

Abstract
Dosage-sensitive sex reversal, adrenal hypoplasia congenita (AHC) critical region on the X chromosome, gene 1 (Dax1) is an orphan nuclear receptor essential for development and function of the mammalian adrenal cortex and gonads. DAX1 was cloned as the gene responsible for X-linked AHC, which is characterized by adrenocortical failure necessitating glucocorticoid replacement. Contrary to these human data, young mice with genetic Dax1 knockout (Dax1−/Y) exhibit adrenocortical hyperfunction, consistent with the historic description of Dax1 as a transcriptional repressor that inhibits steroidogenic factor 1-dependent steroidogenesis. This paradox of molecular function and two apparently opposite phenotypes associated with Dax1 deficiency in mice and humans is compounded by the recent observations that under certain circumstances, Dax1 can serve as a transcriptional activator of steroidogenic factor 1. The recently revealed role of Dax1 in embryonic stem cell pluripotency, together with the observation that its expression in the adult adrenal is restricted to the subcapsular cortex, where presumptive undifferentiated progenitor cells reside, has led us to reexamine the phenotype of Dax1−/Y mice in order to reconcile the conflicting mouse and human data. In this report, we demonstrate that although young Dax1−/Y mice have enhanced steroidogenesis and subcapsular adrenocortical proliferation, as these mice age, they exhibit declining adrenal growth, decreasing adrenal steroidogenic capacity, and a reversal of their initial enhanced hormonal sensitivity. Together with a marked adrenal dysplasia in aging mice, these data reveal that both Dax1−/Y mice and patients with X-linked AHC exhibit adrenal failure that is consistent with adrenocortical subcapsular progenitor cell depletion and argue for a significant role of Dax1 in maintenance of these cells.
Dosage-sensitive sex reversal, adrenal hypoplasia congenita (AHC) critical region on the X chromosome, gene 1 (DAX1) was first identified as the gene altered in X-linked AHC (1–3). The DAX1 gene encodes an atypical nuclear hormone receptor (NR0B1), containing a putative ligand binding domain in its C-terminal region and an atypical 70-amino acid repeat motif in its N-terminal region (2, 4, 5). Expression of Dax1 in the adult mouse is restricted to tissues involved in metabolism, steroidogenesis, and reproduction (6). Dax1 has been defined historically as a transcriptional inhibitor of the nuclear receptor steroidogenic factor 1 (Sf1) (Nr5a1), a critical regulator of development and function of both the hypothalamic-pituitary-adrenal (HPA) and hypothalamic-pituitary-gonadal axes (7). Dax1 inhibits Sf1 transactivation (4, 8) through LXXLL motif recruitment of corepressors, such as nuclear receptor corepressor (8) and COP9 constitutive photomorphogenic homolog subunit 2 (9, 10). Dax1 has also been demonstrated to have additional, less well-characterized properties, such as binding to a stem-loop structure in the steroidogenic acute regulatory enzyme (Star) promoter (11), acting as a shuttling RNA binding protein (12), and inhibiting RNA splicing (13). However, none of these ascribed functions of Dax1 adequately explain the mechanism by which X-linked AHC develops from loss of DAX1.
Patients diagnosed with AHC present with adrenocortical failure, secondary to a hypoplastic/aplastic gland manifesting with symptoms of cortisol and aldosterone deficiency, including hyperpigmentation, salt-wasting crisis with hyponatremia, hyperkalemia, and hypoglycemia (14, 15). There is considerable variability in the DAX1 mutations found in AHC patients with no single event being predominant. Although the disease typically manifests within the first few weeks of life, recent surveillance programs in affected families have revealed a significant variation in the age at which patients present with clinical disease. First, in approximately 60% of affected individuals, X-linked AHC manifests within the first few weeks of life with the remaining 40% of the patients diagnosed in childhood (typically between ages 1 and 10) (14). Moreover, there does not appear to be any consistent phenotypic presentation of a given genotypic mutation. Case reports have confirmed a variety of clinical presentations, including premature adrenarchy, (16, 17), mild phenotypes in heterozygous females (18–20), and late-onset AHC in males (21–23). Lastly, even when the underlying DAX1 mutation is identical, siblings in a given family can manifest at very different ages (24, 25).
Consistent with its role as a steroidogenesis inhibitor, Dax1 knockout (Dax1−/Y) mice, in which exon 2 of the Dax1 gene locus is deleted, have enhanced adrenal steroidogenesis that rescues the adrenal hypofunction manifest in Sf1 haploinsufficient mice (26, 27). These mice also have elevated gonadal steroidogenesis, which renders them infertile partly due to elevated expression of P450 aromatase (Cyp19) (28). However, all the studies performed with Dax1−/Y mice examined relatively young animals (<10 wk). To date, steroidogenic analysis of Dax1−/Y mice over the lifespan of the mouse has not been performed.
In humans, nonsense and frameshift mutations occur very evenly along the entire gene, whereas missense mutations cluster at the C terminus (14, 29, 30), which codes for the repressor domain of DAX1. In vitro analysis of DAX1 mutations reveals that the ability of these mutants to transcriptionally repress SF1 is significantly less than wild-type (Wt) DAX1 (14). However, Dax1 has recently been shown to also function as a coactivator of both Sf1 and the related nuclear receptor, liver receptor homolog 1, in adrenocortical (31) and mES cells (32), respectively. Proteomics studies have identified Dax1 as a binding partner of ES cell factors octamer binding protein 4 and Nanog homeobox, which implicate Dax1 as an important factor within the transcriptional network that maintains stem cell pluripotency (33–35). Indeed, knock down of Dax1 in ES cells results in spontaneous differentiation (36, 37). Taken together, these data reveal an important general role for Dax1 in maintaining or promoting an undifferentiated state, making it reasonable to propose a similar function in cells within the adult adrenal cortex.
A current model implicates Dax1 in the maintenance of adrenocortical progenitor cells, primarily because Dax1 expression within the adrenal is restricted to the subcapsular cortex, which is the location of putative adrenocortical progenitor cells (38). Moreover, we have shown in prior studies that Dax1 expression in the adrenal is activated by glucocorticoids and inhibited by ACTH, placing it within an intraadrenal endocrine system that regulates differentiation (steroidogenesis) of adrenal progenitors in response to limiting corticosterone levels (39–41). In such a model, Dax1 deficiency would be predicted to result in premature progenitor cell differentiation. If the progenitor cells cannot be replenished rapidly enough or have a limited ability to self-renew, adrenal failure would ultimately become manifest. Clinical support for this model is emerging as genetic screening and surveillance programs become incorporated into the care of affected families harboring a DAX1 mutation. In addition to the description of late-onset disease in some patients, a few patients have been reported to have a period of elevated adrenal steroid hormone production that precedes the ultimate development of adrenal failure (25, 42). Additionally, the fact that some children demonstrate early puberty indicates premature elevation of sex steroids (16, 17). Taken together, these phenotypes suggest that, as would be expected based on its in vitro function, loss of DAX1 results in a hyperfunctional adrenal cortex, but only briefly.
In an effort to reconcile the disparate phenotypes of mice and humans deficient in Dax1, and in an attempt to explain the apparent paradox between in vitro and in vivo data, we examine the adrenal form and function of Dax1 deficiency in mice over an extended period of nearly 2 yr.
Materials and Methods
Experimental animals
All experiments involving animals were performed according to institutionally approved and current animal care guidelines. Dax1-deficient mice were obtained previously (26, 27) and maintained on a 129S1/SvImJ background. To obtain Dax1−/Y mice, Wt males were mated with heterozygous females. Experimental animals were housed with two to four animals per cage for up to 100 wk (n = 5 per genotype, per group). Mice were injected 24 h before killing with 1 mg/kg body weight 5-bromo-2-deoxyuridine (BrdU) (Roche Diagnostics, Indianapolis, IN). Mice were transferred to single cages and housed in stress-free conditions until killing by rapid decapitation at 0800 h. Trunk blood was collected in EDTA-coated collection tubes (Sarstedt, Newton, NC). After decapitation, both adrenal glands were removed and placed in ice cold PBS.
ACTH stimulation test
ACTH stimulation test was performed as described previously (43). Briefly, the hypothalamic-pituitary axis was suppressed by ip injection of 5 mg/kg body weight dexamethasone (Sigma, St. Louis, MO) at 1800 h 1 d before and at 0800 h on the day of the assay. At 1000 h, 1 mg/kg body weight ACTH [ACTH-(1–24); Bachem, Torrance, CA] was injected ip, and blood was collected from venous tail puncture at 0, 15, 30, and 60 min after ACTH injection.
Plasma hormone measurements
Serum ACTH and corticosterone levels were determined by RIA using a 125I RIA kit (MP Biomedicals, Solon, OH) according to the manufacturer's instructions. Samples were run in duplicate and quantified using a Gamma Counter. All hormone measurements were within the standard curve of the assay.
Immunohistochemistry
Tissues were fixed for 2–3 h in formalin (4% formaldehyde in PBS) or 4% paraformaldehyde and then dehydrated in graded ethanol solutions before embedding in paraffin. Embedded tissues were sectioned in 7-μm sections and mounted on microscope slides. Sections were then deparaffinized in xylenes (2 × 5 min) and rehydrated in graded ethanol. Antigen retrieval was achieved by boiling in 10 mm citric acid for 15 min. Tissue sections were incubated in antibody diluent for 1 h at room temperature and then in primary antibody [custom anti-Sf1, 1:2000; anti-BrdU, 1:500 (AbD Serotec, Raleigh, NC); antiproliferating cell nuclear antigen (PCNA), 1:1000 (Millipore, Billerica, MA); and custom anti-cyp11β1 and anti-cyp11β2, 1:50 (a generous gift from Celso Gomez-Sanchez, University of Mississippi Medical Center, Jackson, MS)] overnight at 4 C. Sections were then incubated with fluorescent dye-conjugated secondary antibody for 1 h (fluorescein isothiocyanate antirat, 1:1000; Alexa Fluor antirabbit/mouse, 1:1000). For volumetric analysis, every 10th section of an entire adrenal was stained with hematoxylin and eosin and visualized using a light microscope. The area of each section was determined using ImageJ software. The volume of each adrenal was then calculated based on the cross-sectional area and the number of sections per adrenal.
Quantitative RT-PCR
Total RNA was isolated from tissues using TRIzol Reagent (Invitrogen, Carlsbad, CA), and 1 μg was reverse transcribed using the iScript system (Bio-Rad Laboratories, Inc., Hercules, CA). The resulting cDNA was diluted 1:5, and 2 μl were amplified with the appropriate primers using SYBR Green PCR Master Mix and an ABI 7300 Real Time PCR System (Applied Biosystems, Carlsbad, CA). Results were analyzed using the 2−ΔΔC(T) method (44). Primer sequences for each amplified gene are: Glyceraldehyde 3-phosphate dehydrogenase (Gapdh), forward (Fwd) 5′-aatgtgtccgtcgtggatct and reverse (Rev) 5′-cccagctctccccatcacta; 11β-hydroxysteroid dehydrogenase (Cyp11β1), Fwd 5′-gcaccaggtggagagtatgc and Rev 5′-gccattctggcccatttag; 21-hydroxylase (Cyp21a1), Fwd 5′-ctgcttcaccaccctgaga and Rev 5′-agctgcattcggttcctgt; Star, Fwd 5′-gctgctcagtattgacctgaag and Rev 5′-gcgataggacctggttgatg; side-chain cleavage enzyme (P450scc), Fwd 5′-aagtatggccccatttacagg and Rev 5′-tggggtccacgatgtaaact; Sf1, Fwd 5′-tccagtgtccacccttatcc and Rev 5′-cgtcgtacgaatagtccatgc; and 11β-aldosterone synthase (Cyp11β2), Fwd 5′-gcaccaggtggagagtatgc and Rev 5′-ccattctggcccatttagc.
Immunoblotting
Total-cell lysates were prepared in lysis buffer [50 mm Tris-HCl (pH 7.4), 1% Nonidet P-40, 0.25% Na-deoxycholate, 150 mm NaCl, 1 mm EDTA, 1 mm NaF, and protease inhibitor cocktail]. Lysates were allowed to rotate at 4 C for 30 min, and protein contents of the high-speed supernatant were measured using the Bradford protein assay (Bio-Rad Laboratories, Inc.). Equivalent quantities of protein (20–45 μg) were resolved on polyacrylamide-sodium dodecyl sulfate gels, transferred to nitrocellulose membrane (Bio-Rad Laboratories, Inc.), and immunoblotted with specific antibodies to Star (1:2500, a generous gift from D. B. Hales, Southern Illinois University, Carbondale, IL), PCNA (1:1000; Millipore), or β-actin (1:5000; Sigma). Results were visualized using the Supersignal West Dura Extended Duration Substrate kit (Pierce Chemical Co., Rockford, IL) and autoradiography.
Statistics
Statistical significance was determined by using an unpaired Student's t test to compare data from Dax1−/Y mice and Wt mice at each age. Results were considered significant if P < 0.05. Significant differences are denoted with an asterisk.
Results
Dax1−/Y mice have altered acute HPA axis responsiveness and baseline HPA axis profile
To determine whether Dax1-deficient mice have altered steroidogenic profile after 6 wk of age (27), we performed ACTH stimulation tests on 15-, 30-, and 60-wk Wt and Dax1−/Y mice. ACTH stimulation after dexamethasone suppression is a good readout of maximal adrenal steroidogenic capacity or responsiveness (43). Dax1−/Y mice at 15 wk of age have a significantly higher corticosterone output in response to ACTH, particularly evident at 30 and 60 min after ACTH (534 ± 56 vs. 267 ± 81 ng/ml, P < 0.05; 551 ± 50 vs. 322 ± 27 ng/ml, P < 0.05, respectively) (Fig. 1A). However, 30-wk Dax1−/Y mice have a similar corticosterone output to Wt mice (Fig. 1B). Notably, 60-wk Dax1−/Y mice have significantly reduced ACTH-induced corticosterone output at the 60-min interval (261 ± 63 vs. 540 ± 31 ng/ml, P < 0.05) (Fig. 1C). Based on this initial observation, we next investigated the basal hormonal activity of the HPA axis in Dax1-deficient mice more extensively. We measured plasma hormone levels of Wt and Dax1−/Y mice between 15 and 100 wk of age and plotted values for ACTH (Fig. 2A) and corticosterone (Fig. 2B), as well as the ACTH/corticosterone ratio (Fig. 2C). The ACTH/corticosterone ratio is an ideal illustration of actual adrenal steroidogenic responsiveness; in a more sensitive adrenal, a given amount of ACTH will elicit a greater corticosterone response (27). In agreement with our initial observation, 15-wk Dax1−/Y mice have unchanged plasma ACTH levels, elevated corticosterone (153.28 ± 24.72 vs. 43.07 ± 2.87; P < 0.05), and a lower ACTH/corticosterone ratio (3.16 ± 1.02 vs. 8.31 ± 2.05; P < 0.05) than Wt mice, indicating increased adrenal ACTH sensitivity. Mice in the intermediate age groups did not have significant differences in basal levels or ACTH/corticosterone ratio. However, compared with Wt mice, 100-wk Dax1−/Y mice exhibited slightly elevated plasma ACTH, decreased corticosterone, and a significantly greater ACTH/corticosterone ratio (10.44 ± 1.61 vs. 4.47 ± 0.76; P < 0.05) (Fig. 2C). Together, these results indicate age-dependent loss of both adrenal steroidogenic sensitivity and capacity in Dax1-deficient mice.
Fig. 1.
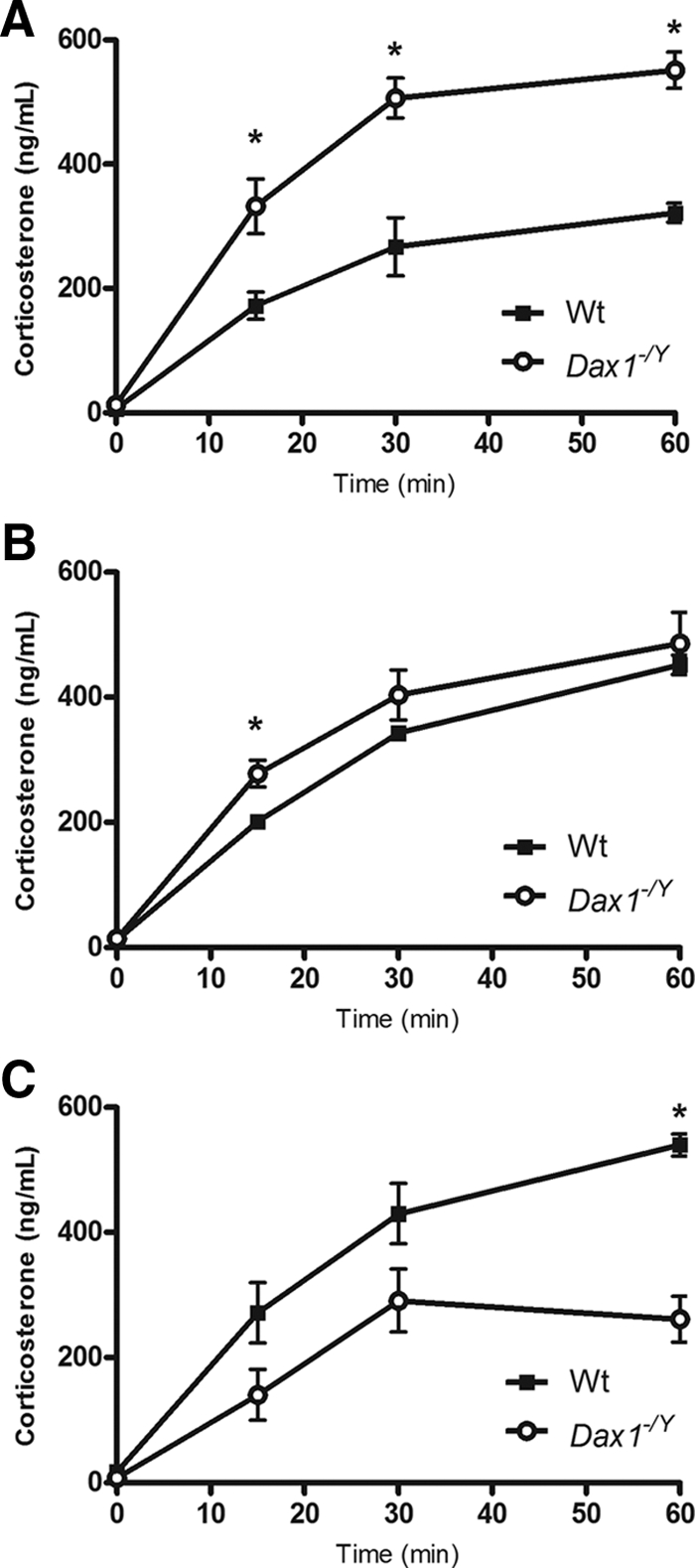
ACTH stimulation test in 15-, 30-, and 60-wk mice. ACTH stimulation test was performed by an overnight dexamethasone suppression followed by injection of ACTH as described in Materials and Methods. Blood samples were collected at 15, 30, and 60 min after ACTH injection. Corticosterone was measured by RIA, and values were plotted for 15- (A), 30- (B), and 60-wk (C) mice. Wt are represented by black squares, and Dax1−/Y are represented by white circles; *, P < 0.05, n = 5.
Fig. 2.

Basal ACTH and corticosterone levels of 15-, 30-, 70-, and 100-wk mice. Trunk blood was collected, and basal serum ACTH (A) and corticosterone (B) was measured using RIA. C, For each age, the average ratio of ACTH (in pg/ml) to corticosterone (in ng/ml) was plotted. Wt are represented by black bars, and Dax1−/Y are represented by white bars; *, P < 0.05, n = 5.
Steroidogenic enzyme gene expression in Dax1−/Y adrenals declines from 15 to 100 wk
To further delineate the age-related changes in steroidogenic output of the Dax1-deficient mice, we evaluated gene expression of several steroidogenic enzymes by quantitative PCR (qRT-PCR). As expected, compared with Wt mice of the same age, 15-wk Dax1−/Y mice have elevated adrenal expression of Star mRNA (2.88 ± 0.31-fold, P < 0.05), side-chain cleavage enzyme (Scc) mRNA (6.61 ± 0.88-fold, P < 0.05), and Cyp11β1 mRNA (3.59 ± 0.41-fold, P < 0.05) (Fig. 3A). In intermediate ages, expression of steroidogenic enzyme mRNA is not significantly different between Dax1−/Y and Wt mice. However, 100-wk Dax1−/Y mice have lower adrenal expression of Star mRNA (0.39 ± 0.02-fold, P < 0.05), Scc mRNA (0.36 ± 0.11-fold, P < 0.05), and Cyp11β1 mRNA (0.36 ± 0.02-fold, P < 0.05) compared with Wt mice. Levels of Sf1, Cyp11β2, and 21Oh are similar in Wt and Dax-deficient mice of all ages tested.
Fig. 3.

Expression of steroidogenic enzymes in Wt and Dax1−/Y mice. A, Total RNA was harvested from freshly isolated mouse adrenals and analyzed by qRT-PCR using primers directed against Scc, Star, Cyp11β1 (11b1), Aldosterone synthase (11b2), 21-hydroxylase (21Oh), and Sf1. For each age, results were plotted relative to Wt levels, which were set to one; *, P < 0.05, n = 5. B, Protein was isolated from freshly harvested adrenals, and immunoblot analysis for Star and β-actin was performed as described in Materials and Methods.
To confirm the results obtained by qRT-PCR, adrenal protein lysates were analyzed by Western blotting. Star protein levels were elevated in the 15- and 30-wk Dax1−/Y adrenals compared with Wt. However, at 80 wk, Star protein levels were dramatically lower than in the Wt counterpart (Fig. 3B). These results indicate that the altered steroidogenic capacity of Dax1−/Y mice is most likely due to altered expression of key steroidogenic enzymes.
Adrenal glands from Dax1-deficient mice display progressively worsening dysplastic adrenal morphology
Because failing adrenals often display abnormal architecture (45), we examined whether loss of Dax1 resulted in altered adrenal morphology in addition to altered hormonal output. Histological analysis revealed that Dax1−/Y adrenals have severe morphological abnormalities when compared with Wt adrenals (Fig. 4, A–H). Dax1−/Y mice at 15 wk of age have a significantly enlarged adrenal cortex (7.29 ± 0.42 vs. 3.05 ± 0.17 mm3; P < 0.05) (Fig. 4I), yet maintained normal adrenal morphological structure (Fig. 4B). Immunostaining for Cyp11β1 and Cyp11β2 verifies that proper adrenal zonation is maintained in 15-wk Dax1−/Y mice (see figure 6B). By 55 wk, although there is no significant difference in adrenal volume between Wt and Dax1−/Y mice, adrenals of Dax1−/Y mice began to display abnormal architecture (Fig. 4F). Dax1−/Y adrenals in 100-wk mice were severely deformed and dysplastic (Fig. 4H), whereas Wt adrenals maintained normal architecture (Fig. 4G). Additionally, Dax1−/Y adrenals contained large eosinophilic cytomegalic cells, which are often present in human X-linked AHC adrenals (15). These results indicate that in addition to simple changes in steroidogenesis or hormone output, adrenals from Dax1-deficient mice demonstrate progressively worsening morphology reminiscent of the architecture of failing adrenal glands.
Fig. 4.

Adrenal histology after hematoxylin and eosin staining. Adrenals, harvested from Wt (A, C, E, and G) and Dax1−/Y (B, D, F, and H) mice at 15 (A and B), 30 (C and D), 55 (E and F), and 100 wk (G and H) were fixed, embedded, and sectioned as described in Materials and Methods. Sections were deparaffinized and stained with hematoxylin and eosin to visualize gross changes in histology or morphology. Volumetric analysis (I) was performed for each adrenal using sequential hematoxylin and eosin-stained sections spanning the entire gland; *, P < 0.05, n = 5.
Cellular proliferation in Dax1−/Y adrenals declines from 15 to 100 wk
Because Dax1 expression is restricted to a region of high proliferation, we sought to determine whether Dax1-deficient mice had altered adrenocortical proliferation using two methodologies: BrdU, which incorporates into newly synthesized DNA, and anti-PCNA, which is expressed specifically in proliferating cells. Using adrenals from mice that had been injected with BrdU 24 h before killing, we did immunohistochemical staining with an antibody directed against BrdU, quantified the number of BrdU foci in each adrenal section, and averaged the results for three sections per adrenal. Adrenals of 15-wk Dax1−/Y mice had significantly more of BrdU-positive foci than their Wt counterparts (13.0 ± 2.5 vs. 2.7 ± 0.2; P < 0.05). Dax1−/Y adrenals in 30-wk mice had more BrdU-positive foci than Wt (6.0 ± 1.0 vs. 2.2 ± 0.2; P < 0.05). At intermediate ages, the number of BrdU-positive foci was not significantly different between the genotypes. However, 100-wk Dax1−/Y mice had fewer BrdU-positive foci than their Wt counterparts (1.9 ± 0.8 vs. 4.1 ± 0.4; P < 0.05) (Fig. 5A). These results were confirmed by performing immunostaining on 15-, 30-, 70-, and 100-wk adrenal sections using an antibody directed against PCNA. Although a greater number of cells in each adrenal section stained positive for PCNA than BrdU, by both methods, an appreciable difference between the genotypes was observed. Adrenals of 15-wk Dax1−/Y mice had significantly higher levels of PCNA-positive foci than their Wt counterparts (110.4 ± 27.4 vs. 42 ± 3.6; P < 0.05) (Fig. 5B). The number of PCNA-positive foci was not significantly different in the 30- and 60-wk adrenals. However, 100-wk Dax1−/Y mice had fewer PCNA-positive foci than their Wt counterparts (9.0 ± 3.8 vs. 24.8 ± 3.8; P < 0.05).
Fig. 5.

Quantification of proliferation markers in Wt and Dax1−/Y mice. BrdU- (A) and PCNA-positive (B) foci per adrenal section were quantified. For each age, foci were counted in three adrenal sections for each animal and results presented as average ± se. Wt are represented by black bars, and Dax1−/Y are represented by white bars; *, P < 0.05, n = 5. C, Protein was isolated from freshly harvested adrenals and subjected to immunoblot analysis for PCNA and β-actin.
To further verify the observed differences in PCNA-positive foci, we performed immunoblot analysis for PCNA using whole adrenal lysate protein from 15-, 30-, and 80-wk Wt and Dax1−/Y mice. There was appreciably more PCNA immunoreactivity in 15- and 30-wk Dax1−/Y adrenals. However, at 80 wk, PCNA protein expression is considerably lower than in Wt adrenals (Fig. 5C).
As shown in Fig. 6A, PCNA staining is considerable more prevalent in Dax1−/Y mice than in Wt littermates and is observed primarily in the subcapsular region, consistent with the expected location of proliferating undifferentiated cells. Importantly, Sf1 expression is maintained throughout the cortices of both Wt and Dax1−/Y adrenals at 15 and 100 wk, indicating that adrenal tissue is not being replaced or infiltrated by nonadrenal tissue in the Dax1−/Y mice. Costaining for PCNA and Cyp11β2, the zona glomerulosa-specific marker, shows that most PCNA-positive cells seem to be localized within this region, although some are clearly outside of this zone (Fig. 6C). Taken together, these results indicate that adrenals from young Dax1-deficient mice have an increased number of rapidly proliferating subcapsular cells, yet demonstrate an age-dependent decrease in cellular proliferation as opposed to the relatively constant level seen in Wt adrenals.
Fig. 6.

Visualization of PCNA reactivity and cortical zonation in young and old Wt and Dax1−/Y mice. Adrenal sections from mice of the indicated ages were subjected to immunofluorescence as described in Materials and Methods using (A) antibodies directed against PCNA (green) and Sf1 (red) and (B) antibodies directed against Cyp11β2 (red) and either cyp11β1 or PCNA (green). Images were merged to show colocalization.
Discussion
The primary goal of this study was to reconcile the interspecies variation observed in mice and humans deficient in DAX1, because their apparently opposite phenotypes have long been a confounding factor for understanding the molecular function of DAX1 in the adrenal cortex. In humans, DAX1 mutations or deletions are the cause of X-linked AHC, a condition that results in extreme adrenal insufficiency. In contrast, previous work in Dax1-deficient mice revealed a milder adrenal phenotype and indeed adrenal hyperfunction (27). In this study, we have revealed a similarity between the adrenal phenotype of aging Dax1-deficient mice and that of human patients with X-linked AHC. As expected based on earlier studies, young Dax1−/Y mice have enhanced adrenal steroidogenic output, which is consistent with the well-known repressor function of Dax1 in steroidogenesis. However, we now show, through hormone monitoring and gene expression analyses, that Dax1−/Y mice have progressively diminishing adrenal steroidogenesis as they age. Additionally, young Dax1−/Y mice display enhanced adrenal subcapsular proliferation, and aging mice exhibit progressively worsening dysplasia accompanied by a marked decrease in proliferation. These results demonstrate for the first time evidence of adrenal failure in Dax1-deficient mice and suggest a common role for Dax1 in adrenocortical progenitor cell maintenance in mouse and human adrenal cortices.
It is interesting to note that although expression of several steroidogenic markers mirrors the change in adrenal volume in the Dax1−/Y mice, not all steroidogenic enzymes are affected. Importantly, Sf1 expression is maintained in the adrenals of Dax1−/Y mice, whereas Star and Scc expression are drastically decreased in older Dax1−/Y mice. These results suggest that the adrenals of the 100-wk Dax1−/Y mice are most likely comprised of tissue that was once steroidogenically active but has become nonfunctional over time. If these adrenals had become infiltrated with nonsteroidogenic tissues, such as fibroblasts, all the steroidogenic markers, including Sf1, would be expected to follow the same pattern. Surprisingly, in light of the dramatic changes in proliferating cells within and around the zona glomerulosa, there is not a significant difference in expression of Cyp11β2 between the two genotypes at any age. However, the dramatically elevated Cyp11β1 expression and adrenal volume in young Dax1−/Y mice suggests expansion of the zona fasciculata. This expression pattern suggests that differentiated adrenocortical cells only transiently reside within the zona glomerulosa and persist for a longer period of time within the zona fasciculata. Further studies using lineage tracing techniques will more clearly elucidate this phenomenon.
DAX1 mutations/deletions in humans are usually not discovered unless there are symptoms of adrenal insufficiency in young males that serve as an indication to sequence the DAX1 allele. Hormone monitoring is not standard practice in asymptomatic children, explaining the paucity of data detailing steroidogenic profiles before adrenal failure. However, recent case reports from surveillance studies in families known to harbor a given DAX1 mutation indicate that some individuals with DAX1 mutations have elevated cortisol or other steroid hormones before the clinical manifestation of adrenal insufficiency (25, 42). These data support a model in which loss of DAX1 results in premature differentiation of the adrenal cortex at the expense of a decrease in progenitor reserve and/or progenitor cell failure.
It is noteworthy that there has been speculation that Dax1−/Y mice might express a hypomorphic Dax1 allele, which could explain interspecies phenotype differences (46). This speculation is largely based on the method used to generate the knockout-deletion of the relatively small exon 2, leaving the larger exon 1 intact. An alternatively spliced isoform, DAX1α/DAX1A, has been identified in human tissues, consisting of all of exon 1 and a very short intronic sequence, which would resemble a protein that is hypothesized to be produced by the knockout mouse (47, 48). However, in human tissues, expression of this shorter isoform is extremely low compared with the longer isoform, raising questions regarding the physiological role of DAX1α in steroidogenic tissue development and function (49). Additionally, to date, neither a mouse orthologue to DAX1α nor a specific hypomorphic Dax1allele in the Dax1−/Y mouse has been identified. Using an antibody that would recognize a truncated protein, our lab showed no evidence of truncated protein expression in the Dax1−/Y adrenals (27). Lastly, X-linked AHC develops in patients with no mutations in exon 1 but with mutation only in the C terminus of DAX1, arguing for a critical function of exon 2 (50). Although the gonadal phenotypes between X-linked AHC patients and Dax1−/Y mice are strikingly similar, there are some noticeable differences, mainly that Dax1−/Y mice do not have evidence of hypogonadotropic hypogonadism (26). Of course, this can reflect either biologic species differences or may simply reflect heterogeneity and variation along the spectrum of phenotypes for DAX1 mutations rather than indicate a hypomorphic allele in the mouse model.
Recently, we reported that Dax1 can function as a coactivator when associated with steroid receptor RNA activator (SRA) (31). Interestingly, this interaction occurs at the N-terminal region of Dax1, suggesting that it could be preserved in cases where only the C terminus of Dax1 is deleted. Perhaps Dax1 interaction with SRA provides the mechanism by which Dax1 is able to activate genes required for maintenance of adrenocortical progenitor cells. Indeed, recent data from our lab has revealed that Dax1 interacts with SRA to up-regulate Oct4 expression in mouse ES cells (32). However, these studies were performed in mouse cells and have not yet been done in human cells. The role of SRA in the context of Dax1 deficiency is not known and would require further investigation.
The results of the current study are consistent with a role for Dax1 in the maintenance of subcapsular adrenocortical progenitor cells. Previous studies have demonstrated that both Wnt signaling and glucocorticoids transcriptionally activate Dax1 (39, 40) and that loss of β-catenin results in adrenocortical failure and cellular depletion (41, 51). This suggests that Dax1 expression is controlled by both developmental (Wnt) and endocrine (glucocorticoids) pathways. Interestingly, ACTH disrupts all Sf1 interactions on the Dax1 promoter, ultimately decreasing Dax1 expression to presumably allow Sf1 to transcriptionally activate genes required for ACTH-dependent steroidogenesis (40). These data, combined with the similar pattern of expression of β-catenin and Dax1 restricted to the subcapsular region, lend credence to a physiologic interaction between these pathways in maintenance of undifferentiated progenitor cells within the adrenal cortex. It is, therefore, reasonable to speculate that loss of Dax1 results in premature differentiation of adrenocortical progenitor cells, which ultimately leads to adrenal failure. In such a model, interspecies differences in lifespan may be the major factor determining the time at which the adrenal ultimately fails.
Loss of a factor leading to exhaustion of organ-specific progenitor cells has been observed in other endocrine tissues: human PROP paired-like homeobox 1 (PROP1) mutations result in dramatic variability of pituitary morphology and longitudinal imaging revealed pituitary enlargement followed by hypoplasia (52, 53). Data from Prop1-deficient mice, in which a hypoplastic pituitary follows expansion of trapped undifferentiated cells, suggests that this may be due to the role of Prop1 in migration of progenitor cells into the anterior pituitary (54). Future studies would be predicted to define additional tissue-specific transcription factors that cooperate in the maintenance of organ-specific progenitor cells in various tissues.
Cytomegaly is a hallmark phenotype of an adrenal cortex from a patient diagnosed with X-linked AHC (15, 55) and in the current study, we observe that adrenal sections from Dax1−/Y mice display dismorphic cytomegaly that increases in intensity with age. Cytomegaly is also a phenotype of other genetic syndromes in which adrenal failure occurs, such as IMAGe syndrome (56). Additionally, several mouse models display a similar histologic phenotype with coincident adrenal failure, including the acd mutant mouse that is defective in telomere maintenance (57) and an adrenal-specific β-catenin knockout mouse (41). It is provocative to speculate that AHC with cytomegaly may represent a compensatory morphological endpoint induced by progenitor cell exhaustion. Indeed, perhaps the most intriguing phenotype observed in the current study is the proliferation difference between Dax1−/Y and Wt mouse adrenal glands, particularly at either end of the age spectrum. Young Dax1−/Y mouse adrenals have a significantly higher proliferation rate (determined by BrdU-positive foci and PCNA staining), whereas it is dramatically lower in old mice. This somewhat surprising result is in stark contrast to the relatively constant rate of proliferation seen in the Wt mice and to previous suggestions that Dax1-deficient adrenals would preferentially differentiate at the expense of proliferation (58). Further analysis will be required to determine whether the data reflect loss of direct inhibition by Dax1 on subcapsular proliferation or activation of compensatory proliferation of progenitor cells that remain after the premature differentiation of a portion of the progenitor cell pool.
Acknowledgments
We thank Dr. J. Larry Jameson (University of Pennsylvania, Philadelphia, PA) for providing Dax1−/Y mice, Dr. D. B. Hales (University of Illinois at Chicago, Chicago, IL) for providing the Star antibody, and Dr. C. Gomez-Sanchez (University of Mississippi Medical Center, Jackson, MS) for providing anti-cyp11β1 and anti-cyp11β2.
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases National Institutes of Health Research Grants R01-DK062027 (to G.D.H.) and R01-DK074819 [to Colin Jefcoate (University of Wisconsin, Madison, WI) and G.D.H.], and by the Training Grant T32-GM008322 Cellular and Molecular Approaches to Systems and Integrative Biology (to J.O.S.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AHC
- Adrenal hypoplasia congenita
- BrdU
- 5-bromo-2-deoxyuridine
- Cyp11β1
- 11β-hydroxysteroid dehydrogenase
- Cyp11β2
- 11β-aldosterone synthase
- DAX1
- dosage-sensitive sex reversal, AHC critical region on the X chromosome, gene 1
- Dax1−/Y
- Dax1 knockout
- Fwd
- forward
- HPA
- hypothalamic-pituitary-adrenal
- PCNA
- proliferating cell nuclear antigen
- qRT-PCR
- quantitative PCR
- Rev
- reverse
- Prop1
- Prop paired-like homeobox 1
- Scc
- side-chain cleavage enzyme
- Sf1
- steroidogenic factor 1
- SRA
- steroid receptor RNA activator
- Star
- steroidogenic acute regulatory enzyme
- Wt
- wild type.
References
- 1. Zanaria E, Muscatelli F, Bardoni B, Strom TM, Guioli S, Guo W, Lalli E, Moser C, Walker AP, McCabe ER. 1994. An unusual member of the nuclear hormone receptor superfamily responsible for X-linked adrenal hypoplasia congenita. Nature 372:635–641 [DOI] [PubMed] [Google Scholar]
- 2. Guo W, Burris TP, Zhang YH, Huang BL, Mason J, Copeland KC, Kupfer SR, Pagon RA, McCabe ER. 1996. Genomic sequence of the DAX1 gene: an orphan nuclear receptor responsible for X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. J Clin Endocrinol Metab 81:2481–2486 [DOI] [PubMed] [Google Scholar]
- 3. Muscatelli F, Strom TM, Walker AP, Zanaria E, Récan D, Meindl A, Bardoni B, Guioli S, Zehetner G, Rabl W. 1994. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature 372:672–676 [DOI] [PubMed] [Google Scholar]
- 4. Ito M, Yu R, Jameson JL. 1997. DAX-1 inhibits SF-1-mediated transactivation via a carboxy-terminal domain that is deleted in adrenal hypoplasia congenita. Mol Cell Biol 17:1476–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lalli E, Bardoni B, Zazopoulos E, Wurtz JM, Strom TM, Moras D, Sassone-Corsi P. 1997. A transcriptional silencing domain in DAX-1 whose mutation causes adrenal hypoplasia congenita. Mol Endocrinol 11:1950–1960 [DOI] [PubMed] [Google Scholar]
- 6. Ikeda Y, Takeda Y, Shikayama T, Mukai T, Hisano S, Morohashi KI. 2001. Comparative localization of Dax-1 and Ad4BP/SF-1 during development of the hypothalamic-pituitary-gonadal axis suggests their closely related and distinct functions. Dev Dyn 220:363–376 [DOI] [PubMed] [Google Scholar]
- 7. Val P, Lefrançois-Martinez AM, Veyssière G, Martinez A. 2003. SF-1 a key player in the development and differentiation of steroidogenic tissues. Nucl Recept 1:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Crawford PA, Dorn C, Sadovsky Y, Milbrandt J. 1998. Nuclear receptor DAX-1 recruits nuclear receptor corepressor N-CoR to steroidogenic factor 1. Mol Cell Biol 18:2949–2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Altincicek B, Tenbaum SP, Dressel U, Thormeyer D, Renkawitz R, Baniahmad A. 2000. Interaction of the corepressor Alien with DAX-1 is abrogated by mutations of DAX-1 involved in adrenal hypoplasia congenita. J Biol Chem 275:7662–7667 [DOI] [PubMed] [Google Scholar]
- 10. Suzuki T, Kasahara M, Yoshioka H, Morohashi K, Umesono K. 2003. LXXLL-related motifs in Dax-1 have target specificity for the orphan nuclear receptors Ad4BP/SF-1 and LRH-1. Mol Cell Biol 23:238–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zazopoulos E, Lalli E, Stocco DM, Sassone-Corsi P. 1997. DNA binding and transcriptional repression by DAX-1 blocks steroidogenesis. Nature 390:311–315 [DOI] [PubMed] [Google Scholar]
- 12. Lalli E, Ohe K, Hindelang C, Sassone-Corsi P. 2000. Orphan receptor DAX-1 is a shuttling RNA binding protein associated with polyribosomes via mRNA. Mol Cell Biol 20:4910–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ohe K, Tamai KT, Parvinen M, Sassone-Corsi P. 2009. DAX-1 and SOX6 molecular interplay results in an antagonistic effect in pre-mRNA splicing. Dev Dyn 238:1595–1604 [DOI] [PubMed] [Google Scholar]
- 14. Reutens AT, Achermann JC, Ito M, Gu WX, Habiby RL, Donohoue PA, Pang S, Hindmarsh PC, Jameson JL. 1999. Clinical and functional effects of mutations in the DAX-1 gene in patients with adrenal hypoplasia congenita. J Clin Endocrinol Metab 84:504–511 [DOI] [PubMed] [Google Scholar]
- 15. Hay ID, Smail PJ, Forsyth CC. 1981. Familial cytomegalic adrenocortical hypoplasia: an X-linked syndrome of pubertal failure. Arch Dis Child 56:715–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Landau Z, Hanukoglu A, Sack J, Goldstein N, Weintrob N, Eliakim A, Gillis D, Sagi M, Shomrat R, Kosinovsky EB, Anikster Y. 2009. Clinical and genetic heterogeneity of congenital adrenal hypoplasia due to NR0B1 gene mutations. Clin Endocrinol 72:448–454 [DOI] [PubMed] [Google Scholar]
- 17. Domenice S, Latronico AC, Brito VN, Arnhold IJ, Kok F, Mendonca BB. 2001. Adrenocorticotropin-dependent precocious puberty of testicular origin in a boy with X-linked adrenal hypoplasia congenita due to a novel mutation in the DAX1 gene. J Clin Endocrinol Metab 86:4068–4071 [DOI] [PubMed] [Google Scholar]
- 18. Bernard P, Ludbrook L, Queipo G, Dinulos M-B, Kletter GB, Zhang Y-H, Phelan JK, McCabe ERB, Harley VR, Vilain E. 2006. A familial missense mutation in the hinge region of DAX1 associated with late-onset AHC in a prepubertal female. Mol Gen Metab 88:272–279 [DOI] [PubMed] [Google Scholar]
- 19. Seminara SB, Achermann JC, Genel M, Jameson JL, Crowley WF., Jr 1999. X-linked adrenal hypoplasia congenita: a mutation in DAX1 expands the phenotypic spectrum in males and females. J Clin Endocrinol Metab 84:4501–4509 [DOI] [PubMed] [Google Scholar]
- 20. Shaikh MG, Boyes L, Kingston H, Collins R, Besley GT, Padmakumar B, Ismayl O, Hughes I, Hall CM, Hellerud C, Achermann JC, Clayton PE. 2008. Skewed X inactivation is associated with phenotype in a female with adrenal hypoplasia congenita. J Med Genet 45:e1–e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tabarin A, Achermann JC, Recan D, Bex V, Bertagna X, Christin-Maitre S, Ito M, Jameson JL, Bouchard P. 2000. A novel mutation in DAX1 causes delayed-onset adrenal insufficiency and incomplete hypogonadotropic hypogonadism. J Clin Invest 105:321–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mantovani G, Ozisik G, Achermann JC, Romoli R, Borretta G, Persani L, Spada A, Jameson JL, Beck-Peccoz P. 2002. Hypogonadotropic hypogonadism as a presenting feature of late-onset X-linked adrenal hypoplasia congenita. J Clin Endocrinol Metab 87:44–48 [DOI] [PubMed] [Google Scholar]
- 23. Ozisik G, Mantovani G, Achermann JC, Persani L, Spada A, Weiss J, Beck-Peccoz P, Jameson JL. 2003. An alternate translation initiation site circumvents an amino-terminal DAX1 nonsense mutation leading to a mild form of X-linked adrenal hypoplasia congenita. J Clin Endocrinol Metab 88:417–423 [DOI] [PubMed] [Google Scholar]
- 24. Calliari LE, Longui CA, Rocha MN, Faria CD, Kochi C, Melo MR, Melo MB, Monte O. 2007. A novel mutation in DAX1 gene causing different phenotypes in three siblings with adrenal hypoplasia congenita. Genet Mol Res 6:177–183 [PubMed] [Google Scholar]
- 25. Peter M, Viemann M, Partsch CJ, Sippell WG. 1998. Congenital adrenal hypoplasia: clinical spectrum, experience with hormonal diagnosis, and report on new point mutations of the DAX-1 gene. J Clin Endocrinol Metab 83:2666–2674 [DOI] [PubMed] [Google Scholar]
- 26. Yu RN, Ito M, Saunders TL, Camper SA, Jameson JL. 1998. Role of Ahch in gonadal development and gametogenesis. Nat Genet 20:353–357 [DOI] [PubMed] [Google Scholar]
- 27. Babu PS, Bavers DL, Beuschlein F, Shah S, Jeffs B, Jameson JL, Hammer GD. 2002. Interaction between Dax-1 and steroidogenic factor-1 in vivo: increased adrenal responsiveness to ACTH in the absence of Dax-1. Endocrinology 143:665–673 [DOI] [PubMed] [Google Scholar]
- 28. Wang ZJ, Jeffs B, Ito M, Achermann JC, Yu RN, Hales DB, Jameson JL. 2001. Aromatase (Cyp19) expression is up-regulated by targeted disruption of Dax1. Proc Natl Acad Sci USA 98:7988–7993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin L, Gu WX, Ozisik G, To WS, Owen CJ, Jameson JL, Achermann JC. 2006. Analysis of DAX1 (NR0B1) and steroidogenic factor-1 (NR5A1) in children and adults with primary adrenal failure: ten years' experience. J Clin Endocrinol Metab 91:3048–3054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Phelan JK, Mccabe ER. 2001. Mutations in NR0B1 (DAX1) and NR5A1 (SF1) responsible for adrenal hypoplasia congenita. Hum Mutat 18:472–487 [DOI] [PubMed] [Google Scholar]
- 31. Xu B, Yang WH, Gerin I, Hu CD, Hammer GD, Koenig RJ. 2009. Dax-1 and steroid receptor RNA activator (SRA) function as transcriptional coactivators for steroidogenic factor 1 in steroidogenesis. Mol Cell Biol 29:1719–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kelly VR, Xu B, Kuick R, Koenig RJ, Hammer GD. 2010. Dax1 up-regulates Oct4 expression in mouse embryonic stem cells via LRH-1 and SRA. Mol Endocrinol 24:2281–2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun C, Nakatake Y, Ura H, Akagi T, Niwa H, Koide H, Yokota T. 2008. Stem cell-specific expression of Dax1 is conferred by STAT3 and Oct3/4 in embryonic stem cells. Biochem Biophys Res Commun 372:91–96 [DOI] [PubMed] [Google Scholar]
- 34. Sun C, Nakatake Y, Akagi T, Ura H, Matsuda T, Nishiyama A, Koide H, Ko MS, Niwa H, Yokota T. 2009. Dax1 binds to Oct3/4 and inhibits its transcriptional activity in embryonic stem cells. Mol Cell Biol 29:4574–4583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim J, Chu J, Shen X, Wang J, Orkin SH. 2008. An extended transcriptional network for pluripotency of embryonic stem cells. Cell 132:1049–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Khalfallah O, Rouleau M, Barbry P, Bardoni B, Lalli E. 2009. Dax-1 knockdown in mouse embryonic stem cells induces loss of pluripotency and multilineage differentiation. Stem Cells 27:1529–1537 [DOI] [PubMed] [Google Scholar]
- 37. Niakan KK, Davis EC, Clipsham RC, Jiang M, Dehart DB, Sulik KK, McCabe ER. 2006. Novel role for the orphan nuclear receptor Dax1 in embryogenesis, different from steroidogenesis. Mol Genet Metab 88:261–271 [DOI] [PubMed] [Google Scholar]
- 38. Kim AC, Barlaskar FM, Heaton JH, Else T, Kelly VR, Krill KT, Scheys JO, Simon DP, Trovato A, Yang WH, Hammer GD. 2009. In search of adrenocortical stem and progenitor cells. Endocr Rev 30:241–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mizusaki H, Kawabe K, Mukai T, Ariyoshi E, Kasahara M, Yoshioka H, Swain A, Morohashi K. 2003. Dax-1 (dosage-sensitive sex reversal-adrenal hypoplasia congenita critical region on the X chromosome, gene 1) gene transcription is regulated by wnt4 in the female developing gonad. Mol Endocrinol 17:507–519 [DOI] [PubMed] [Google Scholar]
- 40. Gummow BM, Scheys JO, Cancelli VR, Hammer GD. 2006. Reciprocal regulation of a glucocorticoid receptor-steroidogenic factor-1 transcription complex on the Dax-1 promoter by glucocorticoids and adrenocorticotropic hormone in the adrenal cortex. Mol Endocrinol 20:2711–2723 [DOI] [PubMed] [Google Scholar]
- 41. Kim AC, Reuter AL, Zubair M, Else T, Serecky K, Bingham NC, Lavery GG, Parker KL, Hammer GD. 2008. Targeted disruption of β-catenin in Sf1-expressing cells impairs development and maintenance of the adrenal cortex. Development 135:2593–2602 [DOI] [PubMed] [Google Scholar]
- 42. Achermann JC, Silverman BL, Habiby RL, Jameson JL. 2000. Presymptomatic diagnosis of X-linked adrenal hypoplasia congenita by analysis of DAX1. J Pediatr 137:878–881 [DOI] [PubMed] [Google Scholar]
- 43. Winnay JN, Xu J, O'Malley BW, Hammer GD. 2006. Steroid receptor coactivator-1-deficient mice exhibit altered hypothalamic-pituitary-adrenal axis function. Endocrinology 147:1322–1332 [DOI] [PubMed] [Google Scholar]
- 44. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 45. Lack EE. 1990. Pathology of the adrenal glands. New York: Churchill Livingstone [Google Scholar]
- 46. Niakan KK, McCabe ER. 2005. DAX1 origin, function, and novel role. Mol Genet Metab 86:70–83 [DOI] [PubMed] [Google Scholar]
- 47. Ho J, Zhang YH, Huang BL, McCabe ER. 2004. NR0B1A: an alternatively spliced form of NR0B1. Mol Genet Metab 83:330–336 [DOI] [PubMed] [Google Scholar]
- 48. Hossain A, Li C, Saunders GF. 2004. Generation of two distinct functional isoforms of dosage-sensitive sex reversal-adrenal hypoplasia congenita-critical region on the X chromosome gene 1 (DAX-1) by alternative splicing. Mol Endocrinol 18:1428–1437 [DOI] [PubMed] [Google Scholar]
- 49. Nakamura Y, Vargas Morris C, Sasano H, Rainey WE. 2009. DAX-1A (NR0B1A) expression levels are extremely low compared to DAX-1 (NR0B1) in human steroidogenic tissues. Horm Metab Res 41:30–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Achermann JC, Meeks JJ, Jameson JL. 2001. Phenotypic spectrum of mutations in DAX-1 and SF-1. Mol Cell Endocrinol 185:17–25 [DOI] [PubMed] [Google Scholar]
- 51. Kim A, Giordano TJ, Kuick R, Serecky K, Hammer GD. 2009. Wnt/βcatenin signaling in adrenocortical stem/progenitor cells: implications for adrenocortical carcinoma. Ann Endocrinol 70:156. [DOI] [PubMed] [Google Scholar]
- 52. Kelberman D, Turton JP, Woods KS, Mehta A, Al-Khawari M, Greening J, Swift PG, Otonkoski T, Rhodes SJ, Dattani MT. 2009. Molecular analysis of novel PROP1 mutations associated with combined pituitary hormone deficiency (CPHD). Clin Endocrinol 70:96–103 [DOI] [PubMed] [Google Scholar]
- 53. Riepe FG, Partsch CJ, Blankenstein O, Monig H, Pfäffle RW, Sippell WG. 2001. Longitudinal imaging reveals pituitary enlargement preceding hypoplasia in two brothers with combined pituitary hormone deficiency attributable to PROP1 mutation. J Clin Endocrinol Metab 86:4353–4357 [DOI] [PubMed] [Google Scholar]
- 54. Ward RD, Raetzman LT, Suh H, Stone BM, Nasonkin IO, Camper SA. 2005. Role of PROP1 in pituitary gland growth. Mol Endocrinol 19:698–710 [DOI] [PubMed] [Google Scholar]
- 55. Weiss L, Mellinger RC. 1970. Congenital adrenal hypoplasia—an X-linked disease. J Med Genet 7:27–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Else T, Hammer GD. 2005. Genetic analysis of adrenal absence: agenesis and aplasia. Trends Endocrinol Metab 16:458–468 [DOI] [PubMed] [Google Scholar]
- 57. Keegan CE, Hutz JE, Else T, Adamska M, Shah SP, Kent AE, Howes JM, Beamer WG, Hammer GD. 2005. Urogenital and caudal dysgenesis in adrenocortical dysplasia (acd) mice is caused by a splicing mutation in a novel telomeric regulator. Hum Mol Genet 14:113–123 [DOI] [PubMed] [Google Scholar]
- 58. Lalli E, Sassone-Corsi P. 2003. DAX-1, an unusual orphan receptor at the crossroads of steroidogenic function and sexual differentiation. Mol Endocrinol 17:1445–1453 [DOI] [PubMed] [Google Scholar]