

Abstract
The voltage-gated potassium channel, Kv1.3, plays an important role in regulating membrane excitability in diverse cell types ranging from T-lymphocytes to neurons. In the present study, we test the hypothesis that the C-terminal PDZ binding domain modulates the function and localization of Kv1.3. We created a mutant form of Kv1.3 that lacked the last three amino acids of the C-terminal PDZ-binding domain (Kv1.3ΔTDV). This form of Kv1.3 did not bind the PDZ domain containing protein, PSD95. We transfected wild type and mutant Kv1.3 into HEK293 cells and determined if the mutation affected current, Golgi localization, and surface expression of the channel. We found that cells transfected with Kv1.3ΔTDV had greater current and lower Golgi localization than those transfected with Kv1.3. Truncation of the C-terminal PDZ domain did not affect surface expression of Kv1.3. These findings suggest that PDZ-dependent interactions affect both Kv1.3 localization and function. The finding that current and Golgi localization changed without a corresponding change in surface expression suggests that PDZ interactions affect localization and function via independent mechanisms.
Keywords: Kv1.3, voltage-gated potassium channel, Golgi apparatus, trafficking, PDZ
Introduction
The Kv1.3 voltage-gated potassium channel is a key determinant of function in many cell types. Kv1.3 plays a central role in immune function by regulating resting membrane potential [1], Ca2+ signaling cascades [2], and antigen-dependent activation of T-lymphocytes [3]. Kv1.3 also plays a role in energy homeostasis. Kv1.3-deficient mice have altered body weight [4, 5] and Kv1.3 influences glucose transport in adipocytes and skeletal muscle [6, 7]. In neurons, Kv1.3 influences resting membrane potential, action potential characteristics, and neurotransmission [4, 8–10].
PDZ (postsynaptic density-95/discs-large/zona-occludens-1)-dependent interactions have emerged as important mechanisms of ion channel regulation. PDZ domain containing proteins interact primarily with the C-termini of channels to modulate their intracellular trafficking, localization and function [11–17]. For example, C-terminal PDZ-dependent interactions modulate endocytic recycling [16] and trafficking from the Golgi apparatus to the plasma membrane of the cystic fibrosis transmembrane conductance regulator (CFTR) [11]. PDZ-dependent interactions at the C-terminus also modulate Kv1 channels [12, 13, 17]. Kv1.3 contains a C-terminal PDZ binding domain, but the functional significance of PDZ domain-containing protein interactions with this channel are not well characterized.
In the present study we test the hypothesis that the C-terminal PDZ binding domain modulates the function and localization of Kv1.3. We demonstrate that truncation of the C-terminal PDZ domain has two effects on Kv1.3 channels expressed in HEK 293 cells. It increased Kv1.3 current and decreased the amount of Kv1.3 localized in the Golgi apparatus. The truncation had no effect on surface expression. These findings suggest that PDZ-dependent interactions affect both Kv1.3 localization and function. The finding that current and Golgi localization were changed without a corresponding change in surface expression suggests that PDZ interactions affect localization and function via independent mechanisms.
Materials and Methods
Animals
The use of animals in the present studies was in accordance with the National Institutes of Health guidelines for the humane care and use of animals in research, and was approved by the Institutional Animal Care and Use Committee of the University of Vermont.
Cell Culture and Transient Transfection
Postganglionic sympathetic neurons were obtained from superior cervical ganglia of neonatal rats. The ganglia (SCG) were dissociated with collagenase/hyaluronidase digest followed by a trypsin digest. Cells were plated onto type I rat tail collagen coated dishes (RT-PCR and western analyses) or coverslips (immunohistochemistry). The day after plating, non-neuronal cells were growth arrested with mitomycin C (1 hour, 10 μg/ml). All neuronal cultures were grown in neuronal growth medium (DMEM/F12 supplemented with 10% NuSerum, 5% FBS, 100 U/ml penicillin/streptomycin, and 50 ng/ml NGF). Vascular smooth muscle cells (VSM) were isolated from explants of adult postpartum Sprague Dawley rat tail arteries. VSM were grown in DMEM supplemented with 10% FBS, and 100 μ/ml penicillin/streptomycin. VSM were used at passage 2. Clonal human embryonic kidney 293 (HEK293) cells were maintained in DMEM/F-12 (Invitrogen) supplemented with 10% fetal bovine serum, 10 U/ml penicillin and streptomycin, and 2 mM L-glutamine. Cells were transiently transfected with 3.0 μg of GFP-Kv1.3/-Flag or GFP-Kv1.3-ΔTDV-Flag, using the Lipofectamine LTX transfection reagent (Invitrogen). Confluent transfected cultures were plated to a low density (25,000 cells/cm2) onto poly-D-lysine-coated tissue culture plates (Corning Glass Works) and subsequently placed in serum-free medium overnight for experimentation the following day.
Mutagenesis
All mutagenesis reactions were performed using the Quickchange Lightning Site-Directed Mutagenesis Kit (Strategene), following the manufacturer’s protocol. The GFP-Kv1.3-ΔTDV truncation was created by inserting two premature stop codons into the wild-type channel using the following primer sequence and its reverse complement: 5′-GTCAACATCATCAAAAAGATATTCTGATGAACTGATGTCTAATAGGGATCCACC-3′. Similar to previous reports [18, 19], the Flag epitope was created by inserting the amino acid sequence, DYKDDDDK, into the first extracellular loop of GFP-Kv1.3/-ΔTDV and incorporating D222 as the first aspartic acid residue in the Flag epitope using the following primer sequence and its reverse complement: 5′-TCTCCGTCGCAGGACTACAAGGACGACGACGACAAGGTGTTTGAGGCTGCC-3′.
Immunofluorescence
Cultures of postganglionic sympathetic neurons and vascular smooth muscle cells were each rinsed in PBS and fixed in 4% formaldehyde in PBS for 12 min. Cultures were permeabilized with 0.2% Triton X-100 in PBS for 5 min and subsequently rinsed in PBS. Cells were blocked for 1 hour at room temperature in 3% goat serum, 0.1% fish skin gelatin in PBS. Cells were co-stained with anti-Kv1.3 mouse monoclonal antibody (0.84 μg/mL; NeuroMab) and anti-GM130 rabbit polyclonal antibody (0.7 μg/mL; Calbiochem) overnight at 4°C, followed by three 5 min washes in PBS. Alexa Fluor Goat anti-mouse (488 nm) and goat anti-rabbit (647) secondary antibodies (4 μg/ml; Invitrogen) were applied for 1 hour at room temperature.
To detect total Kv1.3, transiently transfected HEK293 cells were rinsed in PBS, fixed in 4% formaldehyde in PBS for 20 min, and subsequently permeabilized with acetone for 5 min and rinsed in PBS. Cells were then blocked for 20 min at 37°C in 3% goat serum, 0.1% fish skin gelatin in PBS. Cells were incubated 20 min at 37°C in a rabbit polyclonal GM130 antibody, raised against recombinant protein containing amino acids 371–990 of human GM130 (Lot # D00004465), to identify the Golgi apparatus and subsequently rinsed 3 × 5 min in PBS. Alexa Fluor goat anti-rabbit (647 nm) secondary antibody (4.0 μg/ml; Invitrogen) was applied for 20 min at 37°C.
To detect Kv1.3 on the cell surface, transiently transfected HEK293 cells were live labeled with anti-Flag M2 antibody (1μg/mL; Sigma) at 37°C for 15 min. Cells were then rinsed 3 × 5 min in PBS, and fixed, permeabilized and labeled as described above. Alexa Fluor goat anti-mouse (647 nm) and goat anti-rabbit (568 nm) secondary antibodies (4.0 μg/ml; Invitrogen) were used to detect Kv1.3 surface expression and GM130 localization, respectively. All cells were mounted using ProLong Gold antifade reagent (Invitrogen). All images were taken using the Olympus IX70 microscope and DeltaVision Restoration Imaging System (Applied Precision, LLC) and background subtracted with the appropriate IgG isotype controls (R&D Systems).
Golgi Localization was quantified as follows: Golgi region of interest (ROI) was defined using the GM130 marker and the amount of GFP-Kv1.3-Flag signal intensity present in the Golgi ROI was expressed as a percent of the total Kv1.3 signal for the entire cell (Kv1.3 in Golgi region/total Kv1.3) × 100). Surface expression was quantified as follows: the intensity of the live labeled anti-Flag M2 antibody was normalized to the total GFP-Kv1.3-Flag intensity of the cell (surface Kv1.3/total Kv1.3).
Immunoblot Analysis
Cells were washed with ice-cold phosphate-buffered saline and lysed in radioimmune precipitation assay buffer (50 mM Tris, 150 mM NaCl, 11 mM EDTA, 0.25% deoxycholate, 1% Nonidet P-40, 10% glycerol, 1 mM NaF, 1 mM Na3VO4, 1 mM Na4BAPTA, 1mM dithiothreitol, protease inhibitors (Sigma, catalog no. P8340), phosphatase inhibitors (Calbiochem, catalog nos. 524624 and 524625), pH 8.0). Lysates were centrifuged at 20,000 × g for 5 min, and resulting supernatants were combined with sample buffer and separated by SDS-PAGE. Western blotting detection of Kv1.3 was done with anti-Kv1.3 monoclonal antibody (0.42 μg/mL; NeuroMab). A monoclonal anti-GAPDH antibody (1.0 μg/mL; Millipore) was used to control for loading efficiency. Blots were imaged and quantified with the Odyssey infrared imaging system (Li-Cor Biosciences).
Electrophysiology
Electrophysiological recordings were performed at room temperature, utilizing the whole cell patch clamp technique. Data acquisition and analysis were obtained using the Axopatch 200B (Axon Instruments) patch clamp amplifier and pCLAMP 9.2 (Axon Instruments) software. Electrodes were pulled in two stages from thin wall filament glass capillary tubing (Warner Instruments) and fire polished to a resistance ranging from 1 – 2 MΩ. Voltage clamp recording solutions were as follows (in mM): external (bath) solution 100 NaCl, 5.4 KCl, 1.8 CaCl2. 0.8 MgCl2, 23 glucose, 5 Na Hepes, pH 7.4; internal (pipette) solution: 120 KCl, 3.69 CaCl2, 0.094 MgCl2, 5 BAPTA, 5 EDTA, 5 Na HEPES, 5 glucose, pH 7.2. Cells were held at −60 mV, followed by a 20 ms hyperpolarization to −90 mV, and stepped from −70 mV to +50 mV in 10 mV increments. Leak currents (P/8) were subtracted from all traces.
MBP Pulldown
pMAL-Kv1.3-C-term, pMAL-Kv1.3ΔTDV-C-term, and the empty pMAL vector were transformed into BL21 Gold bacterial cells (Strategene). Cultures were grown in LB supplemented with Glucose/Ampicillin and protein expression was induced by adding 0.3mM IPTG for 2 hours. Cells were harvested at 4000× g for 10 min and resuspended in Column Buffer (20mM Tris-HCl, 200mM NaCl, 1mM EDTA, 1mM sodium azide, 1mM DTT). Cells were lysed and the protein extracts (MBP-Kv1.3, MBP-ΔTDV or MBP) were coated onto Amylose resin (New England Biolabs). HEK293 cells transfected with pGW1-CMV-myc-PSD-95 were lysed in RIPA and precleared with MBP coated Amylose resin by rocking for 30 min at 4°C. Precleared myc-PSD-95 lysates were then rocked with either MBP-Kv1.3, MBP-ΔTDV or MBP coated Amylose resin for 30 min at 4 °C, washed 3x in RIPA, needle aspirated, combined with sample buffer and separated by SDS-PAGE. Western blotting detection of myc-PSD-95 was done with an anti-Myc monoclonal antibody (0.1 μg/mL; Invitrogen) and Kv1.3 was detected using an anti-Kv1.3 monoclonal antibody (0.42 μg/mL; NeuroMab). Blots were imaged and quantified with the Odyssey infrared imaging system (Li-Cor Biosciences).
Flow Cytometry
HEK293 cells were cotransfected with soluble GFP, used as a marker of live cells with intact plasma membranes, and FLAG-Kv1.3 or FLAG-Kv1.3ΔTDV. Transfected HEK cells were treated with 0.5% sodium azide for 30 min at 37 °C to block endocytosis. Surface Kv1.3 was labeled with a monoclonal anti-Flag M2 antibody (1ug/mL; Sigma) directed against an extracellular epitope inserted within the channel. Secondary labeling was done with a fluorescently conjugated goat anti-mouse IgG (0.25ug/mL; Southern Biotech). Kv1.3 surface levels were quantified as the number of live cells (GFP positive) emitting at 667 nm with fluorescence intensity above a threshold value determined using cells labeled with a mouse IgG2a isotype control (R&D Systems). Flow cytometry was done with the Easycyte single laser flow cytometer (Guava Technologies). Analysis of cell populations and histograms was done with FCS Express and WinMDI softwares, respectively.
Cell Surface Biotinylation
HEK cells transiently transfected with either GFP-Kv1.3-Flag or GFP-Kv1.3-Flag-ΔTDV were rinsed 2 × 5 min in Hank’s Buffered Saline Solution (HBSS) at room temperature (RT). Cells were incubated with a 25mM solution of the cell impermeant EZ-Link Sulfo-NHS-SS-Biotin (Thermo Scientific) for 30 min at RT. Cells were then washed 4 × 5 min in HBSS containing 50mM Tris pH 7.4, lysed in RIPA buffer not containing dithiothreitol, and allowed to rock at 4°C for 10 minutes. Lysates were centrifuged at 20,000 × g for 5 min, and a portion of the supernatant was retained for SDS-PAGE analysis. The remaining supernatant was combined with prewashed NeutrAvidin Agarose Resin (Thermo Scientific) and allowed to rock at 4°C for 4 hours. The resin was centrifuged at 2,000 × g for 2 min, washed 3 x in RIPA buffer an needle aspirated. Bound proteins were eluted from resin, separated by SDS-PAGE, and processed according to the Immunoblot Analysis methods listed previously.
Statistical Analysis
Data are presented as means ± standard errors. As noted, either nonparametric Mann-Whitney U tests, unpaired Student’s t-tests assuming unequal variances, or one-way ANOVA with post hoc Bonferroni test were used to determine statistical differences. Differences were considered significant if p < 0.05.
Results
Incorporation of extracellular FLAG epitope for studying Kv1.3 localization
To study the localization of Kv1.3 we introduced a FLAG epitope in the first intrahelical extracellular domain of GFP-Kv1.3 (Fig. 1A). Introduction of this epitope did not affect the function (Fig. 1B) or localization (Fig. 1C) of the channel. The electrophysiological measurements indicate that the mutation did not affect voltage-dependence or the amount of functional channel in the plasma membrane. Previous studies [8, 10, 20] and the images in Fig. 1C indicate that a significant fraction of the channel is localized to an intracellular compartment which we have identified as the Golgi apparatus [8]. Immunohistochemical analyses of colocalization of Kv1.3 with the GM130 Golgi marker, indicate that insertion of the FLAG epitope did not affect the percent of channel in the Golgi region; for GFP-Kv1.3, 28.5 ± 3.8% of the channel was localized to the Golgi region (n=20) and for GFP-Kv1.3-FLAG, 21.7 ± 1.6% was localized to the Golgi region (n = 82; p > 0.05; unpaired t-test).
Fig. 1. Incorporation of extracellular FLAG epitope into GFP-Kv1.3 for studying Kv1.3 localization.
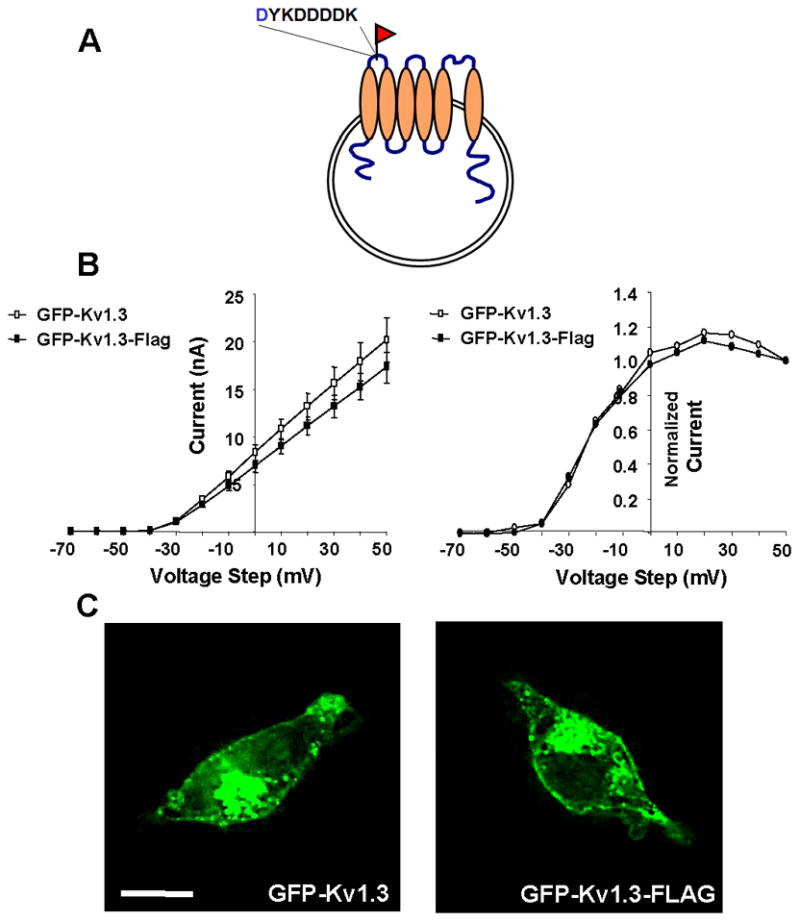
A FLAG epitope was inserted into an extracellular region of wild type and mutant GFP-Kv1.3 to allow detection of Kv1.3 at the cell surface. A) Schematic showing where the FLAG epitope was inserted. B) Steady-state current-voltage relationships for GFP-Kv1.3 (open symbols) and GFP-Kv1.3-FLAG (closed symbols) are shown on left and indicate no effect of the FLAG epitope (n = 8; p > 0.05; unpaired t-test). Activation curves plotted from the tail current of GFP-Kv1.3 (open symbols) and GFP-Kv1.3-FLAG (closed symbols) are shown on the right and indicate that the FLAG epitope did not affect voltage sensitivity (n = 8; p > 0.05; unpaired t-test). C) Fluorescence micrographs of HEK293 cells transfected with GFP-Kv1.3 and GFP-Kv1.3 FLAG. Scale bar represents 10 μM.
Truncation of Kv1.3 C-terminus disrupts PDZ binding domain
The C-terminus of Kv1.3 contains a class I PDZ-binding motif (X-S/T-X-Φ) [21]. We created a mutant form of Kv1.3 that lacked the last three amino acids of this motif (Kv1.3ΔTDV) (Fig. 2A). To confirm that the ΔTDV truncation abolished the binding of PDZ domain containing proteins, we performed a pulldown assay (as described in methods) using the canonical PDZ domain containing protein, PSD-95 [12, 15]. The data in Fig. 2B indicate that PSD-95 bound to the full length C-terminus of Kv1.3, but not to Kv1.3ΔTDV. Densitometric analysis of immunoblots indicate that transfection of equal amounts of DNA resulted in equal levels of wild type and mutant protein (Fig. 2C; Kv1.3 ΔTDV = 92 ± 14% of Kv1.3; n = 8; p = 0.29).
Fig. 2. Truncation of Kv1.3 C-terminal PDZ binding domain.
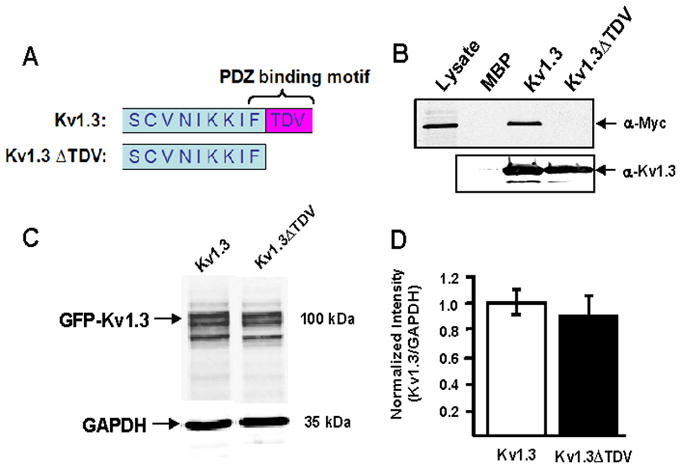
A) Schematic illustrating the deletion of three C-terminal amino acids in Kv1.3. B) Representative immunoblot shows that the indicated deletion disrupts binding to PSD-95, a PDZ domain containing protein, in a pull down assay. The lysate lane confirms expression of myc-PSD-95. The maltose binding protein MBP-Kv1.3 C-terminus (Kv1.3) fusion protein bound to myc-PSD-95. The corresponding mutant fusion protein (Kv1.3ΔTDV) did not. The MBP lane confirms that MBP alone did not bind to myc-PSD-95 in the absence of the Kv1.3 C-terminus. C) Representative immunoblot of Kv1.3 and GAPDH in HEK293 cells transfected with wild type (Kv1.3) and mutant (Kv1.3ΔTDV) channel is shown on the left. Quantitative analysis (right) indicates that wild type and mutant channel protein were equivalently expressed.
Truncation of C-terminal PDZ binding domain increases Kv1.3 current
Electrophysiological analyses were performed on HEK 293 cells transfected with equal amounts of Kv1.3 and Kv1.3ΔTDV DNA to assess the effects of the mutation on the function of the channel. Fig. 3B shows average current traces indicating that the mutation did not qualitatively affect the kinetic properties of the channel. Fig. 3C shows the steady state current-voltage relationships indicating that the mutation increased current amplitudes. This increased current was not caused by a shift in voltage dependence (Fig. 3D).
Fig. 3. Truncation of C-terminal PDZ binding domain increases Kv1.3 current.
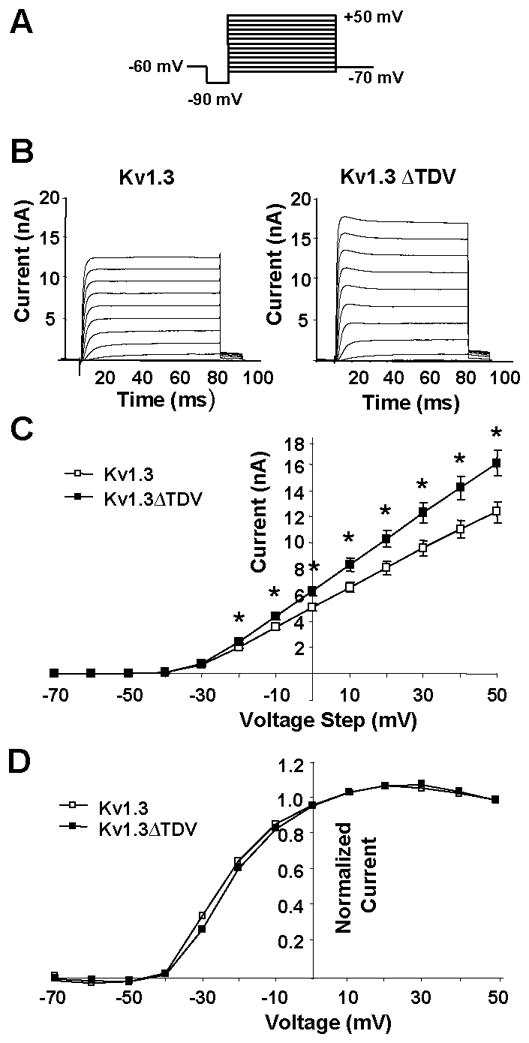
A) Voltage protocol used to elicit ionic currents in transfected HEK cells. Cells were held at −60 mV, followed by a 20 ms hyperpolarization to −90 mV, and stepped from −70 mV to +50 mV in 10 mV increments. B) Ionic currents evoked by stepped voltages from −70 to + 50 mV are shown. Current traces represent averages of currents from multiple cells. Data from Kv1.3 and Kv1.3-ΔTDV transfected HEK293 cells are shown. C) Steady state current-voltage relationships indicate that currents measured in cells transfected with Kv1.3 were less than those measured in cells transfected with Kv1.3-ΔTDV. (* indicates p < 0.05; Mann-Whitney U). D) Activation curves plotted from the tail currents of Kv1.3 and Kv1.3-ΔTDV cells indicate that the ΔTDV did not affect voltage sensitivity (n ≥ 15; p > 0.05; Mann-Whitney U).
Truncation of C-terminal PDZ binding domain decreases Kv1.3 Golgi localization
Our previous studies indicate that Kv1.3 is localized to the Golgi apparatus in postganglionic sympathetic neurons [8]. Fig. 4A confirms this observation and indicates that Kv1.3 is also localized to the Golgi in vascular smooth muscle (Fig. 4B). Trafficking from the Golgi apparatus to the plasma membrane can affect the amount of protein at the cell surface [22], and for ion channels this can increase current. PDZ-dependent interactions are known to affect surface expression of proteins by affecting Golgi retention [11, 23, 24]. For example, decreased Golgi localization of CFTR is associated with increased surface channel and increased ionic current [11]. Since Kv1.3 is localized to the Golgi, the C-terminal PDZ domain could affect Golgi localization of Kv1.3 and thereby affect current. We used HEK 293 cells to determine if disruption of the C-terminal PDZ binding domain affected Golgi localization. Figs. 4C, D and E show representative images of high (61.4%), average (28.3%), and low (10.0%) Golgi localization of Kv1.3. Fig. 4F shows representative images of Kv1.3ΔTDV in HEK 293 cells. Quantitative analysis of this localization is shown in Fig. 4G. These data clearly indicate that the mutation decreased the percentage of Kv1.3 localized to the Golgi region.
Fig. 4. Truncation of C-terminal PDZ binding domain decreases Kv1.3 Golgi localization.
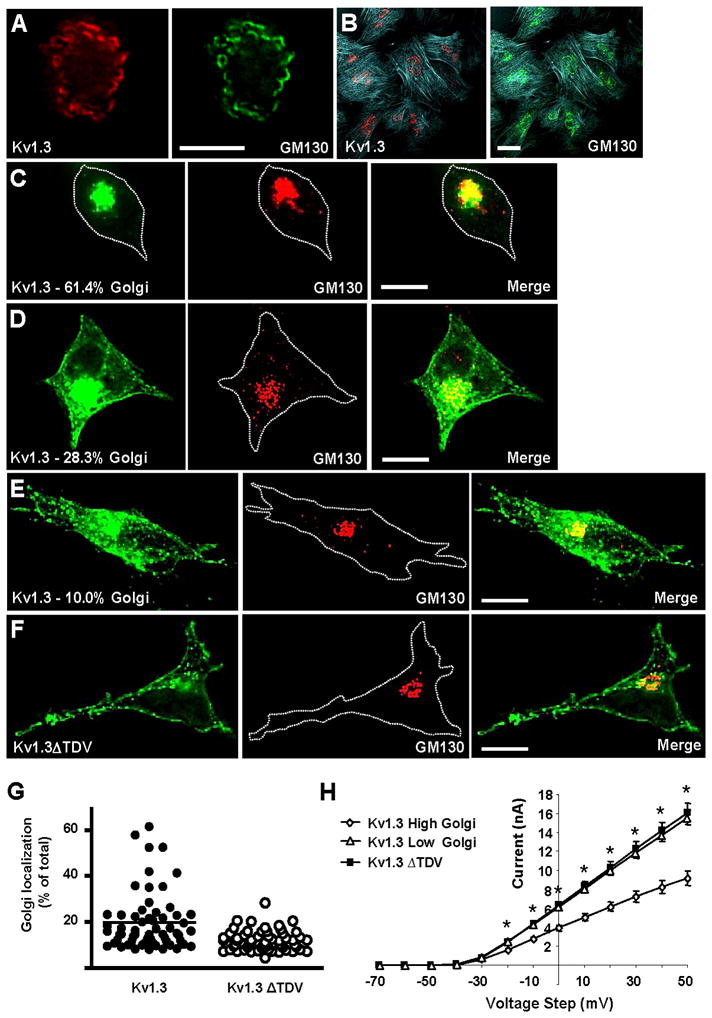
A) Immunohistochemical localization of endogenous Kv1.3 (red) and GM130 (green) in postganglionic sympathetic neurons. B) Immunohistochemical localization of endogenous Kv1.3 (red), GM130 (green) and polymerized actin (blue) in cultured vascular smooth muscle cells. Immunohistochemical localization of endogenous GM130 (red) and localization of GFP fluorescence (green) in HEK293 cells transfected with GFP-Kv1.3 (C, D, and E) and GFP-Kv1.3ΔTDV (F). Representative images of high (C), average (D) and low (E) Kv1.3 Golgi localization are shown. Percentages of Kv1.3 in the Golgi region are indicated on micrograph labels. In micrographs where cell perimeters were difficult to visualize, they were indicated with white dotted lines. G) Vertical scatter plots of Golgi localization. Each data point represents the percent of total Kv1.3 (or Kv1.3ΔTDV) localized to the Golgi region in an individual cell. The horizontal bars indicate the means for each group. H) Steady state current-voltage relationships were measured in cells transfected with Kv1.3 that had low and high Golgi localization and in cells transfected with Kv1.3ΔTDV. *s indicate that the low Golgi population of cells transfected with Kv1.3 had higher currents than the high Golgi population of cells. Note that the Kv1.3ΔTDV currents were not significantly different from the low Golgi Kv1.3 currents.
Does this decrease in Golgi localization contribute to the increase in current observed Fig. 3C? A consideration of the data in Fig. 4G indicates that in HEK 293 cells transfected with wild type Kv1.3, Golgi localization is variable and there appears to be two populations of cells, those with low Golgi localization (Golgi localization less than the median) and those with high localization (Golgi localization greater than the median). If Golgi localization affected current, we would expect that current in the low Golgi cells would be higher than that in the high Golgi cells. Fig. 4H shows that this is the case. The C-terminal deletion eliminated the high Golgi population and the current in these cells was not significantly different from that in the low Golgi population of cells expressing wild type Kv1.3 (Fig. 4H). These data clearly suggest that Golgi localization is a determinant of Kv1.3 current.
Truncation of C-terminal PDZ binding domain does not affect surface expression of Kv1.3
The data in Figs. 3 and 4 suggest that the C-terminal deletion enhanced forward trafficking of Kv1.3 from the Golgi to the plasma membrane, thereby increasing current. These data thus suggest that surface expression, like current (Fig. 4H), should be inversely related to Golgi localization. The data in Fig. 4H also suggest that surface expression of Kv1.3ΔTDV should be comparable to surface expression of Kv1.3 in the low Golgi population. We assessed Kv1.3 surface expression with constructs of GFP-Kv1.3 that contained an extracellular FLAG epitope (see Fig. 1A). Fig. 5A shows total Kv1.3 (green), Kv1.3 detected at the cell surface using a Flag antibody applied to live unpermeabilized cells (blue), and GM130 labeling, obtained after fixation and permeabilization (red). The restriction of the FLAG antibody signal to the plasma membrane, and the absence of this signal in the Golgi apparatus, confirm that the FLAG antibody had access only to channel at the cell surface. Fig. 5C shows that for wild type Kv1.3, surface expression was inversely related to Golgi localization, suggesting that if the channels were not retained in the Golgi, they would localize to the plasma membrane. Although low Golgi Kv1.3 and Kv1.3ΔTDV had similar Golgi localizations (Fig. 4G), surface expression of Kv1.3ΔTDV was less than that of low Golgi Kv1.3. These findings indicate a dissociation between Golgi localization and surface expression of Kv1.3 ΔTDV.
Fig. 5. Truncation of C-terminal PDZ binding domain does not affect surface expression of Kv1.3.

Representative photomicrographs of HEK293 cells transfected with Kv1.3 (A) or Kv1.3ΔTDV (B) (total Kv1.3; green) and live labeled with anti-FLAG-M2 (surface Kv1.3; blue) to detect the extracellular Kv1.3-FLAG epitope, and thus Kv1.3 at the cell surface. After fixation, cells were labeled with anti-GM130 (GM130; red) to define the Golgi region. C) Surface channel was measured immunohistochemically in cells transfected with Kv1.3 or Kv1.3ΔTDV and normalized to that measured in the high Golgi Kv1.3 cells (Kv1.3 high, white bar, n = 41; Kv1.3 low, black bar, n = 41; Kv1.3ΔTDV, gray bar, n = 59). * indicates statistically significant difference from Kv1.3 high Golgi and ★ indicates statistically significant difference from Kv1.3 low Golgi; p < 0.05; one way ANOVA). D) Surface channel was measured by flow cytometry in cells transfected with Kv1.3 or Kv1.3ΔTDV and normalized to surface Kv1.3. E) Surface and total channel protein were measured by cell surface biotinylation in cells transfected with Kv1.3 and Kv1.3ΔTDV. Total and surface Kv1.3 detection was done with anti-Kv1.3 monoclonal antibody (0.42 μg/mL; NeuroMab). A monoclonal anti-GAPDH antibody (1.0 μg/mL; Millipore) was used to control for loading efficiency. F) Immunoblots were quantified and surface expression (surface/GAPDH) of Kv1.3 and Kv1.3ΔTDV were normalized to Kv1.3 (n = 3). G) Total channel protein (total/GAPDH) of Kv1.3 and Kv1.3ΔTDV were normalized to Kv1.3 (n = 3).
These findings also indicate a partial dissociation of current and surface expression for Kv1.3ΔTDV channels. To confirm this unexpected finding, we used two alternate approaches to assess the affects of this mutation on channel surface expression. First, we used flow cytometry in the presence of 0.5% sodium azide, which inhibits endocytosis [25]. The ΔTDV mutation did not affect surface expression as measured by flow cytometry (Fig. 5D), which determines the average surface expression of all cells, which for Kv1.3, includes low and high Golgi populations. Second, we used cell surface biotinylation to quantify surface Kv1.3 levels. These studies also confirmed that the ΔTDV mutation did not affect Kv1.3 surface expression (Figs. 5E and F) or the total amount of channel (Fig. 5G). Thus, using three independent approaches, we find that the mutation did not affect surface expression, even though it did affect ionic current (Fig. 3C).
Discussion
The major findings of this study are: 1) Deletion of the C-terminal PDZ binding domain of Kv1.3 increased current, indicating that this motif is a determinant of the function of this channel. 2) Deletion of the C-terminal PDZ binding domain of Kv1.3 decreased the percent of channel localized to the Golgi region, indicating that this motif is a determinant of the localization of this channel. For wild type but not mutant Kv1.3, decreased Golgi localization was associated with increased surface expression, suggesting that the link between Golgi localization and surface expression is determined by PDZ-dependent interactions. 3) Deletion of the C-terminal PDZ binding domain of Kv1.3 did not affect surface expression of the channel. For wild type, but not mutant Kv1.3, surface expression was correlated with current, suggesting that PDZ-dependent interactions affect the link between surface expression and current.
It is well established that PDZ-dependent interactions affect the function of many ion channels [13, 14, 26–28], but their role in determining Kv1.3 function are not well understood. Our studies suggest that in HEK 293 cells PDZ-dependent interactions limit Kv1.3 current. Our findings differ from those of Marks and Fadool [15] who reported that mutation of the C-terminal PDZ-binding motif of Kv1.3 did not affect current. The reason for this discrepancy is not clear, but is likely due to differences in experimental conditions such as cell confluence and media composition or mutation versus truncation. In our studies, electrophysiological recordings were made on non-confluent cells that had been serum-starved, whereas Marks and Fadool [15] studied cells that were 40–50% confluent that were not serum-starved. In addition, these investigators mutated the C-terminus of Kv1.3 whereas we truncated the C-terminus.
How do PDZ-dependent interactions affect Kv1.3 current? Trafficking to and from the plasma membrane is a well known mechanism for modulating channel function. We hypothesized that PDZ-dependent interactions determined trafficking of Kv1.3 from the Golgi to the plasma membrane. The data in Fig. 4 are consistent with this hypothesis and indicate an inverse relationship between Golgi localization and current. Further analyses, however, indicate that the decrease in Golgi localization of Kv1.3ΔTDV does not result in an increase in surface expression. Thus, the increase in Kv1.3ΔTDV current is not due to an increase in channels at the cell surface. The mechanism underlying the increase in current remains to be established. It is possible that endogenous Kv1 channels contributed to the increase. Although there is evidence indicating that untransfected HEK293 cells express voltage-gated potassium channels [29, 30], the currents generated by these channels are on the order of picoamps, well below the increase in current observed with Kv1.3ΔTDV (Fig. 3). Furthermore, if these endogenous Kv currents were significantly contributing to the current measured, we would expect them to contribute equally in Kv1.3- and Kv1.3ΔTDV-transfected cells. Therefore, it is likely that the increase in current is mediated by transfected Kv1.3 alone. It is also possible that the mutation altered the biophysical properties of the channel. It is further possible that there are inactive or silent channels on the surface, and PDZ-dependent interactions change the number of channels in the inactive pool. Regardless of the underlying mechanisms, the present studies indicate that PDZ-dependent interactions modulate the function of Kv1.3.
The most straightforward interpretation of our data is that the C-terminal PDZ-binding domain of Kv1.3 interacted with PDZ-domain containing proteins endogenously expressed in HEK 293 cells to affect localization and function of Kv1.3. HEK 293 cells are known to express PDZ-domain containing proteins such as CAL [31], which affects Golgi localization and stability of proteins, and SAP 97 [32] which affects Golgi localization and clustering of proteins at the cell surface. The role of these and other PDZ-domain containing proteins warrants further investigation.
Acknowledgments
We greatly appreciate the generous gifts of pEGFP-C1-Kv1.3 from Dr. Jürgen Kupper [Max Planck Institute of Biochemistry, Martinsried, Germany] and pMAL-Kv1.3-C-term from Dr. Todd Holmes [University of California – Irvine]. This work was supported by HL076774 (D.H.D.) and NS050623 (A.D.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Koo GC, Blake JT, Talento A, Nguyen M, Lin S, Sirotina A, Shah K, Mulvany K, Hora D, Jr, Cunningham P, Wunderler DL, McManus OB, Slaughter R, Bugianesi R, Felix J, Garcia M, Williamson J, Kaczorowski G, Sigal NH, Springer MS, Feeney W. Blockade of the voltage-gated potassium channel Kv1.3 inhibits immune responses in vivo. J Immunol. 1997;158:5120–5128. [PubMed] [Google Scholar]
- 2.Panyi G, Varga Z, Gaspar R. Ion channels and lymphocyte activation. Immunol Lett. 2004;92:55–66. doi: 10.1016/j.imlet.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 3.Beeton C, Wulff H, Standifer NE, Azam P, Mullen KM, Pennington MW, Kolski-Andreaco A, Wei E, Grino A, Counts DR, Wang PH, LeeHealey CJSAB, Sankaranarayanan A, Homerick D, Roeck WW, Tehranzadeh J, Stanhope KL, Zimin P, Havel PJ, Griffey S, Knaus HG, Nepom GT, Gutman GA, Calabresi PA, Chandy KG. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc Natl Acad Sci USA. 2006;103:17414–17419. doi: 10.1073/pnas.0605136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fadool DA, Tucker K, Perkins R, Fasciani G, Thompson RN, Parsons AD, Overton JM, Koni PA, Flavell RA, Kaczmarek LK. Kv1.3 channel gene-targeted deletion produces “Super-Smeller Mice” with altered glomeruli, interacting scaffolding proteins, and biophysics. Neuron. 2004;41:389–404. doi: 10.1016/s0896-6273(03)00844-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu J, Koni PA, Wang P, Li G, Kaczmarek L, Wu Y, Li Y, Flavell RA, Desir GV. The voltage-gated potassium channel Kv1.3 regulates energy homeostasis and body weight. Hum Mol Genet. 2003;12:551–559. doi: 10.1093/hmg/ddg049. [DOI] [PubMed] [Google Scholar]
- 6.Li Y, Wang P, Xu J, Desir GV. Voltage-gated potassium channel Kv1.3 regulates GLUT4 trafficking to the plasma membrane via a Ca2+-dependent mechanism. Am J Physiol Cell Physiol. 2006;290:C345–351. doi: 10.1152/ajpcell.00091.2005. [DOI] [PubMed] [Google Scholar]
- 7.Xu J, Wang P, Li Y, Li G, Kaczmarek LK, Wu Y, Koni PA, Flavell RA, Desir GV. The voltage-gated potassium channel Kv1.3 regulates peripheral insulin sensitivity. Proc Natl Acad Sci USA. 2004;101:3112–3117. doi: 10.1073/pnas.0308450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doczi MA, Morielli AD, Damon DH. Kv1.3 channels in postganglionic sympathetic neurons: expression, function, and modulation. Am J Physiol. 2008;295:R733–740. doi: 10.1152/ajpregu.00077.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kupper J, Prinz AA, Fromherz P. Recombinant Kv1.3 potassium channels stabilize tonic firing of cultured rat hippocampal neurons. Pflugers Arch. 2002;443:541–547. doi: 10.1007/s00424-001-0734-4. [DOI] [PubMed] [Google Scholar]
- 10.Ramirez-Navarro A, Glazebrook PA, Kane-Sutton M, Padro C, Kline DD, Kunze D. Kv1.3 channels regulate synaptic transmission in the nucleus of solitary tract. J Neurophysiol. 2011 doi: 10.1152/jn.00494.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng J, Moyer BD, Milewski M, Loffing J, Ikeda M, Mickle JE, Cutting GR, Li M, Stanton BA, Guggino WB. A Golgi-associated PDZ domain protein modulates cystic fibrosis transmembrane regulator plasma membrane expression. J Biol Chem. 2002;277:3520–3529. doi: 10.1074/jbc.M110177200. [DOI] [PubMed] [Google Scholar]
- 12.Kim E, Niethammer M, Rothschild A, Jan YN, Sheng M. Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature. 1995;378:85–88. doi: 10.1038/378085a0. [DOI] [PubMed] [Google Scholar]
- 13.Laufer J, Boehmer C, Jeyaraj S, Knuwer M, Klaus F, Lindner R, Palmada M, Lang F. The C-terminal PDZ-binding motif in the Kv1.5 potassium channel governs its modulation by the Na+/H+ exchanger regulatory factor 2. Cell Physiol Biochem. 2009;23:25–36. doi: 10.1159/000204077. [DOI] [PubMed] [Google Scholar]
- 14.Lunn ML, Nassirpour R, Arrabit C, Tan J, McLeod I, Arias CM, Sawchenko PE, Yates JR, 3rd, Slesinger PA. A unique sorting nexin regulates trafficking of potassium channels via a PDZ domain interaction. Nat Neurosci. 2007;10:1249–1259. doi: 10.1038/nn1953. [DOI] [PubMed] [Google Scholar]
- 15.Marks DR, Fadool DA. Post-synaptic density perturbs insulin-induced Kv1.3 channel modulation via a clustering mechanism involving the SH3 domain. J Neurochem. 2007;103:1608–1627. doi: 10.1111/j.1471-4159.2007.04870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swiatecka-Urban A, Duhaime M, Coutermarsh B, Karlson KH, Collawn J, Milewski M, Cutting GR, Guggino WB, Langford G, Stanton BA. PDZ domain interaction controls the endocytic recycling of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2002;277:40099–40105. doi: 10.1074/jbc.M206964200. [DOI] [PubMed] [Google Scholar]
- 17.Tiffany AM, Manganas LN, Kim E, Hsueh YP, Sheng M, Trimmer JS. PSD-95 and SAP97 exhibit distinct mechanisms for regulating K(+) channel surface expression and clustering. J Cell Biol. 2000;148:147–158. doi: 10.1083/jcb.148.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panyi G, Bagdany M, Bodnar A, Vamosi G, Szentesi G, Jenei A, Matyus L, Varga S, Waldmann TA, Gaspar R, Damjanovich S. Colocalization and nonrandom distribution of Kv1.3 potassium channels and CD3 molecules in the plasma membrane of human T lymphocytes. Proc Natl Acad Sci USA. 2003;100:2592–2597. doi: 10.1073/pnas.0438057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panyi G, Vamosi G, Bacso Z, Bagdany M, Bodnar A, Varga Z, Gaspar R, Matyus L, Damjanovich S. Kv1.3 potassium channels are localized in the immunological synapse formed between cytotoxic and target cells. Proc Natl Acad Sci USA. 2004;101:1285–1290. doi: 10.1073/pnas.0307421100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vicente R, Villalonga N, Calvo M, Escalada A, Solsona C, Soler C, Tamkun MM, Felipe A. Kv1.5 association modifies Kv1.3 traffic and membrane localization. J Biol Chem. 2008;283:8756–64. doi: 10.1074/jbc.M708223200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science. 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 22.Allan VJ, Thompson HM, McNiven MA. Motoring around the Golgi. Nat Cell Biol. 2002;4:E236–242. doi: 10.1038/ncb1002-e236. [DOI] [PubMed] [Google Scholar]
- 23.He J, Bellini M, Xu J, Castleberry AM, Hall RA. Interaction with cystic fibrosis transmembrane conductance regulator-associated ligand (CAL) inhibits beta1-adrenergic receptor surface expression. J Biol Chem. 2004;279:50190–50196. doi: 10.1074/jbc.M404876200. [DOI] [PubMed] [Google Scholar]
- 24.Xu Z, Oshima K, Heller S. PIST regulates the intracellular trafficking and plasma membrane expression of Cadherin 23. BMC Cell Biol. 2010;11:80. doi: 10.1186/1471-2121-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nesti E, Everill B, Morielli AD. Endocytosis as a mechanism for tyrosine kinase-dependent suppression of a voltage-gated potassium channel. Mol Biol Cell. 2004;15:4073–4088. doi: 10.1091/mbc.E03-11-0788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim HJ, Yang DK, So I. PDZ domain-containing protein as a physiological modulator of TRPV6. Biochem Biophys Res Commun. 2007;361:433–438. doi: 10.1016/j.bbrc.2007.07.027. [DOI] [PubMed] [Google Scholar]
- 27.Lee HJ, Zheng JJ. PDZ domains and their binding partners: structure, specificity, and modification. Cell Commun Signal. 2010;8:8. doi: 10.1186/1478-811X-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- 29.Jiang B, Sun X, Cao K, Wang R. Endogenous Kv channels in human embryonic kidney (HEK-293) cells. Mol Cell Biochem. 2002;238:69–79. doi: 10.1023/a:1019907104763. [DOI] [PubMed] [Google Scholar]
- 30.Varghese A, TenBroek EM, Coles J, Jr, Sigg DC. Endogenous channels in HEK cells and potential roles in HCN ionic current measurements. Prog Biophys and Mol Bio. 2006;90:26–37. doi: 10.1016/j.pbiomolbio.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 31.Cheng J, Cebotaru V, Cebotaru L, Guggino WB. Syntaxin 6 and CAL mediate the degradation of the cystic fibrosis transmembrane conductance regulator. Mol Biol Cell. 2010;21:1178–1187. doi: 10.1091/mbc.E09-03-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petitprez S, Zmoos AF, Ogrodnik J, Balse E, Raad N, El-Haou S, Albesa M, Bittihn P, Luther S, Lehnart SE, Hatem SN, Coulombe A, Abriel H. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ Res. 2011;108:294–304. doi: 10.1161/CIRCRESAHA.110.228312. [DOI] [PubMed] [Google Scholar]