

Abstract
T cell activation is controlled by incompletely defined opposing stimulation and suppression signals that together sustain the balance between optimal host defense against infection and peripheral tolerance. Herein, we explored the impacts of Foxp3+ regulatory T cell (Treg) suppression in priming antigen-specific T cell activation under non-infection and infection conditions. We find the transient ablation of Foxp3+ Tregs unleashes the robust expansion and activation of peptide stimulated CD8+ T cells that provide protection against Listeria monocytogenes (Lm) infection in an antigen-specific fashion. By contrast, Treg-ablation had non-significant impacts on the CD8+ T cell response primed by infection with recombinant Lm. Similarly, non-recombinant Lm administered with peptide stimulated the expansion and activation of CD8+ T cells that paralleled the response primed by Treg-ablation. Interestingly, these adjuvant properties of Lm did not require CD8+ T cell stimulation by IL-12 produced in response to infection, but instead were associated with sharp reductions in Foxp3+ Treg suppressive potency. Therefore, Foxp3+ Tregs impose critical barriers that when overcome naturally during infection or artificially with ablation allows the priming of protective antigen-specific CD8+ T cells.
INTRODUCTION
Defining the signals that control the expansion and activation of protective antigen-specific T cells are important prerequisites for designing therapies aimed at targeting these adaptive immune components for boosting host defense against infection. Although some molecular signals capable of stimulating CD8+ T cell activation in vitro have been identified (1, 2), the essential components required for priming protective CD8+ T cells in vivo in the context of infection or immunization remain incompletely defined. For example, although cytokines such as IL-12, type I IFN, or IL-21 each together with cognate antigen and costimulation stimulate the expansion of CD8+ T cells in vitro, these cytokines are jointly non-essential for priming the expansion of protective pathogen-specific CD8+ T cells after in vivo infection with the intracellular bacterium Listeria monocytogenes (Lm) (3–6). This discordance is likely attributed to the more complex balance between immune stimulation and suppression signals that regulate T cell activation in vivo, and other differences in the relative activation of immune components during infection compared with non-infection conditions. These include the stimulation of immune cells through a growing list of pattern recognition receptors that recognize defined pathogen-associated ligands, the activation of more complex inflammatory cytokine cascades, and up-regulation of costimulation and cytokine signals by antigen-presenting cells that are each difficult to recapitulate using in vitro models of T cell stimulation (7–11). Therefore, dissociating the parameters that permit the priming of pathogen-specific T cells during infection from non-infection conditions using relevant in vivo models are required.
Regulatory T cells (Tregs), identified as the Foxp3+ subset of CD4+ T cells, play pivotal roles in controlling the balance between immune stimulation and suppression during both non-infection and infection conditions (12–14). Mice with naturally occurring or experimentally induced sustained defects in Foxp3+ develop fatal systemic autoimmunity that is associated with the activation of self-reactive T cells and antigen-presenting cells (15–19). By extension, the potency of many immune adjuvants coincides with their ability to either directly dampen Treg suppression or indirectly by reducing the impacts of Treg suppression on target cells (20–25). Although these associations suggest overriding Treg suppression may represent an important prerequisite for priming protective T cells in vivo, the specific limitations imposed by Foxp3+ Tregs on the expansion and activation of antigen-specific T cells have not been clearly identified.
To interrogate the impacts of Treg suppression in priming T cells in vivo, we enumerated the effects of Foxp3+ cell-ablation on the expansion of antigen-specific CD8+ T cells initially after stimulation with purified peptide designed to mimic antigen exposure under non-inflammatory conditions. Related experiments investigated the relative impacts of Treg suppression on the pathogen-specific T cell response primed by infection with recombinant Lm engineered to express the same antigen, non-recombinant Lm administered with purified peptide, and potential shifts in Treg suppressive potency after this infection. We used Foxp3DTR transgenic mice that express the human high-affinity diphtheria toxin (DT) receptor or Foxp3GFP reporter mice that express GFP each with Foxp3 that allows the targeted ablation or isolation, respectively, of Tregs based on their lineage-defining marker (19, 26). These experiments demonstrate Foxp3+ Treg suppression imposes critical limitations for priming protective CD8+ T cells after stimulation during non-inflammatory conditions that are overcome by acute infection.
MATERIALS AND METHODS
Mice
C57BL/6 (B6) and Ly5.2/Cr (CD45.1+) mice were purchased from The National Cancer Institute. Foxp3DTR and Foxp3GFP were kindly provided by Dr. Alexander Rudensky (Memorial Sloan Kettering) (19, 26), and each backcrossed over 15 generations to the B6 background. WT OT-1 TCR transgenic mice maintained on a CD90.1 CD45.2 background, and IL-12 receptor-deficient OT-1 transgenic mice maintained on a CD90.2 CD45.2 background have been described (27, 28). CD19Cre and ROSAiDTR mice were purchased from The Jackson Laboratory and intercrossed to generate CD19Cre ROSAiDTR mice (29, 30). For cell-ablation, purified DT (Sigma-Aldrich) dissolved in saline was injected intraperitoneally (50 μg/kg) one day before and on the day of peptide stimulation and/or Lm infection. For IL-12 neutralization, purified anti-IL-12p40 (clone 17.8) or isotype control rat IgG2a antibody was inoculated intraperitoneally (1.0 mg per mouse) one day prior to peptide stimulation and/or Lm infection (31, 32). All experiments were performed under University of Minnesota IACUC approved protocols.
Cell staining, stimulation, and adoptive cell transfer
Fluorophore-conjugated antibodies for cell surface and intracellular staining were purchased from eBioscience or BD-Biosciences. CD8+ cells harvested from OT-1 TCR transgenic mice were transferred intravenously into Foxp3DTR or Foxp3WT B6 control mice (104 cells per mouse). OVA257–264 peptide (United Biochemical Research Inc., > 95% purity) was inoculated intravenously (200 μg/mouse) one day after CD8+ T cell transfer from OT-1 TCR transgenic mice. For tracking the response among endogenous CD8+ T cells, H-2Kb dimer × (BD Bioscience) loaded with OVA257–264 peptide was used for staining as described (6, 33). For cytokine production, splenocytes were stimulated with ex-vivo with OVA257–264 peptide (1 μM) for 5 hours in cultures supplemented with GolgiPlug (BD Bioscience). JAWS II cells (ATCC, CLR-11904) were grown in media supplemented with 20% FCS, 1% Hepes, and 5 ng/mL GM-CSF (34, 35). For stimulation, JAWS II cells were either pulsed with OVA257–264 peptide (5 μM, 2 hours at 37°C) or no peptide control in complete media, washed 3 times with serum-free media, and injected intravenously into mice (1 × 106 cells per mouse in 200 μl).
Infections
Recombinant Lm-OVA (36) or non-recombinant Lm-10403s (37) were each grown in Brain Heart Infusion media at 37°C, back-diluted to log-phase (OD600 0.1), washed and re-suspended in sterile saline, and injected intravenously (104 CFUs per mouse, 0.2 LD50). The number of CFUs in organ homogenates was enumerated three days after infection as described (6, 33).
Suppression assays
GFP+ Tregs were isolated from Foxp3GFP reporter mice by first enriching for CD4+ T cells using negative selection (Miltenyi Biotec), and then sorting for GFP+ cells (FACSAria). Responder T cells from naïve CD45.1+ mice were labeled with CFSE (5 μM for 10 minutes), co-cultured in 96-well round bottom plates (5 × 104 cells/100 μl) with purified GFP+ Tregs at a 1:1 ratio, and serial 2-fold dilutions of Treg to responder T cells. Shifts in Treg suppressive potency were enumerated by comparing the proliferation (CFSE dilution) in responder CD8+ CD45.1+ cells after co-culture with GFP+ Tregs isolated from mice before and after Lm infection, and stimulation with anti-mouse CD3 (1 μg/ml) for three days.
Statistics
The percent and total cell numbers, and log10 number of recoverable CFUs after infection were first determined to be normally distributed. The differences in each group were then analyzed using the unpaired Student's t test (Prism, GraphPad) with P < 0.05 taken as statistical significance.
RESULTS
Regulatory T cells impede peptide stimulated CD8+ T cell expansion and activation
To identify the limitations imposed by Treg suppression for priming T cell in vivo, the impacts of Foxp3+ cell-ablation on the expansion of CD8+ T cells with specificity to a defined non-self antigen were evaluated. We used adoptively transferred CD8+ T cells with specificity to the H-2Kb OVA257–264 peptide derived from OT-I TCR transgenic mice that are identified among cells in recipient mice based on CD90.1 expression (27). After stimulation with OVA257–264 peptide in Treg-sufficient mice, these cells expand only modestly (< 50-fold) that is consistent with results from previous reports (3) (Fig 1A). By contrast, DT treatment one-day prior and on the day of peptide inoculation triggered significantly more (> 100-fold, P < 0.001) expansion of OVA-specific CD90.1+ CD8+ cells in Foxp3DTR compared with Foxp3WT controls (Fig 1A). The expansion of these CD8+ T cells occurred in an antigen-specific fashion because only background levels were recovered from Treg-ablated mice without peptide stimulation (Fig 1A). In parallel with expansion, peptide stimulated CD8+ T cells also proliferated more rapidly with accelerated kinetics of CFSE dilution in Treg-ablated compared with Treg-sufficient mice (Fig 1B). Furthermore, the impacts of transient Treg-ablation on the proliferation and expansion of peptide stimulated CD8+ T cells were not limited only to adoptively transferred cells because increased numbers of antigen-specific among endogenous CD8+ T cells were also identified by staining with OVA257-264 peptide loaded H-2Kb dimers in Treg-ablated compared with Treg-sufficient controls (Supplementary Fig 1). Importantly, OVA-specific CD8+ T cell expansion was uniquely triggered by the ablation of Foxp3+ Tregs and not due to non-specific immune activation caused by DT induced cell death because only background levels were found after peptide stimulation and DT treatment in CD19CreROSAiDTR mice where >30-fold increased apoptotic cell death occurs (29, 30) (Supplementary Fig 2).
Figure 1.

Foxp3+ Treg-ablation primes the expansion and activation of peptide stimulated T cells. A. Percent and number of OVA-specific CD90.1+ CD8+ T cells among splenocytes day 5 after stimulation with cognate peptide in the presence (Foxp3-WT) or absence of Tregs (Foxp3-DTR). Each group of mice received DT treatment one day before and on the day of peptide inoculation. B. CFSE expression among OVA-specific CD90.1+ CD8+ T cells 60 hrs (day 2.5) and 120 hrs (day 5) after stimulation with cognate peptide in Treg-sufficient (Foxp3-WT) or Treg-ablated mice (Foxp3-DTR) (black line histogram), compared with unstimulated cells (filled histogram) or unlabelled cells (gray line histogram). C. Expression of CD25 and IFN-γ production by CD90.1+ (line histogram) or bulk CD8+ T cells (filled histogram) day 5 after peptide stimulation in Treg-sufficient (Foxp3-WT) or mice ablated of Tregs on day before and on the day of peptide inoculation (Foxp3-DTR). D. Percent and number of CD90.1+ CD8+ cells among splenocytes at the indicated time points after peptide stimulation. These data are representative of three experiments containing 10–12 mice per group. Bar, one standard error.
We next compared the impacts of Treg-suppression on the activation of peptide simulated CD8+ T cells. The relative expression of T cell activation markers such as CD25 or CD62L, and production of IFN-γ were found to each parallel the more robust expansion of peptide-stimulated cells in Treg-ablated mice (Fig 1A, 1C). Compared with the few OVA-specific CD8+ T cells recovered from Treg-sufficient mice that were predominantly CD25lo, CD62Lhi, and produced little to no cytokine, the expanded population of these cells primed in Treg-ablated mice up-regulated CD25 and IFN-γ production, and down-regulated CD62L expression (Fig 1A, 1C). These findings indicate Foxp3+ Tregs suppress both the expansion and activation of peptide stimulated CD8+ T cells in vivo. In related experiments, we extended these studies to investigate the impact of transient Treg-ablation on the kinetics whereby CD8+ T cells expand and contract after peptide stimulation. Compared with the level of OVA-specific CD8+ T cells day 5 after peptide stimulation in Treg-ablated mice, reductions in both percent and total cell numbers became apparent beginning day 10 that steadily declined through day 30 (Fig 1D). As expected, only background levels of OVA-specific CD90.1+ CD8+ T cells were found in peptide stimulated Treg-sufficient control mice at each of these time points. Together, these results demonstrate Treg-ablation primes the activation, expansion, and subsequent contraction of peptide stimulated CD8+ T cells.
Peptide stimulated CD8+ T cells primed in the absence of Tregs are protective
To more definitively evaluate the functional properties of peptide stimulated CD8+ T cells primed in Treg-ablated mice, we compared their ability to confer protection against virulent Lm engineered to stably express a truncated form of OVA that contains the OVA257-264 peptide (Lm-OVA) (36). Significantly reduced numbers of bacterial CFUs were recovered from mice initially stimulated with OVA257-264 peptide in the absence of Tregs compared with Treg-sufficient controls day 3 after Lm-OVA infection (> 100-fold reductions in both the spleen and liver, P < 0.005) (Fig 2). Importantly, these protective effects occurred in an antigen-specific fashion because the differences in susceptibility were eliminated when an isogenic strain of virulent Lm (10403s) that does not express OVA was used for infection (Fig 2). Thus, Foxp3+ Tregs impede the priming of peptide stimulated CD8+ T cells that protect against Lm infection.
Figure 2.
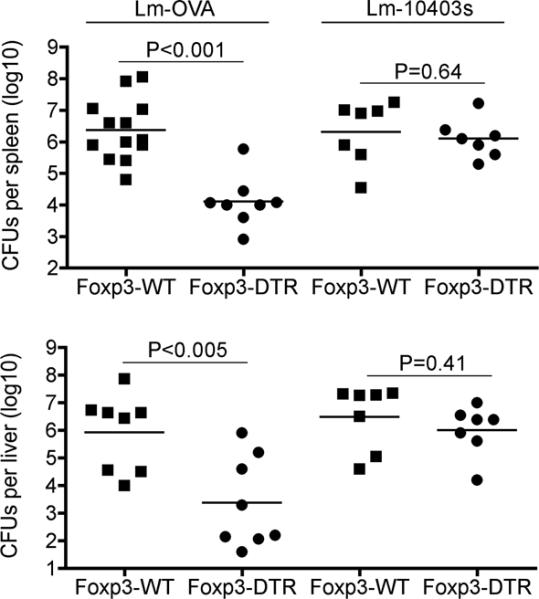
Peptide-stimulated CD8+ T cells primed in the absence of Tregs protect against Lm infection. Bacterial CFUs in the spleen (top) or liver (bottom) day 3 after infection with either Lm-OVA or Lm-10403s for the indicated mice stimulated with OVA257–264 peptide (and treated with DT one day prior and on the day of peptide inoculation) 30 days prior to infection. These data are representative of three experiments containing 8–12 mice per group.
In related experiments, we compared the secondary expansion kinetics of OVA-specific CD8+ T cells after Lm-OVA infection. Consistent with their ability to confer protection against Lm infection in an antigen-specific fashion, OVA-specific CD8+ T cells re-expanded rapidly in mice initially primed with peptide in the absence of Tregs (Fig 3A). Although differences in Lm-OVA pathogen burden and T cell precursor frequency at the time of infection preclude a direct comparison of the secondary OVA-specific CD8+ T cell response after Lm-OVA infection between Treg-ablated and Treg-sufficient control mice, the robust levels of IFN-γ, IL-2, and TNF-α produced by cells isolated from mice primed initially with OVA257-264 peptide in the absence of Foxp3+ Tregs illustrate the highly activated nature of these cells (Fig 3B). Reciprocally, reduced IFN-γ production by cells isolated from mice initially stimulated in the presence of Tregs is consistent with a functionally more tolerant phenotype previously described for CD8+ T cells stimulated in vivo with purified peptide (3, 38, 39).
Figure 3.
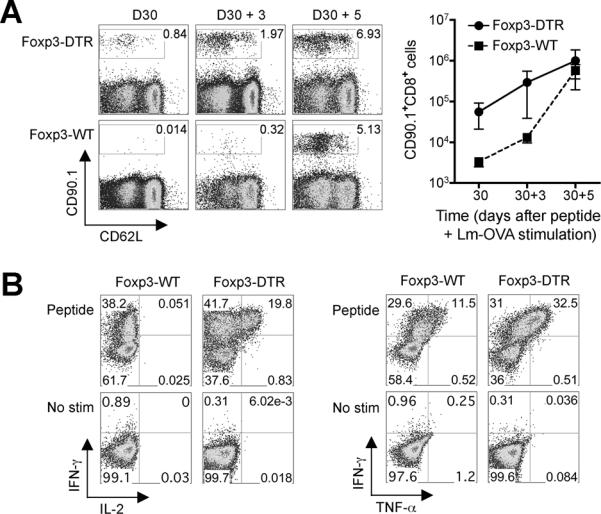
Secondary expansion and cytokine production for peptide-stimulated CD8+ T cells primed initially in Treg-ablated or Treg-sufficient mice. A. Percent and number of CD90.1+ CD8+ cells among splenocytes prior to (D30), or days 3 (D30 + 3) and 5 (D30 + 5) after Lm-OVA infection in mice treated with peptide 30 days prior. B. Representative FACS plots demonstrating percent IFN-γ, IL-2, and TNF-α producing CD90.1+ CD8+ T cells day 5 after secondary Lm-OVA infection (D30 + 5 after peptide stimulation) directly ex-vivo (no stim) or after peptide stimulation. These data are representative of three experiments containing 8–12 mice per group. Bar, one standard error.
Lm infection overrides the impacts of Treg suppression
The primary expansion kinetics and protective capacity of peptide stimulated CD8+ T cells primed in the absence of Tregs bear striking resemblance with the pathogen-specific T cell response primed by Lm infection (2, 6, 40). These parallels led us to investigate the impact of Treg-suppression on the OVA257-264-specific CD8+ T cell response after primary infection with recombinant Lm-OVA where OVA is expressed as a surrogate Lm-antigen (36). In sharp contrast to stimulation with purified peptide, Treg-ablation did not significantly influence the expansion (Fig 4A), or relative expression of CD25 or CD62L, or IFN-γ production by OVA-specific CD8+ T cells after Lm-OVA infection (Fig 4B). In turn, infection with non-recombinant Lm-10403s administered with OVA257-264 peptide also primed the robust expansion of OVA-specific CD8+ T cells that mirrored the effects of Foxp3+ Treg ablation (Fig 5A compared with Fig 1A). The expansion of these cells was antigen-specific and required stimulation with OVA-peptide because only background levels were found in non-recombinant Lm infected mice without peptide stimulation (Fig 5A). Furthermore, purified peptide with Lm-10403s infection also stimulated the activation (CD25 expression and IFN-γ production) of OVA-specific CD8+ T cells to levels comparable to that achieved with Treg-ablation (Fig 5B compared with Fig 1C). Together, these findings indicate Lm infection overrides suppression imposed by Foxp3+ Tregs, and primes the expansion and activation of peptide stimulated CD8+ T cells.
Figure 4.

Foxp3+ Treg-ablation does not impact the expansion and activation of antigen-specific CD8+ T cells primed by recombinant Lm-OVA infection. A. Percent and number of OVA-specific CD90.1+ CD8+ T cells among splenocytes day 5 after Lm-OVA infection in Foxp3-WT or Foxp3-DTR mice each treated with DT one-day prior and on the day of infection. B. Expression of CD25 and IFN-γ production by CD90.1+ (line histogram) or bulk CD8+ T cells (filled histogram) day 5 after Lm-OVA infection in Treg-ablated (Foxp3-DTR) or Treg-sufficient (Foxp3-WT) controls. These data are representative of three experiments containing 10–12 mice per group.
Figure 5.

Non-recombinant Lm-10403s primes the expansion and activation of peptide stimulated T cells. A. Percent and number of CD90.1+ CD8+ T cells among splenocytes day 5 after stimulation with OVA peptide alone (no infection) or peptide stimulation plus Lm infection (Lm-10403s). B. Expression of CD25 and IFN-γ production by CD90.1+ (line histogram) or bulk CD8+ T cells (filled histogram) recovered from mice day 5 after peptide stimulation alone (no infection) or peptide stimulation plus Lm infection (Lm-10403s). C. Percent OVA-specific CD90.1+ among CD8+ T cells day 5 after stimulation with OVA peptide plus Lm-10403s in mice treated with anti-IL-12 neutralizing or isotype (rat IgG2a) antibodies (top). Percent IL-12 receptor deficient OT-1 or WT OT-1 (each CD45.2+) among CD8+ T cells after adoptive transfer into CD45.1+ mice, and day 5 after stimulation with OVA peptide and Lm-10403s (bottom). Percent IFN-γ production for each group of OT-1 cells (line histogram) or bulk CD8+ T cells (filled histogram) after peptide stimulation. These data are representative of three experiments containing 8–12 mice per group.
Since inflammatory cytokines such as IL-12 produced in response to Lm infection can directly stimulate the activation of protective CD8+ T cells (3, 41), related experiments explored if these adjuvant effects of Lm on the expansion and activation of peptide stimulated CD8+ T cells required IL-12. We found administration of neutralizing antibody against IL-12 p40 compared with isotype control antibody one day prior to OVA-peptide stimulation and Lm infection had no significant impact on the expansion or activation of OVA-specific CD8+ T cells (Fig 5C). To more specifically investigate the direct impacts of IL-12 stimulation on CD8+ T cells, and address the possibility that in vivo neutralization with anti-IL-12 antibody was incomplete or non-specific because IL-23 also shares the same IL-12 p40 subunit, complementary experiments compared the expansion and activation of CD8+ T cells from IL-12-receptor deficient OT-1 and IL-12 receptor sufficient (WT) OT-1 TCR transgenic mice each on the CD45.2 background after adoptive transfer into CD45.1 recipients. Similar to the results after anti-IL-12 neutralization, no significant differences in expansion or IFN-γ production were found for IL-12-receptor-deficient compared with WT OT-1 cells after stimulation with OVA peptide and Lm infection (Fig 5C). Therefore, while IL-12 can stimulate T cell activation and is produced in response to Lm infection (3, 32, 42), the adjuvant effects of Lm on the expansion and activation of peptide stimulated CD8+ T cells do not require IL-12 stimulation.
Reduced Treg suppressive potency after Lm infection
Given the potential for pathogen-associated ligands to control Treg suppression through cell-intrinsic TLR stimulation or indirectly by activating antigen-presenting cells (20–24, 43–45), we investigated if overriding the effects of Treg suppression after Lm infection may reflect infection-induced shifts in Foxp3+ cell potency. To test this hypothesis, we compared the suppressive potency of Tregs isolated before and after Lm infection in Foxp3GFP mice where Foxp3+ cells can be purified by FACS based on GFP expression (26). We found infection did not interfere with the isolation of Tregs because GFP+ Foxp3+ cells were purified to the same extent (98–99% purity) before and at each time point after infection (Fig 6A). To identify potential shifts in suppression, the relative potency whereby purified GFP+ Tregs inhibit the proliferation of CFSE labeled responder cells in co-culture after stimulation with anti-CD3 antibody was enumerated. We found GFP+ Tregs isolated from mice within the first three days after Lm infection compared with Tregs from naïve mice were less potent at suppressing the proliferation of responder cells (Fig 6B, 6C). In turn, CFSE dilution among responder cells progressively increased after co-culture with GFP+ Tregs from mice within the first three days after Lm infection compared with cells from naïve mice (Fig 6B, 6C). By titrating the ratio of Treg to responder cells in co-culture that allows a semi-quantitative assessment for these shifts in suppressive potency, we find ~ 4-fold reductions in suppressive potency for Tregs isolated from mice day 3 after Lm infection compared with cells from naïve mice because two 2-fold dilutions of Tregs from naïve mice achieved the same level of suppression as Tregs from Lm infected mice (Fig 6B, 6C). Thus, while TLR-stimulation through pathogen-associated ligands and inflammatory cytokines can trigger either increased or diminished Treg suppression potency after stimulation in vitro (20–24, 43–45), the cumulative effects of in vivo infection with intact Lm and the ensuing immune response cause reductions in Foxp3+Treg suppressive potency.
Figure 6.

Reduced Treg suppressive potency after Lm infection. A. Purification of Foxp3+ Tregs as GFP+ CD4+ cells in Foxp3GFP reporter mice after Lm infection. Percent GFP or Foxp3 expression among CD4+ splenocytes at each time point after infection before sorting (Pre-sort) and among all cells after sorting (Post-sort) for GFP+ CD4+ cells. B. CFSE dilution among CD8+ CD45.1+ responder cells after co-culture with GFP+ Tregs at the indicated ratio and stimulation with anti-CD3 antibody (line histogram), no stimulation controls (dark filled histogram), or stimulation without Tregs (light filled histogram). C. Percent CD8+ CD45.1+ responder cells in each CFSE generation after co-culture with GFP+ Tregs from mice at the indicated time point after infection at a 1:8 ratio. These data are representative of three experiments containing Tregs from 6–8 mice per infection time point. Bar, one standard error.
Antigen-pulsed dendritic cells do not override the impacts of Treg-suppression
The potential mechanisms whereby Foxp3+ Tregs mediate immune suppression are broadly divided into those that directly suppress effector T cell activation or others that suppress the activation of dendritic and other antigen-presenting cells (46, 47). However, dissociating the relative importance of each is complicated by the activation of both effector T cells and antigen-presenting cells after Foxp3+ cell ablation in vivo (19). To investigate the potential importance whereby Treg suppression of dendritic cells controls T cell priming, we enumerated the impacts of Foxp3 cell-ablation on the expansion and activation of OVA-specific CD8+ T cells after stimulation with peptide pulsed JAWS II dendritic cells (34, 35). These cells are a representative model for activated dendritic cell because JAWS II cells maintained in vitro constitutively up-regulate the expression of molecules required for CD8+ T cell stimulation such as H-2Kb (MHC I) and CD80 (costimulation) to a similar or even greater extent compared with CD11c+ cells isolated ex vivo from Treg-ablated or Lm infected mice (Fig 7A). Comparatively, each of these activation parameters on JAWS II cells or CD11c+ cells isolated directly ex vivo after Lm infection or Treg ablation are significantly elevated compared with CD11c+ cells from Treg-sufficient uninfected control mice (Fig 7A). Interestingly, the in vivo impacts of Treg-suppression were sustained even when OVA257–264 peptide pulsed JAWS II dendritic cells were used for stimulation because significantly more OVA-specific CD8+ T cells were recovered from Treg-ablated compared with Treg-sufficient controls (Fig 7B). In turn, expanded OVA-specific CD8+ T cells stimulated with peptide pulsed dendritic cells in Treg-ablated mice also up-regulated CD25 expression and were primed to produce significantly more IFN-γ compared with the few cells recovered from Treg-sufficient mice or after stimulation with control unpulsed JAWS II cells (Fig 7C). These findings demonstrate that although Foxp3 cell-ablation and Lm-infection each activate CD11c+ cells (19, 48, 49), stimulation with activated antigen-pulsed dendritic cells does not override the in vivo impacts of Treg suppression on the expansion and activation of antigen-specific CD8+ T cells. Taken together, these results indicate Foxp3+ Treg suppression of dendritic cells, compared with effector T cells, play less significant roles in impeding antigen-specific CD8+ T cell priming.
Figure 7.

Antigen-pulsed dendritic cells do not override the impacts of Treg-suppression. A. Relative expression of MHC-I (H-2Kb) or CD80 on CD11c+ cells isolated directly ex vivo from naïve Treg sufficient mice (gray filled histogram and symbols), or mice day 5 after Lm infection (red line histogram and symbols) or ablation of Foxp3+ Tregs (blue line histogram and symbols) compared with JAWS II cells maintained in vitro (black line histogram and symbols). B. Percent and number of OVA-specific CD90.1+ CD8+ T cells among splenocytes day 5 after stimulation with peptide pulsed or unpulsed JAWS II dendritic cells in the presence (Foxp3-WT) or absence of Tregs (Foxp3-DTR). Expression of CD25 and IFN-γ production by CD90.1+ (line histogram) or bulk CD8+ T cells (filled histogram) day 5 after peptide stimulation in Treg-sufficient (Foxp3-WT) or mice ablated of Tregs one day before and on the day of peptide inoculation (Foxp3-DTR). These data are representative of three experiments containing 8–12 mice per group. Bar, one standard error.
DISCUSSION
Although the Foxp3+ subset of regulatory CD4+ T cells were initially identified based on their essential role in maintaining peripheral tolerance and suppressing potentially detrimental self-reactive immune responses (15–17), accumulating evidence indicate the importance of Foxp3+ Tregs readily extends to controlling the immune response against non-self pathogen-associated antigens. In this regard, while the relative impacts of Treg ablation on infection susceptibility have been characterized in numerous models of experimental infection or immunization (12, 13, 50–54), the specific limitations imposed by Foxp3-expressing cells on the priming and expansion of antigen-specific T cells after stimulation with purified antigens have not been clearly identified. Herein, we enumerated the impacts of Treg ablation on the expansion and activation of CD8+ T cells with specificity for a defined non-self antigen after stimulation with cognate peptide. We find transient ablation of Foxp3+ Tregs during stimulation with purified peptide without adjuvant was sufficient to prime the robust expansion and activation of antigen-specific CD8+ T cells (Fig 1). Furthermore, CD8+ T cells stimulated with peptide in the absence of Tregs contract from peak expansion levels, are sustained through day 30 after stimulation, and provide protection in an antigen-specific fashion against Lm infection (Fig 2). Interestingly, while stimulation with OVA257–264 peptide in Treg-ablated mice confers protection against subsequent Lm-OVA infection, the similar number of bacterial CFUs after Lm-OVA compared with Lm-10403s infection in peptide stimulated Treg-sufficient mice suggest that the modest expansion of OVA-specific CD8+ T cells primed by peptide in the presence of Tregs does not confer significant protective effects. These results are consistent with the functionally tolerant phenotype of CD8+ T cells stimulated in vivo with purified peptide without adjuvant (3, 38). Similarly, the delayed re-expansion of OVA-specific T cells after Lm-OVA infection in these mice is also consistent with the previously reported reductions in CD8+ T cell expansion after secondary stimulation with purified peptide plus LPS (3). However, the ability of these cells to re-expand at all indicates that stimulation with purified peptide under these experimental conditions were insufficient to prime a completely anergic response (55). Although our studies were not designed to address the degree of tolerance in Treg-sufficient mice that most likely reflect differences in the amount of peptide antigen used for stimulation (39), the more robust expansion and activation of CD8+ T cells in Foxp3+ cell-ablated mice clearly demonstrate Tregs actively suppress and impose critical barriers for priming protective antigen-specific T cells under these non-inflammatory, non-infection stimulation conditions.
The impacts triggered by transient ablation of Foxp3-expressing cells on the activation of peptide stimulated T cells we demonstrate are in agreement with the enhanced vaccine-induced immunogenicity associated with anti-CD25 antibody co-administration (54), and reductions in Treg suppression triggered by many immune adjuvants and purified pathogen-associated ligands that stimulate immune cells through Toll-like and other pattern recognition receptors (20–25). In this regard, while immune adjuvants have been characterized primarily for their ability to stimulate pro-inflammatory cytokines and/or up-regulate costimulation signals on antigen-presenting cells (7–11), our finding that transient Treg-ablation during stimulation with purified peptide alone is sufficient to prime the expansion and activation of protective CD8+ T cells illustrates the critical barriers imposed Foxp3+ cells. Therefore, in addition to direct stimulation of antigen presenting cells, overriding immune suppression dictated by Tregs represent important considerations in designing adjuvants for stimulating T cells in vivo.
In complementary experiments designed to enumerate the relative importance of Treg-suppression in priming antigen-specific CD8+ T cells under infection conditions, the relative impacts of Treg ablation on the expansion and activation of CD8+ T cells primed by Lm infection were enumerated. In sharp contrast to stimulation with purified peptide where Treg ablation results in greater than 100-fold increased expansion of OVA-specific CD8+ T cells, Treg-ablation caused no significant effects on the expansion of these same cells after infection with recombinant Lm that stably expresses a truncated form of OVA (Fig 4) (36). Thus, the priming of antigen-specific T cells after Lm infection, unlike peptide stimulation, are not subject to active suppression by Foxp3+ Tregs. Related experiments tracking antigen-specific CD8+ T cells after stimulation with purified peptide and infection with non-recombinant Lm revealed that these T cell stimulatory effects are not limited only for cells with specificity to Lm-expressed antigen, but also extend to those stimulated with peptide antigen during acute Lm infection (Fig 5). Thus, Lm infection overrides suppression imposed by Foxp3+ Tregs that restricts the expansion and activation of T cells after stimulation under non-inflammatory conditions.
Given the ability of Lm infection to stimulate IL-12 production, and the potency whereby this cytokine stimulates the activation of protective T cells (3, 32, 41, 42), the contribution of IL-12 on the adjuvant effects of Lm infection were also evaluated. Complementary approaches using either neutralizing antibody or adoptively transferred T cells with targeted defects in the IL-12 receptor each revealed non-essential roles for IL-12 in stimulating CD8+ T cell activation after Lm infection. Together with the recent observation that pathogen-specific CD8+ T cell responses are un-ablated even in mice with combined defects in IL-12, IL-21, and type I IFN-receptor representing all known third signals for CD8+ T cells (1, 6), these results suggest Lm stimulates other immune pathways that prime protective CD8+ T cells. Based on the Lm induced reductions in Treg-suppressive potency (Fig 6), and the efficiency whereby Lm overrides the impacts of Treg-suppression on the priming of peptide stimulated CD8+ T cells (Fig 5), we propose blunting the impacts of Foxp3+ Treg suppression play dominant roles in priming protective T cells after this in vivo infection.
The reduction in Treg potency after Lm infection that we demonstrate is consistent with the efficiency whereby cell-intrinsic stimulation through TLR2 and TLR5 that respond to Lm-associated antigens shift the suppressive potency of CD25+CD4+ Tregs (23, 24, 43, 45, 56). However, while these studies revealed divergent effects on Treg suppressive potency after TLR stimulation using purified ligands, we find the cumulative impacts of infection and the ensuing inflammatory response results in approximate four-fold reductions in suppressive potency for Foxp3+ Tregs on a per cell basis. These results using a model of acute bacterial infection are also consistent with reductions in suppressive potency for Foxp3+ or CD25+ CD4+ T cells at various stages after infection with other pathogens that primarily cause persistent infection (53, 57–59). Therefore, while Treg suppression plays important roles in shaping the immune response that controls the severity of infection (12, 13), these findings also illustrate the potential whereby infection triggers changes in Treg suppression that controls effector T cell activation. In the case of acute Lm infection, reduced Treg suppressive potency likely permits the expansion of pathogen-specific T cells that eradicates infection. Moving forward, identifying the molecular basis whereby Tregs suppress T cell-activation under non-infection conditions and the specific signals triggered by infection that overrides Treg suppression are important areas for additional investigation required for designing therapies aimed at manipulating the fluid and shifting balance between immune stimulation and suppression controlled by Tregs.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Alexander Rudensky for providing Foxp3DTR and Foxp3GFP mice, Paul Champoux and Genya Gekker for assistance with flow cytometry and cell sorting, and Dr. Stephen McSorley and other faculty in the Center for Immunology for helpful discussion.
This work was supported by NIH grants F30DK084674 (JHR) and R01AI087830 (SSW).
REFERENCES
- 1.Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Current opinion in immunology. 2010;22:333–340. doi: 10.1016/j.coi.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haring JS, Badovinac VP, Harty JT. Inflaming the CD8+ T cell response. Immunity. 2006;25:19–29. doi: 10.1016/j.immuni.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. The Journal of experimental medicine. 2003;197:1141–1151. doi: 10.1084/jem.20021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casey KA, Mescher MF. IL-21 promotes differentiation of naive CD8 T cells to a unique effector phenotype. J Immunol. 2007;178:7640–7648. doi: 10.4049/jimmunol.178.12.7640. [DOI] [PubMed] [Google Scholar]
- 5.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. 2005;174:4465–4469. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 6.Ertelt JM, Johanns TM, Rowe JH, Way SS. Interleukin (IL)-21-independent pathogen-specific CD8(+) T-cell expansion, and IL-21-dependent suppression of CD4(+) T-cell IL-17 production. Immunology. 2010;131:183–191. doi: 10.1111/j.1365-2567.2010.03287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nature immunology. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 8.Pasare C, Medzhitov R. Toll-like receptors and acquired immunity. Seminars in immunology. 2004;16:23–26. doi: 10.1016/j.smim.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 9.van Duin D, Medzhitov R, Shaw AC. Triggering TLR signaling in vaccination. Trends in immunology. 2006;27:49–55. doi: 10.1016/j.it.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science (New York, N.Y. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature immunology. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 12.Belkaid Y, Rouse BT. Natural regulatory T cells in infectious disease. Nature immunology. 2005;6:353–360. doi: 10.1038/ni1181. [DOI] [PubMed] [Google Scholar]
- 13.Belkaid Y. Regulatory T cells and infection: a dangerous necessity. Nature reviews. 2007;7:875–888. doi: 10.1038/nri2189. [DOI] [PubMed] [Google Scholar]
- 14.Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nature immunology. 2010;11:7–13. doi: 10.1038/ni.1818. [DOI] [PubMed] [Google Scholar]
- 15.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nature genetics. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 16.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nature immunology. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 17.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nature immunology. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 18.Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, Wagner H, Huehn J, Sparwasser T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. The Journal of experimental medicine. 2007;204:57–63. doi: 10.1084/jem.20061852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nature immunology. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 20.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science (New York, N.Y. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 21.Kubo T, Hatton RD, Oliver J, Liu X, Elson CO, Weaver CT. Regulatory T cell suppression and anergy are differentially regulated by proinflammatory cytokines produced by TLR-activated dendritic cells. J Immunol. 2004;173:7249–7258. doi: 10.4049/jimmunol.173.12.7249. [DOI] [PubMed] [Google Scholar]
- 22.Peng G, Guo Z, Kiniwa Y, Voo KS, Peng W, Fu T, Wang DY, Li Y, Wang HY, Wang RF. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science (New York, N.Y. 2005;309:1380–1384. doi: 10.1126/science.1113401. [DOI] [PubMed] [Google Scholar]
- 23.Sutmuller RP, den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ, Joosten LA, Akira S, Netea MG, Adema GJ. Toll-like receptor 2 controls expansion and function of regulatory T cells. The Journal of clinical investigation. 2006;116:485–494. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:7048–7053. doi: 10.1073/pnas.0601554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jarnicki AG, Conroy H, Brereton C, Donnelly G, Toomey D, Walsh K, Sweeney C, Leavy O, Fletcher J, Lavelle EC, Dunne P, Mills KH. Attenuating regulatory T cell induction by TLR agonists through inhibition of p38 MAPK signaling in dendritic cells enhances their efficacy as vaccine adjuvants and cancer immunotherapeutics. J Immunol. 2008;180:3797–3806. doi: 10.4049/jimmunol.180.6.3797. [DOI] [PubMed] [Google Scholar]
- 26.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 27.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 28.Xiao Z, Casey KA, Jameson SC, Curtsinger JM, Mescher MF. Programming for CD8 T cell memory development requires IL-12 or type I IFN. J Immunol. 2009;182:2786–2794. doi: 10.4049/jimmunol.0803484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buch T, Heppner FL, Tertilt C, Heinen TJ, Kremer M, Wunderlich FT, Jung S, Waisman A. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nature methods. 2005;2:419–426. doi: 10.1038/nmeth762. [DOI] [PubMed] [Google Scholar]
- 30.Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic acids research. 1997;25:1317–1318. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cousens LP, Peterson R, Hsu S, Dorner A, Altman JD, Ahmed R, Biron CA. Two roads diverged: interferon alpha/beta- and interleukin 12-mediated pathways in promoting T cell interferon gamma responses during viral infection. The Journal of experimental medicine. 1999;189:1315–1328. doi: 10.1084/jem.189.8.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Way SS, Havenar-Daughton C, Kolumam GA, Orgun NN, Murali-Krishna K. IL-12 and type-I IFN synergize for IFN-gamma production by CD4 T cells, whereas neither are required for IFN-gamma production by CD8 T cells after Listeria monocytogenes infection. J Immunol. 2007;178:4498–4505. doi: 10.4049/jimmunol.178.7.4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rowe JH, Johanns TM, Ertelt JM, Way SS. PDL-1 blockade impedes T cell expansion and protective immunity primed by attenuated Listeria monocytogenes. J Immunol. 2008;180:7553–7557. doi: 10.4049/jimmunol.180.11.7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang X, Shen C, Rey-Ladino J, Yu H, Brunham RC. Characterization of murine dendritic cell line JAWS II and primary bone marrow-derived dendritic cells in Chlamydia muridarum antigen presentation and induction of protective immunity. Infection and immunity. 2008;76:2392–2401. doi: 10.1128/IAI.01584-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Otsu S, Gotoh K, Yamashiro T, Yamagata J, Shin K, Fujioka T, Nishizono A. Transfer of antigen-pulsed dendritic cells induces specific T-Cell proliferation and a therapeutic effect against long-term Helicobacter pylori infection in mice. Infection and immunity. 2006;74:984–993. doi: 10.1128/IAI.74.2.984-993.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foulds KE, Zenewicz LA, Shedlock DJ, Jiang J, Troy AE, Shen H. Cutting edge: CD4 and CD8 T cells are intrinsically different in their proliferative responses. J Immunol. 2002;168:1528–1532. doi: 10.4049/jimmunol.168.4.1528. [DOI] [PubMed] [Google Scholar]
- 37.Portnoy DA, Jacks PS, Hinrichs DJ. Role of hemolysin for the intracellular growth of Listeria monocytogenes. The Journal of experimental medicine. 1988;167:1459–1471. doi: 10.1084/jem.167.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dubois PM, Pihlgren M, Tomkowiak M, Van Mechelen M, Marvel J. Tolerant CD8 T cells induced by multiple injections of peptide antigen show impaired TCR signaling and altered proliferative responses in vitro and in vivo. J Immunol. 1998;161:5260–5267. [PubMed] [Google Scholar]
- 39.Aichele P, Brduscha-Riem K, Zinkernagel RM, Hengartner H, Pircher H. T cell priming versus T cell tolerance induced by synthetic peptides. The Journal of experimental medicine. 1995;182:261–266. doi: 10.1084/jem.182.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pamer EG. Immune responses to Listeria monocytogenes. Nature reviews. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 41.Miller MA, Skeen MJ, Ziegler HK. A synthetic peptide administered with IL-12 elicits immunity to Listeria monocytogenes. J Immunol. 1997;159:3675–3679. [PubMed] [Google Scholar]
- 42.Seki E, Tsutsui H, Tsuji NM, Hayashi N, Adachi K, Nakano H, Futatsugi-Yumikura S, Takeuchi O, Hoshino K, Akira S, Fujimoto J, Nakanishi K. Critical roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. J Immunol. 2002;169:3863–3868. doi: 10.4049/jimmunol.169.7.3863. [DOI] [PubMed] [Google Scholar]
- 43.Zanin-Zhorov A, Cahalon L, Tal G, Margalit R, Lider O, Cohen IR. Heat shock protein 60 enhances CD4+ CD25+ regulatory T cell function via innate TLR2 signaling. The Journal of clinical investigation. 2006;116:2022–2032. doi: 10.1172/JCI28423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. The Journal of experimental medicine. 2003;197:403–411. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crellin NK, Garcia RV, Hadisfar O, Allan SE, Steiner TS, Levings MK. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J Immunol. 2005;175:8051–8059. doi: 10.4049/jimmunol.175.12.8051. [DOI] [PubMed] [Google Scholar]
- 46.Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 47.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nature reviews. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muraille E, Giannino R, Guirnalda P, Leiner I, Jung S, Pamer EG, Lauvau G. Distinct in vivo dendritic cell activation by live versus killed Listeria monocytogenes. European journal of immunology. 2005;35:1463–1471. doi: 10.1002/eji.200526024. [DOI] [PubMed] [Google Scholar]
- 49.Shedlock DJ, Whitmire JK, Tan J, MacDonald AS, Ahmed R, Shen H. Role of CD4 T cell help and costimulation in CD8 T cell responses during Listeria monocytogenes infection. J Immunol. 2003;170:2053–2063. doi: 10.4049/jimmunol.170.4.2053. [DOI] [PubMed] [Google Scholar]
- 50.Scott-Browne JP, Shafiani S, Tucker-Heard G, Ishida-Tsubota K, Fontenot JD, Rudensky AY, Bevan MJ, Urdahl KB. Expansion and function of Foxp3-expressing T regulatory cells during tuberculosis. The Journal of experimental medicine. 2007;204:2159–2169. doi: 10.1084/jem.20062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lund JM, Hsing L, Pham TT, Rudensky AY. Coordination of early protective immunity to viral infection by regulatory T cells. Science (New York, N.Y. 2008;320:1220–1224. doi: 10.1126/science.1155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steeg C, Adler G, Sparwasser T, Fleischer B, Jacobs T. Limited role of CD4+Foxp3+ regulatory T cells in the control of experimental cerebral malaria. J Immunol. 2009;183:7014–7022. doi: 10.4049/jimmunol.0901422. [DOI] [PubMed] [Google Scholar]
- 53.Johanns TM, Ertelt JM, Rowe JH, Way SS. Regulatory T cell suppressive potency dictates the balance between bacterial proliferation and clearance during persistent Salmonella infection. PLoS pathogens. 2010;6 doi: 10.1371/journal.ppat.1001043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moore AC, Gallimore A, Draper SJ, Watkins KR, Gilbert SC, Hill AV. Anti-CD25 antibody enhancement of vaccine-induced immunogenicity: increased durable cellular immunity with reduced immunodominance. J Immunol. 2005;175:7264–7273. doi: 10.4049/jimmunol.175.11.7264. [DOI] [PubMed] [Google Scholar]
- 55.Hammerbeck CD, Mescher MF. Antigen controls IL-7R alpha expression levels on CD8 T cells during full activation or tolerance induction. J Immunol. 2008;180:2107–2116. doi: 10.4049/jimmunol.180.4.2107. [DOI] [PubMed] [Google Scholar]
- 56.Way SS, Thompson LJ, Lopes JE, Hajjar AM, Kollmann TR, Freitag NE, Wilson CB. Characterization of flagellin expression and its role in Listeria monocytogenes infection and immunity. Cellular microbiology. 2004;6:235–242. doi: 10.1046/j.1462-5822.2004.00360.x. [DOI] [PubMed] [Google Scholar]
- 57.Suvas S, Kumaraguru U, Pack CD, Lee S, Rouse BT. CD4+CD25+ T cells regulate virus-specific primary and memory CD8+ T cell responses. The Journal of experimental medicine. 2003;198:889–901. doi: 10.1084/jem.20030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hisaeda H, Tetsutani K, Imai T, Moriya C, Tu L, Hamano S, Duan X, Chou B, Ishida H, Aramaki A, Shen J, Ishii KJ, Coban C, Akira S, Takeda K, Yasutomo K, Torii M, Himeno K. Malaria parasites require TLR9 signaling for immune evasion by activating regulatory T cells. J Immunol. 2008;180:2496–2503. doi: 10.4049/jimmunol.180.4.2496. [DOI] [PubMed] [Google Scholar]
- 59.Rausch S, Huehn J, Kirchhoff D, Rzepecka J, Schnoeller C, Pillai S, Loddenkemper C, Scheffold A, Hamann A, Lucius R, Hartmann S. Functional analysis of effector and regulatory T cells in a parasitic nematode infection. Infection and immunity. 2008;76:1908–1919. doi: 10.1128/IAI.01233-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.