

Abstract
The initial engagement of the T cell receptor (TCR) through interaction with cognate peptide-MHC is a requisite for T cell activation and confers antigen specificity. While this is a key event in T cell activation, the duration of these interactions may affect the proliferative capacity and differentiation of the activated cells. Here, we developed a system to evaluate the temporal requirements for antigenic stimulation during an immune response, in vivo. Using antibodies that target specific antigens in the context of MHC, we were able to manipulate the duration of antigen availability to both CD4 and CD8 T cells during an active infection. During the primary immune response, the magnitude of the CD4 and CD8 T cell response was dependent on the duration of antigen availability. Both CD4 and CD8 T cells required sustained antigenic stimulation for maximal expansion. Memory cell differentiation was also dependent on the duration of antigen exposure, albeit to a lesser extent. However, memory development did not correlate with the magnitude of the primary response, suggesting that the requirements for continued expansion of T cells and memory differentiation are distinct. Finally, a shortened period of antigen exposure was sufficient to achieve optimal expansion of both CD4 and CD8 T cells during a recall response. It was also revealed that limiting exposure to antigen late during the response may enhance the CD4 T cell memory pool. Collectively, these data indicated that antigen remains a critical component of the T cell response after the initial APC-T cell interaction.
Introduction
Antigen recognition by the T cell receptor (TCR)2 expressed by CD4 and CD8 T cells is the initial step in T cell activation that leads to clonal expansion and acquisition of effector function. Following the expansion phase, 90–95% of the T cells at the peak of the response undergo apoptosis (1). The remaining population of cells differentiates to become memory T cells that provide long lasting protection to the host. Formation of an immunological synapse allows signal integration to occur through TCR engagement with peptide-MHC complexes leading to T cell activation (2,3). While TCR triggering is required for the initial activation of a T cell, the role that the duration of the antigenic stimulus plays in the differentiation of a T cell during an immune response remains less clear.
CD8 T cells require only transient stimulation with antigen to initiate proliferation (4–6), whereas CD4 T cells appear to require a longer encounter with antigen for division to occur (7). Nonetheless, both CD8 and CD4 T cells require sustained periods of stimulation to differentiate into effector and memory T cells (7–9). In vivo studies suggest that 36–60 hours of antigen availability is sufficient for T cells to undergo the programming necessary for the acquisition of effector function and differentiation to memory cells (6,10–12). However, more prolonged antigen exposure may be required for optimal expansion and effector differentiation (13,14). Moreover, successive T cell encounters with DC following the initial priming event correlates with the induction of effector function (15). In contrast, other data indicate that antigenic stimulation beyond two days results in a diminished population of effector CD4 T cells (16,17). Thus, there remains ambiguity in terms of the durational requirements of antigen recognition by T cells for mounting an effective immune response. We have previously shown that minimally limiting the amount of antigen during the priming of CD4 T cells allows maximal expansion but results in defective effector differentiation and memory development (18). Together, these studies indicate that T cell programming occurs very early during the T cell response and that antigen, even beyond the initial APC-T cell interaction may continue to influence the process. In support of this, it has also been demonstrated that the contraction phase of the immune response is programmed early on during the response (19,20).
While the aforementioned studies have revealed the relevance of antigen presence beyond initial T cell activation during the immune response on T cell programming, further investigation is warranted. For example, Listeria monocytogenes (Lm) infection followed by antibiotic treatment has been used as an in vivo model to assess the role of antigen duration on T cell programming (10–12,21). This results in the clearance of the pathogen thereby eliminating the source of the antigen. One caveat of these studies is that the duration of antigen availability is only addressed indirectly by altering the duration of the infection. Thus, while bacterial clearance occurs rapidly following treatment, the potential for residual antigen, which can continue to stimulate T cells, exists. Additionally, antibiotic treatment also curtails the inflammatory response. Inflammation, in and of itself, can influence the T cell response (22–26). Thus, it is difficult to discriminate the effects of antigen diminution from the reduction in inflammation. To circumvent these issues, an elegant study assessed the duration of antigen and T cell programming in vivo using an alternative approach (6) in which antigen-bearing dendritic cells (DC) could be selectively depleted. The results indicate that a period of 6–12 hours of antigenic stimulation is required for the differentiation of effector cells and development of memory cells, at least for the high avidity TCR transgenic CD8 T cell, OT-I. In contrast, the magnitude of the CD8 T cell response correlated with the duration of antigen availability. However, this study did not address the in vivo endogenous response to infection.
In an effort to better understand the temporal requirements for antigen on T cell differentiation during an immune response we set out to develop a model where we could modulate antigen availability to CD4 and CD8 T cells in vivo, while leaving all other parameters intact. To this end, we took advantage of two MHC-peptide specific blocking antibodies to examine the effects of limiting the duration of antigen availability to both CD4 and CD8 T cells during the primary and recall responses to pathogen. Through the antibody mediated regulation of antigen during the course of infection we were able to directly evaluate the temporal effects of antigen on T cells throughout the course of the immune response. We show that both CD4 and CD8 T cells share similar requirements for antigen availability during the primary immune response. However, antigen is only briefly required to drive the secondary response to infection, although differences between CD4 and CD8 T cells were noted. These findings hold implications with regard to the development of antigen-based immunotherapies with respect to the requirements of antigen duration necessary for adequate T cell responses and the development of memory T cells.
Materials and Methods
Mice
C57BL/6J (CD45.1 and CD45.2) mice were purchased from Charles River Laboratories/National Cancer Institute (Wilmington, MA). TEa TCR transgenic mice (27) whose CD4 T cells recognize the Eα peptide (amino acid 52–68) from the I-Eα MHC class II molecule in the context of I-Ab were generously provided by R. Noelle (Dartmouth Medical School, Lebanon, NH) and bred and maintained on a recombinase-activating gene (RAG)-deficient background. For adoptive transfer experiments, 1×104 naïve TEa-RAG−/− CD4 T cells were injected intravenously (i.v.) into congenic B6 recipients. In some cases the TEa cells were CFSE-labeled as previously described(18). Animal protocols were approved by the University of Connecticut Health Center Animal Care Committee.
Isolation of lymphocytes and flow cytometry
Single cell suspensions were prepared from spleens and lymph nodes by mechanical disruption of the tissues between frosted glass slides. Cells were filtered over Nytex mesh (Tetko, Kansas City, MO) and pelleted by centrifugation. Following treatment with tris-ammonium chloride to lyse red blood cells, cells were washed and resuspended in HBSS. For isolation of lymphocytes from lung tissue, mice were anesthetized and perfused with PBS-heparin prior to tissue harvest to clear blood from the tissues. Lymphocytes were then isolated by cutting the tissue into small pieces, followed by digestion in collagenase for one hour at 37°C. Following collagenase digestion, the tissue was passed over a 40 micron cell strainer. For staining, cells were resuspended in 0.2% BSA, 0.01% NaN3 in PBS (FACS Buffer) at a concentration of ~1×107 cells/ml and stained with indicated antibodies. All antibodies were obtained from BD Pharmingen (San Diego, CA), eBioscience (San Diego, CA), BioLegend (San Diego, CA), or Caltag (Burlingame, CA). Following staining, cells were washed in FACS Buffer and fixed with 2% paraformaldehyde/PBS. Multiparameter flow cytometry was performed with a FACSCalibur or LSR II (BD Biosciences, San Jose, CA) cytometer and data were analyzed using FlowJo Software (Tree Star, Ashland, OR). In some experiments, peptide:MHC class I tetramers were used for identification of endogenous populations of antigen specific CD8+ T cells. Single cell suspensions were incubated for one hour at room temperature with the tetramer, Fc block, and antibodies against indicated cell surface proteins. H-2Kb tetramers containing the ovalbumin-derived peptide SIINFEKL or the VSV nucleoprotein-derived peptide RGYVYQGL were produced as previously described (28,29).
Infections
Recombinant VSV containing the DNA sequence for eGFP, the SIINFEKL peptide of ovalbumin, and the Eα peptide (VSV-GSE) was generated as previously described (30). Mice were infected i.v. with 1×105 PFU VSV-GSE or VSV-OVA (31), where indicated. For recall experiments, mice were infected with the Indiana strain of VSV followed by a recall challenge with the New Jersey strain of VSV, or vice-versa. For influenza virus infections, mice were inoculated intranasally with 1×103 PFU of WSN-ova1 (32).
Peptide immunization
In some experiments, memory TEa CD4 T cells were generated via peptide immunization as previously described (33). Briefly, 1×104 TEa CD4 T cells were adoptively transferred into congenic hosts and the following day 100 μg of Eα peptide (amino acid 52–68) (Invitrogen Life Technologies) along with 50μg of anti-OX40 (OX86 clone) was injected i.p.. 18 hours later mice were injected with 50μg Salmonella typhimurium LPS (Sigma-Aldrich).
In vivo inhibition of antigen presentation
Purified Y-Ae mAb (34) was purchased from the National Cell Culture Center (NCCC) (Minneapolis, MN) and diluted in PBS. To evaluate the effects of antigen availability during the course of the immune response, 100–500μg Y-Ae was administered i.p. at a concentration of 1 mg/ml at the indicated time points. For CD8 T cell blocking experiments, 500ug of the 25-D1.16 mAb (35) (NCCC) was administered at the indicated times. Mouse IgG2b and IgG1 (NCCC) were used as isotype controls for Y-Ae and 25-D1.16, respectively.
Intracellular cytokine staining
Lymphocytes were isolated from the spleen five days after infection and restimulated in vitro with 10μg/ml of Eα peptide in the presence of GolgiStop (BD Pharmingen) for 5 hours at 37°C. Following in vitro restimulation, lymphocytes were stained for surface markers, fixed, and permeabilized in PermWash (BD Pharmingen) and stained with anti-IL-2, anti-IFN-γ, or isotype control mAb (BD Pharmingen).
Results
Duration of antigen availability affects the magnitude of the primary CD8 T cell response to systemic virus infection
To investigate the temporal requirements for antigen in the activation of CD8 and CD4 T cells, we developed a model where antigen availability following infection could be manipulated in vivo. To regulate the duration of antigen exposure to CD8 T cells we used the 25-D1.16 mAb, which recognizes the ovalbumin derived peptide SIINFEKL bound to H-2K b (35). For CD4 T cell studies we used the Y-Ae mAb which inhibits antigen presentation to Eα–specific CD4 T cells in vivo (18). Because we wished only to block antigen-presentation to a subset of responding CD8 T cells, we verified that 25-D1.16 treatment in vivo did not deplete antigen bearing cells (Supplemental Figure 1). Spleen cells were coated with the SIINFEKL peptide and transferred to new hosts that were then treated with 500ug of 25-D1.16 or control mAb. Three days later the survival of the cells was measured and the results indicated that 25-D1.16 mAb treatment did not result in any loss of transferred cells (Supplemental Figure 1). Thus, any effects observed should be due to inhibition of antigen recognition. Our previous findings also show that the Y-Ae mAb does not deplete APC or inhibit concomitant bystander responses (18).
To test the temporal requirements for antigen during the CD8 T cell response we infected mice with recombinant vesicular stomatitis virus containing the gene encoding chicken ovalbumin (VSV-ova) and the response was measured using H-2Kb-Ova tetramers. Mice were treated with 500μg of 25-D1.16 mAb, at either day 0,3,4,5, or 6 post-infection, and the Ova-specific CD8 T cell response was measured in the spleen at the peak of the response on day 7 after infection (Figure 1A). The response was inhibited to the greatest extent (~80%) when mice were treated at the time of infection with the blocking antibody (Figure 1). In addition, the expansion of Ova-specific CD8 T cells was reduced to the same extent in mice treated with the blocking antibody at day 1 and 2 (data not shown) or at days 3 and 4 after infection compared to control treated mice (Figure 1A). Interestingly, there was no statistically significant difference between the extent of blocking when mAb was given on day zero or at four days post-infection. In contrast to this, blocking antigen presentation at five or six days after infection had no effect on the magnitude of the CD8 T cell response compared to control antibody treated mice. 25-D1.16 mAb treatment had no effect on the concomitant CD8 T cell response to the VSV nucleoprotein indicating the specificity of the inhibition as well as the lack of any general effects on APC function (Figure 1C). Total numbers of ova-specific CD8 T cells were also reduced suggesting an effect on proliferation (Figure 1D). These results indicated that the presence of antigen is required for up to five days post-infection for maximal expansion of antigen-specific CD8 T cells.
Figure 1. Antigen is required for several days for optimal CD8 T cell response to virus infection.
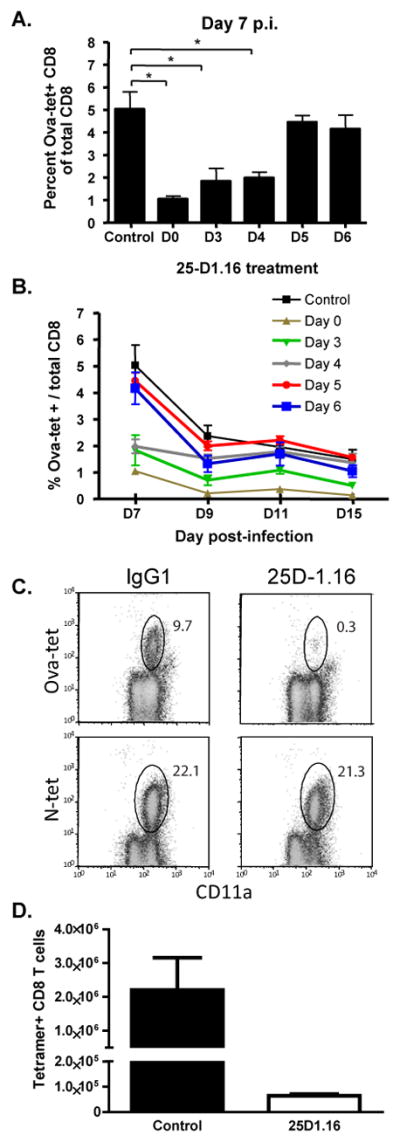
Mice were infected with 105 pfu of VSV-Ova-NJ and treated with 500ug of the blocking 25-D1.16 mAb at the indicated times. Control mice received mouse IgG1 isotype mAb. Mice were then bled at days 7, 9, 11, and 15 post-infection. OVA-specific CD8 T cells were quantitated by flow cytometry using an OVA:Kb tetramer. (A) Blood analysis at day 7; (B) Kinetics of the OVA-specific CD8 T cell response; (C) Analysis of ova-specific and VSV-nucleoprotein (N) -specific CD8 T cells after antibody treatment at day 0; (D) Total numbers of ova-specific splenic CD8 T cells seven days after infection and treatment with the indicated mAbs. The x-axis in A&B indicates the time post-infection that mice were treated with control or blocking antibody. Graphs represent the mean +/− SEM of Ova-tetramer positive cells as a percentage of the entire CD8 T cell population from 3–4 mice per group. These data are representative of three similar experiments. *, p< 0.01.
Following expansion, T cells undergo a period of contraction, in which 90–95% of the T cells undergo apoptosis, while the remaining population goes on to develop into long-lived memory cells (1). To determine if antigen availability had an impact on the contraction of the responding T cells, mice were bled at 9, 11, and 15 days after infection. At day 9 post-infection, Ova-specific CD8 T cells in the blood from mice treated at day 0 or day 3 post-infection were significantly lower than control treated mice (Figure 1B), while at days 11 and 15 post-infection, only mice that were treated at the time of infection with the blocking antibody had significantly lower numbers of Ova-specific CD8 T cells (Figure 1B). While the percentage of Ova-specific CD8 T cells in the blood from mice that were treated at day 3 post-infection was lower compared to the control treated mice, the difference did not reach significance. Thus, while expansion of responding CD8 T cells was reduced when the duration of antigen was limited, the number of Ova-specific CD8 T cells during the contraction phase normalized in the mice, suggesting that the duration of antigen availability during the priming of T cells affects the magnitude of the CD8 T cell response but also may affect the overall survival of the responding cells.
Antigen is required late after influenza virus infection for optimal CD8 T cell expansion
We also analyzed the temporal requirements for antigen after intranasal infection with influenza virus expressing the ovalbumin derived SIINFEKL epitope (WSN-ova). Treatment with 25-D1.16 at the time of infection decreased the peak response at day 10 post-infection 10-fold in the lungs, the mediastinal lymph nodes (MLN) and the spleen (Figure 2A, B). Remarkably, mAb treatment on days 2–7 inhibited the response 75–90% in the lung and MLN, with the spleen response being more variable. In fact, little difference was noted between blocking on day 2 as compared to day 7 suggesting either that antigen presentation began late or that later CD8 T cell-APC interactions subsequent to initial activation were important for continued expansion of CD8 T cells. We believe the latter to be true since T cell activation could be detected within the first two days after infection (data not shown). It should be noted that mAb treatment did not affect virus replication in the lungs (Supplementary table).
Figure 2. Blocking antigen presentation with 25-D1.16mAb after influenza virus infection inhibits the expansion of CD8 T cells.
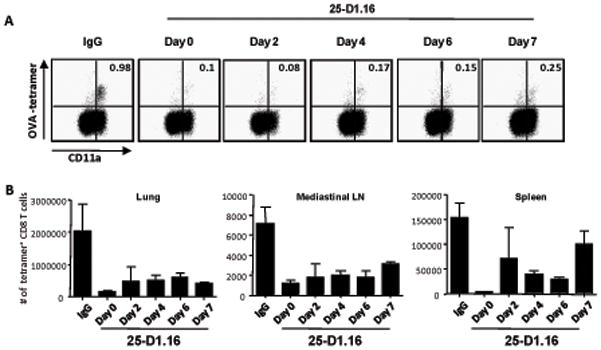
Mice were infected with 1000 pfu of WSN-OVA1 and treated with 500ug of the blocking 25-D1.16 mAb or IgG at the indicated times. On day 10 post infection cells from the lung, mediastinal lymph node (MLN) and spleen were stained with OVA:Kb tetramer and antibodies to CD8 and CD11a then analyzed by flow cytometry. A. Representative plots of gated MLN CD8+ lymphocytes showing staining for CD11a and OVA:Kb tetramer. Values indicate the percentages of tetramer+ cells among CD8+ T cells. B. Graph represents the total number of OVA-specific CD8+ cells in the indicated tissues; n=3. This experiment was performed 2 additional times with similar results. Days indicate the day on which mice were treated with mAb.
Our results showed that there was a reduction in the magnitude of the Ova-specific CD8 T cell population at the peak of the immune response if antigen presentation was blocked within 7 days of influenza virus infection. This effect may be due to inhibition of further expansion of responding cells at the time of mAb administration or due to the early contraction of the antigen-specific population due to reduced antigen presentation subsequent to antibody administration. To distinguish between these possibilities we compared the size of the virus-specific CD8 T cell response at days 6 and 9 (~peak) post-infection (Figure 3). In the lung and spleen very few tet+ cells were present 6 days after infection (day 6 PBS) likely due to insufficient time having passed to allow migration of activated CD8 T cells to the tissues. Whether the mice were treated with 25-D1.16 on day 3 or day 6 after infection, by day 9 after infection an increase in tet+ cells in the spleen and lung was evident, but did not reach maximum (PBS). However, in the MLN, the magnitude of the Ova-specific response on day 6 (Day 6 PBS) was similar to the magnitude at day 9 when 25-D1.16 mAb was administered at either day 3 or day 6 (Figure 3). This result suggested that, upon administration at later times after infection, 25-D1.16 mAb reduced the expansion of previously primed SIINFEKL-specific CD8 T cells and did not cause the depletion or contraction of the responding CD8 T cell population.
Figure 3. Inhibition of the CD8 T cell response by 25-D1.16mAb is through the abrogation of expansion of previously primed cells.
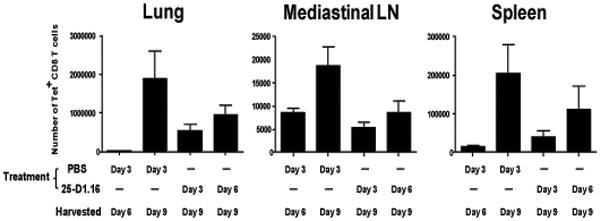
Mice were infected with 1000 pfu WSN-OVA and treated with PBS or 500ug of the blocking 25-D1.16 mAb at the indicated times post infection. Mice from the different groups were sacrificed at the indicated times and lymphocytes isolated from the lung, MLN and spleen, stained with OVA:Kb tetramer and antibodies to CD8 and CD11a then analyzed by flow cytometry. Graphs depict mean +/− SEM of the total number of CD11hitet+ CD8 cells in the indicated tissues of mice from the various groups; n=4.
Prolonged antigen presentation controls the magnitude of the CD4 T cell response
In vitro and in vivo data indicate that CD4 T cells require sustained antigenic stimulation for the programming of proliferation and effector differentiation (13). Few studies have evaluated the role of antigen duration for CD4 T cell differentiation in vivo during infection. At least for infection with Lm, it appears that antigen duration may play a role in CD4 T cell differentiation (11,12). To address the role of antigen duration on the differentiation of CD4 T cells, we utilized an adoptive transfer system and the I-Ab-Eα peptide specific Y-Ae mAb (34,36) to regulate antigen availability in vivo. We have previously shown that the Y-Ae mAb inhibits antigen presentation to TCR transgenic CD4 T cells (TEa) specific to the Eα peptide in vivo (18). Subsequent to adoptive transfer of 1×104 TEa TCR tg T cells into congenic mice, the mice were infected with VSV-GSE, a VSV recombinant expressing the Eα peptide. To regulate antigen exposure during infection, mice were treated with 100μg of the Y-Ae mAb at different times following infection. Y-Ae treatment at the time of infection resulted in almost complete inhibition of the TEa - specific CD4 T cell response to VSV-GSE with very few TEa – specific CD4 T cells detected in the blood five days after infection (Figure 4A and B). This correlated with an inhibition of proliferation on three and five days after infection as detected by the level of CFSE dilution (Supplemental figure 2). Moreover, in mice treated with the blocking antibody at either two or three days after infection the expansion of the TEa CD4 T cells was significantly reduced compared to control treated mice at day five post-infection. Similarly, analysis of lymphoid and nonlymphoid tissues at 6 days after infection also corresponded with reduced expansion of TEa CD4 T cells in mice where antigen was limited up to and including 72 hours after infection (Figure 4C). Thus, the CD4 T cells required at least 72 hours of antigen availability to undergo expansion equivalent to that seen in control treated mice indicating that prolonged antigenic stimulation is necessary for maximal expansion of CD4 T cells responding to virus infection.
Figure 4. Antigen specific CD4 T cells require sustained antigenic stimulation for optimal expansion and cytokine production.
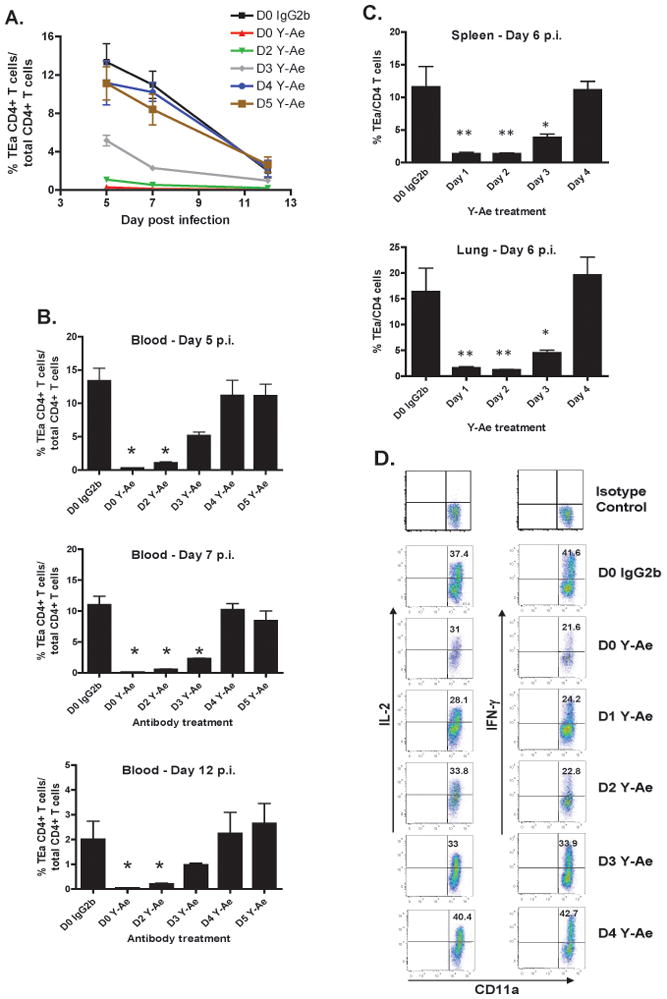
(A) Kinetics of TEa TCR transgenic CD4 T cells in the blood in response to VSV-GSE at different times during the primary immune response under conditions of varying antigen availability. 1×104 TEa T cells were transferred one day before infection with 1×105 pfu of VSV-GSE. Separate groups of mice received 500 μg of the Y -Ae antibody on different days of the response. At the time of infection, or at 24 hour intervals after infection for up to five days, mice were treated with Y-Ae. The populations of TEa cells were analyzed at the indicated times by flow cytometry using a congenic marker. Data represents the mean +/− SEM of four mice/group and are representative of several independent experiments. (B) Individual time points from A. The x-axis indicates the time after infection when mice were treated with the Y-Ae mAb. *, p <0.05. (C) Magnitude of the TEa response in spleen and lung at day 6 post-infection following Y-Ae treatment at day 1,2,3, or 4 post-infection. Mice were treated as in A, with the exception that they received 100ug of Ab. *, p <0.05. **, p<0.01. Data for A-C is derived from 3–4 mice per time point and treatment group and is representative of 3–4 experiments. (D) Limiting the duration of antigen availability results in reduced cytokine production by CD4 T cells. IL-2 and IFN-γ expression by TEa cells in the spleen 5 days after infection. Following adoptive transfer and infection as in (A), mice were injected with Y-Ae or mouse IgG2b control antibody at the indicated times after infection. 5 days after infection gated TEa cells were analyzed for IL-2 and IFN-γ production by intracellular staining after Eα peptide stimulation in vitro. Values represent the mean of 2 mice per group.
To assess whether the attenuated expansion of the TEa CD4 T cells in mice that had limited antigen exposure during the primary immune response altered effector function, we measured IL-2 and IFN-γ production by the TEa cells isolated from the spleen five days after infection. As shown in Figure 4D, cytokine production by TEa cells was dependent on the duration of antigen availability and strongly correlated with the magnitude of the response. Mice treated with Y-Ae at the time of infection, or one or two days following infection, exhibited decreased production of both IL-2 and the effector cytokine IFN-γ compared to TEa T cells that had access to antigen for longer periods of time. Thus, prolonged access to antigen by CD4 T cells resulted in increased capacity to produce cytokines, indicating that progressive differentiation of the CD4 T cells was dependent on the presence of antigen.
Collectively, these data demonstrated that during infection, both CD4 and CD8 T cells require sustained antigenic stimulation to undergo maximal expansion.
Duration of antigen availability during the primary response affects the development of memory T cells
Having established the temporal requirements of antigenic stimulation during the primary immune response, we next asked to what extent limiting antigen exposure early during the primary response affected the development of memory T cells. To this end, VSV-ova infected mice were treated with the 25-D1.16 antibody during the primary infection. Ten weeks later, memory compartments in the spleen and lung were analyzed for the presence of Ova-specific CD8 T cells. Analysis of these tissues indicated that mice treated at the time of infection (D0) with 25-D1.16 mAb had an approximately six-fold reduction in the number of memory cells isolated from the spleen and a 13-fold reduction in the lung, while mice treated at day 3 post-infection had approximately a 2- and 2.5-fold reduction in memory cells in the spleen and lungs, respectively (Figure 5A). In addition, mice limited to antigen exposure for four days during the primary response, which resulted in reduced expansion at the peak of the primary response (Figure 2), had a similar percentage of Ova-specific memory CD8 T cells compared to control treated mice (Figure 5A). We also examined memory development after 25-D1.16 mAb treatment of influenza virus infected mice (Figure 5B). Blockade from days 0–3 greatly reduced the number of memory cells generated by thirty one days after infection. A reduction was also noted when mAb was administered at day 6 after primary infection, but the difference did not reach statistical significance. Beyond that time, no difference in memory development was noted. Thus, while the proliferative capacity of CD8 T cells was strictly regulated by the continued presence of antigen during the primary phase of the immune response, the requirement for optimal memory cell differentiation was less stringent. Therefore, truncating the period of antigen availability to CD8 T cells during the primary response had overlapping, yet independent effects on expansion and memory generation of CD8 T cells. These data demonstrate that for the generation of memory T cells, a threshold must be reached in regards to the duration of antigen exposure and that expansion does not absolutely correlate with the generation of memory cells.
Figure 5. The duration of antigen availability during the primary immune response influences the generation of antigen-specific memory CD8 T cells.
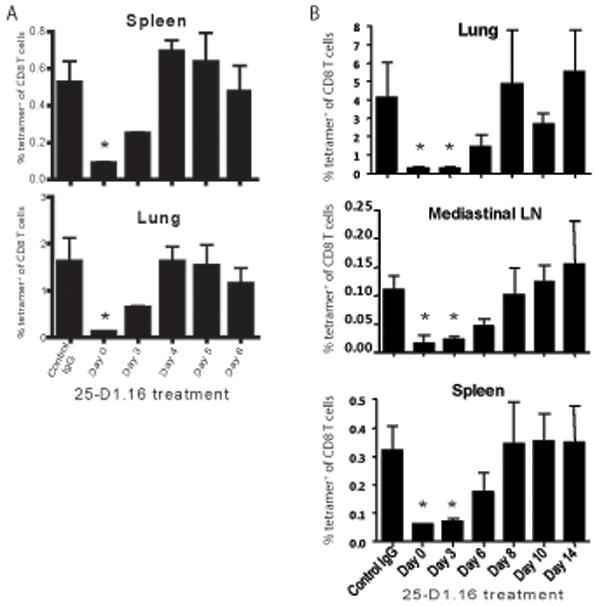
Mice were infected with VSV-OVA-NJ (A) or influenza virus (B) and treated with the 25-D1.16 mAb at the times indicated after infection. Control mice were treated with a mouse IgG1 mAb at the time of infection. At 10 weeks (A) or day 31 (B) post infection the indicated tissues were harvested and analyzed by flow cytometry for the presence of Ova-specific CD8 T cells. Data represents the percentage of Ova-tetramer positive CD8 T cells relative to the total CD8 T cell population from the indicated tissue. The x-axis represents the time after infection that mice were treated with mAb. Data is representative of two independent experiments with 3–4 mice per group. *, p<0.05.
Next, we determined how antigen availability during the primary response affected CD4 T cell differentiation to memory cells. To this end, 1×104 TEa CD4 T cells were adoptively transferred into congenic B6 mice that were subsequently infected with VSV-GSE. In mice treated with an isotype control antibody at the time of infection, a small population of memory cells was detected in the spleen and lung at four weeks after infection (Figure 6). In addition, reducing antigen availability for a period as short as 24 hours after infection did not inhibit the generation of memory CD4 T cells. Surprisingly, in mice where antigenic stimulation was limited to 72 or 96 hours (Day 3 or Day 4 in Figure 6) the memory population in both the spleen and lung were greater than in control mice or when antigen was limited to 24 or 48 hours of availability. While the difference in the lung of mice treated four days after infection were statistically significant compared to control treated mice, the difference did not reach statistical significance in the spleen. However, this trend was similar in independent experiments, and suggested that for the differentiation of memory CD4 T cells, a lengthened period of antigenic stimulation during the primary immune response may be deleterious to memory generation. Furthermore, these data suggested that there was a window of “antigenic opportunity” which resulted in optimal expansion, effector function, and differentiation to memory CD4 T cells.
Figure 6. Duration of antigen availability during the primary immune response influences the generation of antigen-specific memory CD4 T cells.
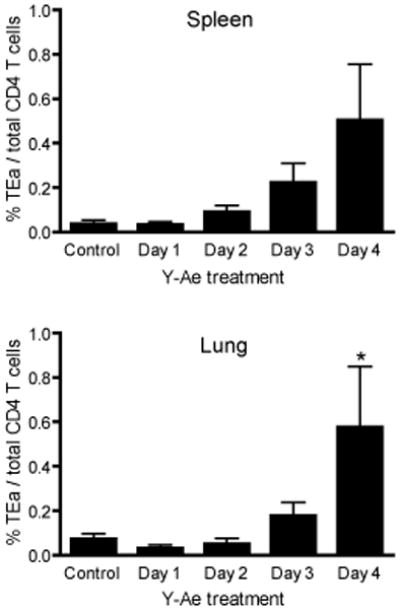
1×104 TEa CD4 T cells were adoptively transferred into congenic B6 mice. One day later mice were infected with VSV-GSE. At the times indicated after infection mice were treated with 100ug of Y-Ae mAb. Control mice were treated with 100ug of mouse IgG2b antibody. Four weeks later, cells from the spleen and lung were analyzed for the presence of donor TEa cells by flow cytometry. Graph indicates the mean +/− SEM from 5 mice per group (Day 1, n=3) and is representative of two independent experiments. *, p< 0.05.
Reduced requirements for antigen availability during the recall response
As compared to naive T cells, memory T cells are thought to require less overall stimulation to mount a response. To formally test this, mice were initially immunized with VSV-Ova-NJ and 60 days later were challenged with a different serotype of recombinant VSV-Ova (VSV-Ova-Ind) to avoid neutralizing antibody specific for the initial virus. Just prior to the recall infection, or at days 1–4 after the secondary challenge, mice were treated with 500 μg of the 25-D1.16 mAb. Five days after challenge, spleen and lung were analyzed for the expansion of Ova-specifc CD8 T cells. In contrast to the primary response where more than 4 days of antigen availability was necessary to drive optimal expansion, up to 24 hours of antigenic stimulation was sufficient for Ova-specific memory CD8 T cells to generate a robust recall response to VSV-Ova in both lymphoid and nonlymphoid tissues (Figure 7). Reducing antigenic stimulation beyond 24 hours had no effect on the magnitude of the recall response. These data indicated that the threshold for expansion was reached during the first 24 hours of the recall response.
Figure 7. Memory CD8 T cells require a reduced period of antigenic stimulation for optimal expansion.
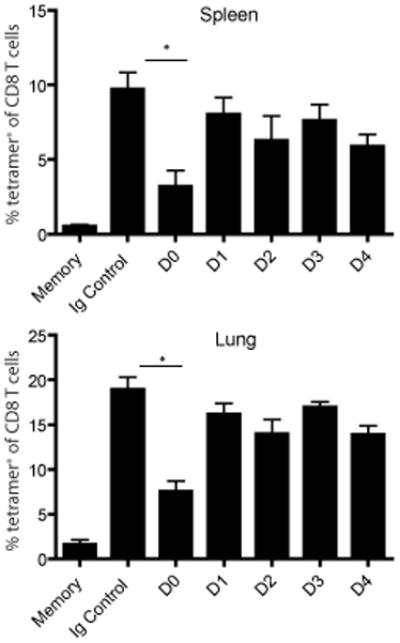
Mice were immunized with 1×105 VSV-OVA-NJ 60 days prior to secondary challenge with 1×105 VSV-OVA-Ind and treated with 500ug of 25-D1.16 mAb at the time of infection (D0) or at 24 hour intervals after the secondary challenge (D1-D4). Control mice (Ig Control) were treated with 500ug of mouse IgG1 mAb. Memory indicates the population of Ova-tet+ cells from mice that only received primary infection. Five days after secondary infection, Ova-tetramer positive CD8 T cells from the spleen and lung were assessed via flow cytometry. Graphs represent the mean +/− SEM from 4–5 mice per group of Ova-tetramer positive cells as a percentage of the entire CD8 T cell population. Representative data from two independent experiments. *, p < 0.01.
We also examined the TEa CD4 T cell response during secondary challenge with VSV. Seven months after adoptive transfer of TEa CD4 T cells and primary infection with VSV-GSE-Ind, mice were recalled with VSV-GSE-NJ. Prior to secondary challenge, mice were bled to ensure even distribution of memory cells across the groups of mice that would be treated with the Y-Ae blocking antibody. The percentage of Eα-specific memory CD4 T cells in the blood was similar in all groups (Figure 8). Mice were then challenged with 1×106 PFU of VSV-GSE-NJ and treated with the blocking antibody at the indicated times for up to 4 days after infection. Analysis of blood 4 days after secondary infection (Figure 8) and spleen and lung five days after secondary infection (Figure 8) revealed that mice treated with Y-Ae at the time of infection or 24 hours after infection resulted in attenuated expansion of the TEa T cells. Compared with the primary response, the period of antigenic stimulation required during the recall response was shortened by 2 days, as antigen was required for up to 4 days during the primary response and up to 2 days during the recall response. Thus, as with the CD8 T cell recall response, maximum CD4 T cell expansion occurred within a shortened period of antigenic stimulation.
Figure 8. Memory CD4 T cells require a reduced period of antigenic stimulation during secondary encounter with antigen.
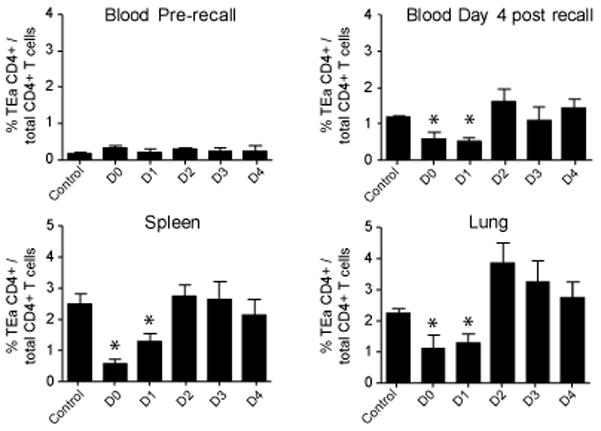
1×104 TEa CD4 T cells were adoptively transferred into B6 congenic mice that were subsequently infected with 1×105 PFU of VSV-GSE-Ind. Seven months later mice were recalled with 1×106 PFU of VSV-GSE-NJ. At the indicated times after recall infection, mice were treated with 500ug of Y-Ae mAb, or mouse IgG2b at the time of infection (Control). Upper left panel: memory TEa cells in the blood prior to recall infection. Cells were identified by flow cytometric analysis of CD4+CD45.2+ T cells. The x-axis denotes the mice assigned to each group for the recall response. Upper right panel: analysis of TEa cells in the blood 4 days after the recall response. Bottom panels: Five days after the recall response, the TEa CD4 T cell response in the spleen and lung was analyzed by flow cytometry. *, p < 0.05.
Interestingly, the magnitude of the TEa recall response (Figure 8) was not as robust as the primary response (Figure 4). This was not due to precursor frequency, as memory TEa CD4 T cells were readily detectable seven months after primary infection (Figure 8), whereas naïve T cells in the blood following adoptive transfer of 1×104 cells are virtually undetectable. In addition, the CD8 T cell response to VSV in the same mice was greater during the recall compared to the primary response (data not shown). Thus, it was possible that the memory TEa CD4 T cells were particularly refractory to the secondary infection due to the enhanced memory CD8 T cell response. Although perhaps unlikely, it is also possible that non-neutralizing cross-reactive antibodies specific for VSV G protein could confer a limited level of protection(37). To eliminate the memory CD8 T cell response or any secondary antibody response against the virus during a recall challenge, we generated memory CD4 TEa T cells in mice by peptide/adjuvant immunization, as previously described (33). Four weeks after immunization, memory cells were barely detectable in the blood (data not shown). Mice were then infected with VSV-GSE-Ind to generate a TEa recall response. In this scenario, VSV-GSE will induce a primary CD8 T cell response, allowing the evaluation of the memory CD4 T cell response in the absence of a memory CD8 T cell response. Similar to the previous studies, mice were injected with the Y-Ae antibody at different times during the infection. Five days after infection, lymphoid and nonlymphoid tissues were analyzed for expansion of the TEa memory population. Indeed, the magnitude of the response in control treated mice was approximately 5- and 20- fold greater in the spleen and lung, respectively, compared to control mice that were primed and recalled with VSV (Figures 9 – IgG2b and Figure 8 - Control). This experiment demonstrated that memory TEa CD4 T cells can, in fact, generate a robust recall response. The response was effectively blocked when Y-AE was administered at the time of infection (Figure 9) but inhibition was rapidly lost as in the previous experiment (Figure 8). Interestingly, in the peptide immunized mice, treatment with the Y-Ae mAb 4 days after the recall response resulted in a significant increase in TEa effector memory cells in the spleen and peripheral lymph nodes relative to control antibody treated mice (Figure 9). This result was reminiscent of the increase in memory CD4 T cells observed in mice treated with the Y-Ae antibody 4 days after primary infection (Figure 6), and suggested that prolonged APC-CD4 T cell interactions could have a negative impact on the response. Thus, enhanced populations of CD4 T cells may be generated by defining the optimal period of antigen availability.
Figure 9. Duration of antigen availability and the recall response of antigen-specific memory CD4 T cells generated by peptide immunization.
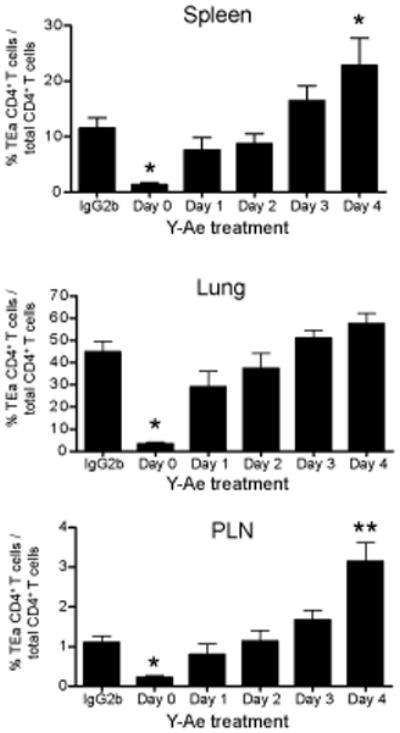
1×104 naïve TEa CD4 T cells were adoptively transferred into congenic B6 mice. Memory TEa cells were generated as described in the materials and methods. Four weeks after immunization, memory TEa cells were recalled by infection with VSV-GSE-Ind. Five days after infection, cells from the spleen, lung, and peripheral lymph nodes (PLN) were analyzed for the presence of TEa cells (gated on CD4+CD45.2+ cells). Graphs represent the mean +/− SEM of TEa CD4 T cells relative to the total CD4 population from 4 mice per group. Data is representative of two independent experiments. *, p<0.05. **, p<0.01.
Discussion
The strength of stimulation following activation of a T cell is a critical component in determining the outcome of a T cell response (38). Sufficient stimulation results in proliferation and differentiation into effector and memory T cells, whereas insufficient stimulation results in an abortive response. Several factors including the antigen dose, TCR avidity (39–43), costimulation (44), and the duration of these interactions (7,45) determine the overall strength of stimulation. Integration of these signals by the T cell initiates a program for proliferation and differentiation. Here we have looked in isolation at one component of this complex set of interactions, the duration of antigen availability, and how it affects the dynamics of the T cell response during infection.
These studies examined the temporal requirements of antigen availability for CD4 and CD8 T cell differentiation during an active infection. We developed a model where the duration of antigen presentation could be manipulated via the administration of MHC-peptide specific antibodies. This model provides many advantages over previous studies. First, the blocking mAbs, 25-D1.16 (35) and Y-Ae (34) are specific for a single MHC-peptide and thus do not alter other T cell responses during the infection. Thus, the overall infectious cycle and level of inflammation remains intact. Lastly, manipulating the duration of antigen is easily controlled and can be tailored to the timeframe investigated. In our system, it would appear that sufficient ova-epitope after VSV or influenza virus infections is available to drive robust CD8 T cell expansion. Moreover, the epitope is sustained late during the response as evidenced by our ability to inhibit the response at relatively late times after infection. This however, may not always be the case as recently shown for the CD8 T cell response to certain influenza virus epitopes, where the apparent lack of persistent epitope expression resulted in diminished CD8 T cell recruitment(46).
To study CD4 T cell responses in conjunction with the blocking mAb, we used an adoptive transfer system since the endogenous Eα-specific CD4 T cell response is essentially undetectable using conventional means (data not shown). While this did not allow for a direct comparison of the endogenous CD4 and CD8 T cell responses, similarities were identified between the two subsets of cells. Both CD4 and CD8 T cells required sustained antigen availability during the primary immune response for optimal expansion to occur. CD4 T cells required between three and four days of antigen availability (Figure 4), whereas CD8 T cells required between four and five days of antigen availability (Figure 2) during VSV infection and even longer after influenza virus infection for optimal expansion. Our previous work also showed a similar effect following Lm infection(47). These results showed that antigen per se continues to drive the T cell response long after initial infection and T cell activation. These late APC encounters serve to enhance expansion as well as augment functionality since late interactions were required for optimal cytokine production by CD4 T cells. In the case of the CD8 T cell response to influenza virus infection, late costimulation through CD27 is also required for optimal expansion(48). Blocking CD27/CD70 interactions on day 6 or 8 after influenza virus infection results in a decrease in the number of antigen-specific CD8 T cells apparently through Fas mediated apoptosis driven by CD4 T cells. In our system, any CD4 T cell response will remain intact so the effects we observed were directly mediated through CD8 T cell-APC interactions.
For both CD4 and CD8 T cells, the recall response required a substantially shorter duration of antigen availability compared to the respective primary response. Thus, upon challenge, memory T cells initiated and sustained division with a substantially reduced duration of antigenic exposure as compared to naïve T cells. These studies formally established the duration of antigenic stimulation required for a recall response to occur. Our results agreed with the general tenet that memory T cells are more readily activated than naive T cells. This is clearly true for effector functions which are rapidly induced following memory T cell encounter with antigen. Induction of proliferation could be considered the second phase of reactivation resulting in generation of a new numerically increased cohort of secondary memory cells. In fact, previous work indicates that the rate of proliferation is similar for naive versus memory CD8 T cells(49). Nevertheless, our results suggested that TCR triggering is either more sensitive to lower levels of antigen or that initial TCR triggering results in sustained activation in the absence of antigen. Previous studies support these concepts in that TCR associated signal transduction components are modified in memory T cells as compared to naive T cells(50–52). Nevertheless, despite heightened TCR responsiveness, costimulation is required for memory T cell reactivation in some cases. Blockade of CD28 greatly inhibits the CD8 and CD4 T cell recall response to influenza virus infection(53,54) while CD40L blockade inhibits the secondary CD8 T cell response to VSV infection(55). In addition, the lack of expression of certain costimulators (e.g. CD70) by particular DC subsets may preclude efficient memory CD8 T cell reactivation(56). Thus, heightened TCR responsiveness in memory T cells may not necessarily correlate with costimulation independence. In part, this hypothesis may be explained by the increase in TCR avidity that is observed in polyclonal memory CD8 T cells(57,58), while functional avidity maturation occurs in transgenic T cells expressing monoclonal TCRs(59).
For the CD4 recall response, the preexisting memory Eα–specific CD4 population expanded in response to secondary challenge, yet the response was not as robust as the memory CD8 T cell response. This finding is in agreement with recent reports where memory CD4 T cells do not respond as well during secondary antigen encounter (60,61) and may be due to reduced levels of IL-2 production in combination with increased levels of IFN-γ production (60). Although we did not measure cytokine production in the secondary response, we showed that the TEa recall response was greatly enhanced when mice were immunized with peptide/adjuvant and then challenged with VSV-expressing Eα. While it is possible that the two forms of immunization (infection vs. adjuvant) could induce physiologically distinct memory cells, this result could be due to the presence of memory CD8 T cells after VSV infection. Although the CD8 T cell primary and secondary responses to VSV infection are CD4 T cell independent (55), rapid viral clearance by memory CD8 T cells could limit the secondary CD4 T cell response in part through reduction of antigen. Of particular interest was the finding that limiting antigen late in the primary TEa response to VSV infection resulted in an increase in the CD4 T cell memory population. A similar result was observed during VSV recall of memory cells generated by peptide/adjuvant immunization. These data suggested that continued antigenic stimulation had a negative impact on memory development. These events could be related to induction of cell death which can result from sustained antigenic stimulation (62). On the other hand, our previous work shows that even a minor change in antigen expression (through injection of 1ug of Y-Ae) early in the response is sufficient to block memory development while not affecting initial expansion(18). Thus, for effective generation of T cell memory there is a favorable window of activation related to antigen levels. Determining the optimal period of antigenic stimulation of a T cell may lead to more rationale vaccine designs.
As stated above, for optimal expansion, endogenous CD8 T cells required the presence of antigen for at least four days following VSV infection and at least seven days following influenza virus infection. This difference may be attributed to the level of antigen expressed at different times after infection with the different viruses and may also be manifested in the differences in the time required for the CD8 T cell responses to reach apogee (~day 7 for VSV and ~day 10 for influenza virus). Thus, priming for the influenza virus response appears more protracted perhaps due to the necessity for local viral replication to occur in the lung epithelium while VSV can productively infect many cell types. Clearly, the overall quantity and the location of antigen can affect response outcome but nevertheless limiting antigen exposure resulted in an overall decrease in the magnitude of the response. This is in contrast to earlier work showing that CD8 T cell responses are unchanged after antibiotic treatment 24 hours post-Lm infection(10–12). These differences may be accounted for by the reduced inflammation mediated by antibiotic treatment and truncated infection. Additionally, it is likely that residual antigen persists beyond bacterial clearance, further complicating data interpretation. In our model, antigen availability was modulated by inhibiting antigen presentation to the responding antigen-specific T cells. All other parameters of the infection and immune responses were maintained. Thus, in our model, we were able to specifically examine the effects of antigen availability while maintaining all other parameters. Although CD8 T cell expansion was reduced when antigen duration was shortened, memory differentiation was apparent. In agreement with other studies, the duration of antigen availability affects the magnitude of the primary response (9,13) but does not necessarily correlate with the development of memory (6,20). Interestingly, while limiting the antigen duration for up to four days resulted in attenuated expansion, there were divergent effects on the memory populations generated. In mice treated at day four post-infection, the magnitude of expansion was similar to mice treated with blocking antibody 24 hours earlier (Figure 1). Remarkably, the resulting memory population in the two groups was markedly different (Figure 5). This was not the result of changes in the ratio of short-lived versus memory precursor effector populations which was largely unaffected by mAb treatment (data not shown and (47)). Thus, these data suggested that by day four post-infection, the CD8 T cells had received sufficient stimulation for programmed differentiation, but required additional interactions with DCs for continued proliferation. This result also suggested that the differentiation of cells may be asynchronous and continued throughout the primary response. It will be interesting to determine if there are functional differences in the memory populations generated under the varying durations of antigen exposure. We would surmise that such memory cells would exhibit normal functional capabilities since even in the absence of accumulation of T cells during the primary response, functional memory cells are generated(6).
While the specific mechanism by which addition of the blocking antibodies is regulating the immune response was not defined, several lines of evidence point to a likely explanation. T cell priming occurs in three phases with multiple T cell-DC interactions taking place during the first 48 hours of the response (63). Other studies have also shown that T cells undergo multiple T cell-DC interactions during an immune response and that these interactions are important in the differentiation of effector T cells (64). In situ analysis of the CD8 T cell immune response reveals that antigen-specific CD8 T cells form localized clusters with DC in the spleen at five days after Lm infection, and administration of 25-D1.16 prevents cluster formation (65). Moreover, a recent study using real-time imaging techniques, shows that blocking MHC class II in vivo promoted the dissociation of T cell-DC interactions (14). Therefore, the 25-D1.16 and Y-Ae mAbs are most likely disrupting stable T cell-DC interactions and potentially preventing subsequent interactions from developing. The precise DC subset and location of inhibition is likely dependent on the infection type and the time of blocking mAb administration. For example, early after influenza virus infection, DC migrating from the lung to the draining mediastinal LN are important for T cell priming. Thus, early administration of mAb could result in inhibition of early T cell-DC interactions in the LN. Conversely, late after infection activated CD8 T cells that have migrated to the lung tissue may interact with antigen bearing APC or with infected parenchymal cells resulting in further T cell expansion (56,66–71).
In summary, we have developed a model to assess the antigen requirements for both CD4 and CD8 T cells in vivo during an active infection. Prolonged antigen availability was required for maximal T cell expansion as shortening the period of antigen availability resulted in a decrease in the magnitude of the primary response. The duration of antigen availability also influenced memory T cell development. However, the magnitude of the response did not necessarily correlate with memory generation suggesting that independent mechanisms regulate T cell proliferation and differentiation. For recall responses, the antigen requirements were lessened compared to the primary response as T cells required a shorter period of antigen availability for maximal expansion to occur. Lastly, optimal CD4 T cell populations may be generated by determining the optimal window of antigen availability. Understanding these temporal requirements for antigen availability by T cells during an immune response and its effect on effector and memory differentiation will be critical for the design of future vaccines. The period of antigen availability needs to be considered and may need to be customized to individual CD4 and CD8 T cell responses to elicit the maximum protective capacity of a vaccine.
Supplementary Material
Footnotes
This work was funded by National Institutes of Health grants AI41576 and AI76457(to L.L.), P01 I56172 (to L.L., A.T.V., L.S.C.), AI42858 (to A.T.V.) and T32 AI07080 (to D.A.B., T.O.B. and J.P.M.).
Abbreviations used in this paper: VSV, vesicular stomatitis virus, Listeria monocytogenes, Lm; TCR, T cell receptor
References
- 1.Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002;111:837–851. doi: 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- 2.Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 3.Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 4.Kaech SM, Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol. 2001;2:415–422. doi: 10.1038/87720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Stipdonk MJ, Lemmens EE, Schoenberger SP. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat Immunol. 2001;2:423–429. doi: 10.1038/87730. [DOI] [PubMed] [Google Scholar]
- 6.Prlic M, Hernandez-Hoyos G, Bevan MJ. Duration of the initial TCR stimulus controls the magnitude but not functionality of the CD8+ T cell response. J Exp Med. 2006;203:2135–2143. doi: 10.1084/jem.20060928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iezzi G, Karjalainen K, Lanzavecchia A. The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity. 1998;8:89–95. doi: 10.1016/s1074-7613(00)80461-6. [DOI] [PubMed] [Google Scholar]
- 8.Van Stipdonk MJ, Hardenberg G, Bijker MS, Lemmens EE, Droin NM, Green DR, Schoenberger SP. Dynamic programming of CD8(+) T lymphocyte responses. Nat Immunol. 2003;4:361–365. doi: 10.1038/ni912. [DOI] [PubMed] [Google Scholar]
- 9.Curtsinger JM, Johnson CM, Mescher MF. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J Immunol. 2003;171:5165–5171. doi: 10.4049/jimmunol.171.10.5165. [DOI] [PubMed] [Google Scholar]
- 10.Mercado R, Vijh S, Allen SE, Kerksiek K, Pilip IM, Pamer EG. Early programming of T cell populations responding to bacterial infection. J Immunol. 2000;165:6833–6839. doi: 10.4049/jimmunol.165.12.6833. [DOI] [PubMed] [Google Scholar]
- 11.Corbin GA, Harty JT. Duration of infection and antigen display have minimal influence on the kinetics of the CD4+ T cell response to Listeria monocytogenes infection. J Immunol. 2004;173:5679–5687. doi: 10.4049/jimmunol.173.9.5679. [DOI] [PubMed] [Google Scholar]
- 12.Williams MA, Bevan MJ. Shortening the infectious period does not alter expansion of CD8 T cells but diminishes their capacity to differentiate into memory cells. J Immunol. 2004;173:6694–6702. doi: 10.4049/jimmunol.173.11.6694. [DOI] [PubMed] [Google Scholar]
- 13.Obst R, van Santen HM, Mathis D, Benoist C. Antigen persistence is required throughout the expansion phase of a CD4(+) T cell response. J Exp Med. 2005;201:1555–1565. doi: 10.1084/jem.20042521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Celli S, Lemaitre F, Bousso P. Real-time manipulation of T cell-dendritic cell interactions in vivo reveals the importance of prolonged contacts for CD4+ T cell activation. Immunity. 2007;27:625–634. doi: 10.1016/j.immuni.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 15.Celli S, Garcia Z, Bousso P. CD4 T cells integrate signals delivered during successive DC encounters in vivo. J Exp Med. 2005;202:1271–1278. doi: 10.1084/jem.20051018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jelley-Gibbs DM, Lepak NM, Yen M, Swain SL. Two distinct stages in the transition from naive CD4 T cells to effectors, early antigen-dependent and late cytokine-driven expansion and differentiation. J Immunol. 2000;165:5017–5026. doi: 10.4049/jimmunol.165.9.5017. [DOI] [PubMed] [Google Scholar]
- 17.Jelley-Gibbs DM, Dibble JP, Filipson S, Haynes L, Kemp RA, Swain SL. Repeated stimulation of CD4 effector T cells can limit their protective function. J Exp Med. 2005;201:1101–1112. doi: 10.1084/jem.20041852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blair DA, Lefrancois L. Increased competition for antigen during priming negatively impacts the generation of memory CD4 T cells. Proc Natl Acad Sci U S A. 2007;104:15045–15050. doi: 10.1073/pnas.0703767104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Badovinac VP, Porter BB, Harty JT. Programmed contraction of CD8(+) T cells after infection. Nat Immunol. 2002;3:619–626. doi: 10.1038/ni804. [DOI] [PubMed] [Google Scholar]
- 20.Badovinac VP, Porter BB, Harty JT. CD8+ T cell contraction is controlled by early inflammation. Nat Immunol. 2004;5:809–817. doi: 10.1038/ni1098. [DOI] [PubMed] [Google Scholar]
- 21.Tseng KE, Chung CY, H’ng WS, Wang SL. Early infection termination affects number of CD8+ memory T cells and protective capacities in listeria monocytogenes-infected mice upon rechallenge. J Immunol. 2009;182:4590–4600. doi: 10.4049/jimmunol.0801125. [DOI] [PubMed] [Google Scholar]
- 22.Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK, Mescher MF. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J Immunol. 1999;162:3256–3262. [PubMed] [Google Scholar]
- 23.Busch DH, Kerksiek KM, Pamer EG. Differing roles of inflammation and antigen in T cell proliferation and memory generation. J Immunol JID - 2985117R. 2000;164:4063–4070. doi: 10.4049/jimmunol.164.8.4063. [DOI] [PubMed] [Google Scholar]
- 24.Badovinac VP, Tvinnereim AR, Harty JT. Regulation of antigen-specific CD8(+) T cell homeostasis by perforin and interferon-gamma. Science. 2000;290:1354–1358. doi: 10.1126/science.290.5495.1354. [DOI] [PubMed] [Google Scholar]
- 25.Haring JS, V, Badovinac P, Harty JT. Inflaming the CD8+ T cell response. Immunity. 2006;25:19–29. doi: 10.1016/j.immuni.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 26.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grubin CE, Kovats S, deRoos P, Rudensky AY. Deficient positive selection of CD4 T cells in mice displaying altered repertoires of MHC class II-bound self-peptides. Immunity. 1997;7:197–208. doi: 10.1016/s1074-7613(00)80523-3. [DOI] [PubMed] [Google Scholar]
- 28.Altman JD, Moss PAH, Goulder PJR, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. doi: 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- 29.Masopust D, Jiang J, Shen H, Lefrançois L. Direct analysis of the dynamics of the intestinal mucosa CD8 T cell response to systemic virus infection. J Immunol. 2001;166:2348–2356. doi: 10.4049/jimmunol.166.4.2348. [DOI] [PubMed] [Google Scholar]
- 30.Lawson ND, Stillman EA, Whitt MA, Rose JK. Recombinant vesicular stomatitis viruses from DNA. Proc Natl Acad Sci USA. 1995;92:4477–4481. doi: 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SK, Reed DS, Olson S, Schnell MJ, Rose JK, Morton PA, Lefrançois L. Generation of mucosal cytotoxic T cells against soluble protein by tissue-specific environmental and costimulatory signals. Proc Natl Acad Sci USA. 1998;95:10814–10819. doi: 10.1073/pnas.95.18.10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Topham DJ, Castrucci MR, Wingo FS, Belz GT, Doherty PC. The role of antigen in the localization of naive, acutely activated, and memory CD8(+) T cells to the lung during influenza pneumonia. J Immunol. 2001;167:6983–6990. doi: 10.4049/jimmunol.167.12.6983. [DOI] [PubMed] [Google Scholar]
- 33.McAleer JP, Zammit DJ, Lefrancois L, Rossi RJ, Vella AT. The lipopolysaccharide adjuvant effect on T cells relies on nonoverlapping contributions from the MyD88 pathway and CD11c+ cells. J Immunol. 2007;179:6524–6535. doi: 10.4049/jimmunol.179.10.6524. [DOI] [PubMed] [Google Scholar]
- 34.Rudensky AY, Rath S, Preston-Hurlburt P, Murphy DB, Janeway CA., Jr On the complexity of self. Nature. 1991;353:660–662. doi: 10.1038/353660a0. [DOI] [PubMed] [Google Scholar]
- 35.Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity. 1997;6:715–726. doi: 10.1016/s1074-7613(00)80447-1. [DOI] [PubMed] [Google Scholar]
- 36.Murphy DB, Rath S, Pizzo E, Rudensky AY, George A, Larson JK, Janeway CA., Jr Monoclonal antibody detection of a major self peptide. MHC class II complex. J Immunol. 1992;148:3483–3491. [PubMed] [Google Scholar]
- 37.Lefrancois L. Protection against lethal viral infection by neutralizing and nonneutralizing monoclonal antibodies: distinct mechanisms of action in vivo. J Virol. 1984;51:208–214. doi: 10.1128/jvi.51.1.208-214.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gett AV, Sallusto F, Lanzavecchia A, Geginat J. T cell fitness determined by signal strength. Nat Immunol. 2003;4:355–360. doi: 10.1038/ni908. [DOI] [PubMed] [Google Scholar]
- 39.Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273:104–106. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 40.Henrickson SE, Mempel TR, Mazo IB, Liu B, Artyomov MN, Zheng H, Peixoto A, Flynn MP, Senman B, Junt T, Wong HC, Chakraborty AK, von Andrian UH. T cell sensing of antigen dose governs interactive behavior with dendritic cells and sets a threshold for T cell activation. Nat Immunol. 2008;9:282–291. doi: 10.1038/ni1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature. 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- 42.Rees W, Bender J, Teague TK, Kedl RM, Crawford F, Marrack P, Kappler J. An inverse relationship between T cell receptor affinity and antigen dose during CD4(+) T cell responses in vivo and in vitro. Proc Natl Acad Sci U S A. 1999;96:9781–9786. doi: 10.1073/pnas.96.17.9781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature. 2009;458:211–214. doi: 10.1038/nature07657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tuosto L, Acuto O. CD28 affects the earliest signaling events generated by TCR engagement. Eur J Immunol. 1998;28:2131–2142. doi: 10.1002/(SICI)1521-4141(199807)28:07<2131::AID-IMMU2131>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 45.Sarkar S, Teichgraber V, Kalia V, Polley A, Masopust D, Harrington LE, Ahmed R, Wherry EJ. Strength of stimulus and clonal competition impact the rate of memory CD8 T cell differentiation. J Immunol. 2007;179:6704–6714. doi: 10.4049/jimmunol.179.10.6704. [DOI] [PubMed] [Google Scholar]
- 46.La Gruta NL, Rothwell WT, Cukalac T, Swan NG, Valkenburg SA, Kedzierska K, Thomas PG, Doherty PC, Turner SJ. Primary CTL response magnitude in mice is determined by the extent of naive T cell recruitment and subsequent clonal expansion. J Clin Invest. 2010;120:1885–1894. doi: 10.1172/JCI41538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Obar JJ, Lefrancois L. Early signals during CD8 T cell priming regulate the generation of central memory cells. J Immunol. 2010;185:263–272. doi: 10.4049/jimmunol.1000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dolfi DV, Boesteanu AC, Petrovas C, Xia D, Butz EA, Katsikis PD. Late signals from CD27 prevent Fas-dependent apoptosis of primary CD8+ T cells. J Immunol. 2008;180:2912–2921. doi: 10.4049/jimmunol.180.5.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stock AT, Jones CM, Heath WR, Carbone FR. Cutting edge: central memory T cells do not show accelerated proliferation or tissue infiltration in response to localized herpes simplex virus-1 infection. J Immunol. 2006;177:1411–1415. doi: 10.4049/jimmunol.177.3.1411. [DOI] [PubMed] [Google Scholar]
- 50.Bachmann MF, Gallimore A, Linkert S, Cerundolo V, Lanzavecchia A, Kopf M, Viola A. Developmental regulation of Lck targeting to the CD8 coreceptor controls signaling in naive and memory T cells. J Exp Med. 1999;10:1521–1530. doi: 10.1084/jem.189.10.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chandok MR, Farber DL. Signaling control of memory T cell generation and function. Semin Immunol. 2004;16:285–293. doi: 10.1016/j.smim.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 52.Hussain SF, Anderson CF, Farber DL. Differential SLP-76 expression and TCR-mediated signaling in effector and memory CD4 T cells. J Immunol. 2002;168:1557–1565. doi: 10.4049/jimmunol.168.4.1557. [DOI] [PubMed] [Google Scholar]
- 53.Borowski AB, Boesteanu AC, Mueller YM, Carafides C, Topham DJ, Altman JD, Jennings SR, Katsikis PD. Memory CD8+ T cells require CD28 costimulation. J Immunol. 2007;179:6494–6503. doi: 10.4049/jimmunol.179.10.6494. [DOI] [PubMed] [Google Scholar]
- 54.Ndejembi MP, Teijaro JR, Patke DS, Bingaman AW, Chandok MR, Azimzadeh A, Nadler SG, Farber DL. Control of memory CD4 T cell recall by the CD28/B7 costimulatory pathway. J Immunol. 2006;177:7698–7706. doi: 10.4049/jimmunol.177.11.7698. [DOI] [PubMed] [Google Scholar]
- 55.Marzo AL, Vezys V, Klonowski KD, Lee SJ, Muralimohan G, Moore M, Tough DF, Lefrancois L. Fully functional memory CD8 T cells in the absence of CD4 T cells. J Immunol. 2004;173:969–975. doi: 10.4049/jimmunol.173.2.969. [DOI] [PubMed] [Google Scholar]
- 56.Belz GT, Bedoui S, Kupresanin F, Carbone FR, Heath WR. Minimal activation of memory CD8+ T cell by tissue-derived dendritic cells favors the stimulation of naive CD8+ T cells. Nat Immunol. 2007;8:1060–1066. doi: 10.1038/ni1505. [DOI] [PubMed] [Google Scholar]
- 57.Busch DH, Pamer EG. T cell affinity maturation by selective expansion during infection. J Exp Med. 1999;189:701–710. doi: 10.1084/jem.189.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turner MJ, Jellison ER, Lingenheld EG, Puddington L, Lefrancois L. Avidity maturation of memory CD8 T cells is limited by self-antigen expression. J Exp Med. 2008;205:1859–1868. doi: 10.1084/jem.20072390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Slifka MK, Whitton JL. Functional avidity maturation of CD8(+) T cells without selection of higher affinity TCR. Nat Immunol. 2001;2:711–717. doi: 10.1038/90650. [DOI] [PubMed] [Google Scholar]
- 60.MacLeod MK, McKee A, Crawford F, White J, Kappler J, Marrack P. CD4 memory T cells divide poorly in response to antigen because of their cytokine profile. Proc Natl Acad Sci U S A. 2008;105:14521–14526. doi: 10.1073/pnas.0807449105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Han S, Asoyan A, Rabenstein H, Nakano N, Obst R. Role of antigen persistence and dose for CD4+ T-cell exhaustion and recovery. Proc Natl Acad Sci U S A. 2010;107:20453–20458. doi: 10.1073/pnas.1008437107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lanzavecchia A, Sallusto F. Progressive differentiation and selection of the fittest in the immune response. Nat Rev Immunol. 2002;2:982–987. doi: 10.1038/nri959. [DOI] [PubMed] [Google Scholar]
- 63.Mempel TR, Henrickson SE, von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- 64.Celli S, Garcia Z, Bousso P. CD4 T cells integrate signals delivered during successive DC encounters in vivo. J Exp Med. 2005;202:1271–1278. doi: 10.1084/jem.20051018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khanna KM, McNamara JT, Lefrancois L. In situ imaging of the endogenous CD8 T cell response to infection. Science. 2007;318:116–120. doi: 10.1126/science.1146291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Legge KL, Braciale TJ. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity. 2003;18:265–277. doi: 10.1016/s1074-7613(03)00023-2. [DOI] [PubMed] [Google Scholar]
- 67.Belz GT, Smith CM, Kleinert L, Reading P, Brooks A, Shortman K, Carbone FR, Heath WR. Distinct migrating and nonmigrating dendritic cell populations are involved in MHC class I-restricted antigen presentation after lung infection with virus. Proc Natl Acad Sci U S A. 2004;101:8670–8675. doi: 10.1073/pnas.0402644101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim TS, Braciale TJ. Respiratory dendritic cell subsets differ in their capacity to support the induction of virus-specific cytotoxic CD8+ T cell responses. PLoS One. 2009;4:e4204. doi: 10.1371/journal.pone.0004204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ballesteros-Tato A, Leon B, Lund FE, Randall TD. Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8(+) T cell responses to influenza. Nat Immunol. 2010;11:216–224. doi: 10.1038/ni.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Suarez-Ramirez JE, Wu T, Lee YT, Aguila CC, Bouchard KR, Cauley LS. Division of labor between subsets of lymph node dendritic cells determines the specificity of the CD8 recall response to influenza infection. Eur J Immunol. 2011 doi: 10.1002/eji.201141546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hufford MM, Kim TS, Sun J, Braciale TJ. Antiviral CD8+ T cell effector activities in situ are regulated by target cell type. J Exp Med. 2011;208:167–180. doi: 10.1084/jem.20101850. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.