNA-dependent RNA polymerases. Curr. Opin. Struct. Biol. 2006;16:27–34. doi: 10.1016/j.sbi.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Takahashi K, Shiraki K, Sakamoto K, Smee DF, Barnard DL, Gowen BB, Julander JG, Morrey JD. T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections. Antiviral Res. 2009;82:95–102. doi: 10.1016/j.antiviral.2009.02.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong P, Campagnola G, Peersen OB. A quantitative stopped-flow fluorescence assay for measuring polymerase elongation rates. Anal. Biochem. 2009;391:45–55. doi: 10.1016/j.ab.2009.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong P, Peersen OB. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA. 2010;107:22505–22510. doi: 10.1073/pnas.1007626107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu B, Johnston VK, Gutshall LL, Nguyen TT, Gontarek RR, Darcy MG, Tedesco R, Dhanak D, Duffy KJ, Kao CC, Sarisky RT. Arresting initiation of hepatitis C virus RNA synthesis using heterocyclic derivatives. J. Biol. Chem. 2003;278:16602–16607. doi: 10.1074/jbc.M210891200. [DOI] [PubMed] [Google Scholar]
- Hobdey SE, Kempf BJ, Steil BP, Barton DJ, Peersen OB. Poliovirus polymerase residue 5 plays a critical role in elongation complex stability. J. Virol. 2010;84:8072–8084. doi: 10.1128/JVI.02147-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong AD, McNair L, Jacobson I, George S. Recent progress in the development of selected hepatitis C virus NS3.4A protease and NS5B polymerase inhibitors. Curr. Opin. Pharmacol. 2008;8:522–531. doi: 10.1016/j.coph.2008.09.007. [DOI] [PubMed] [Google Scholar]
- Legrand-Abravanel F, Nicot F, Izopet J. New NS5B polymerase inhibitors for hepatitis C. Exp. Opin. Invest. Drugs. 2010;19:963–975. doi: 10.1517/13543784.2010.500285. [DOI] [PubMed] [Google Scholar]
- Malet H, Masse N, Selisko B, Romette JL, Alvarez K, Guillemot JC, Tolou H, Yap TL, Vasudevan S, Lescar J, Canard B. The flavivirus polymerase as a target for drug discovery. Antiviral Res. 2008;80:23–35. doi: 10.1016/j.antiviral.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Mendenhall M, Russell A, Juelich T, Messina EL, Smee DF, Freiberg AN, Holbrook MR, Furuta Y, de la Torre JC, Nunberg JH, Gowen BB. T-705 (favipiravir) inhibition of arenavirus replication in cell culture. Antimicrob. Agents Chemother. 2011;55:782–787. doi: 10.1128/AAC.01219-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestas SP, Sholders AJ, Peersen OB. A fluorescence polarization-based screening assay for nucleic acid polymerase elongation activity. Anal. Biochem. 2007;365:194–200. doi: 10.1016/j.ab.2007.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CW, Blundell TL. Structural biology in fragment-based drug design. Curr. Opin. Struct. Biol. 2010;20:497–507. doi: 10.1016/j.sbi.2010.04.003. [DOI] [PubMed] [Google Scholar]
- Ng KK, Arnold JJ, Cameron CE. Structure-function relationships among RNA-dependent RNA polymerases. Curr. Top. Microbiol. Immunol. 2008;320:137–156. doi: 10.1007/978-3-540-75157-1_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niyomrattanakit P, Chen YL, Dong H, Yin Z, Qing M, Glickman JF, Lin K, Mueller D, Voshol H, Lim JY, Nilar S, Keller TH, Shi PY. Inhibition of dengue virus polymerase by blocking of the RNA tunnel. J. Virol. 2010;84:5678–5686. doi: 10.1128/JVI.02451-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich S, Golbik RP, Geissler R, Lilie H, Behrens SE. Mechanisms of activity and inhibition of the hepatitis C virus RNA-dependent RNA polymerase. J. Biol. Chem. 2010;285:13685–13693. doi: 10.1074/jbc.M109.082206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath A, Padmanabhan R. Molecular targets for flavivirus drug discovery. Antiviral Res. 2009;81:6–15. doi: 10.1016/j.antiviral.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarisky RT. Non-nucleoside inhibitors of the HCV polymerase. J. Antimicrob. Chemother. 2004;54:14–16. doi: 10.1093/jac/dkh319. [DOI] [PubMed] [Google Scholar]
- Tomei L, Altamura S, Bartholomew L, Biroccio A, Ceccacci A, Pacini L, Narjes F, Gennari N, Bisbocci M, Incitti I, Orsatti L, Harper S, Stansfield I, Rowley M, De Francesco R, Migliaccio G. Mechanism of action and antiviral activity of benzimidazole-based allosteric inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 2003;77:13225–13231. doi: 10.1128/JVI.77.24.13225-13231.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traut TW. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- Vignuzzi M, Stone JK, Andino R. Ribavirin and lethal mutagenesis of poliovirus: molecular mechanisms, resistance and biological implications. Virus Res. 2005;107:173–181. doi: 10.1016/j.virusres.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Wang M, Ng KK, Cherney MM, Chan L, Yannopoulos CG, Bedard J, Morin N, Nguyen-Ba N, Alaoui-Ismaili MH, Bethell RC, James MN. Non-nucleoside analogue inhibitors bind to an allosteric site on HCV NS5B polymerase. Crystal structures and mechanism of inhibition. J. Biol. Chem. 2003;278:9489–9495. doi: 10.1074/jbc.M209397200. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zou G, Chen YL, Dong H, Lim CC, Yap LJ, Yau YH, Shochat SG, Lescar J, Shi PY. Functional analysis of two cavities in flavivirus NS5 polymerase. J. Biol. Chem. 2011;286:14362–14372. doi: 10.1074/jbc.M110.214189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.