

Abstract
Recent evidence suggests an innovative application of chemical modulators targeting the S1P4 receptor as novel mechanism-based drugs for the treatment of influenza virus infection. Modulation of the S1P4 receptor may also represent an alternative therapeutic approach for clinical conditions where reactive thrombocytosis is an undesired effect or increased megakaryopoiesis is required. With the exception of our recent research program disclosure, we are not aware of any selective S1P4 antagonists reported in the literature to date. Herein, we describe complementary structure-activity relationships (SAR) of the high-throughput screening (HTS)-derived hit 5-(2,5-dichlorophenyl)-N-(2,6-dimethylphenyl)furan-2-carboxamide and its 2,5-dimethylphenyl analog. Systematic structural modifications of the furan ring showed that both steric and electronic factors in this region have a significant impact on the potency. The furan moiety was successfully replaced with a thiophene or phenyl ring maintaining potency in the low nanomolar range and high selectivity against the other S1P receptor subtypes. By expanding the molecular diversity within the hit-derived class, our SAR study provides innovative small molecule potent and selective S1P4 antagonists suitable for in vivo pharmacological validation of the target receptor.
Keywords: S1P4 receptor antagonists; S1P1-3,5 receptor family; megakaryocyte differentiation; viral infections
Sphingosine-1-phosphate (S1P) is a pleiotropic lysophospholipid involved in a wide range of cellular functions including proliferation, apoptosis, differentiation and migration.1–6 High levels of S1P are present in lymph (30 nM to 300 nM) and plasma (100 nM to 1 μM), whereas those found in tissues are significantly lower.7 Although platelets possess the highest sphingosine kinase activity, accumulating evidence suggests that the plasma levels of S1P are mainly the result of its release from erythrocytes due to their great number, capability to store S1P produced from other tissues, and lack of S1P-degrading enzymes. Furthermore, mast cells have been described as additional vascular source of S1P upon IgE dependent activation, while the vascular endothelium could be the main source of lymph S1P.7,8
S1P can function intracellularly as a second messenger or extracellularly by binding with nanomolar affinities to five S1P G-protein coupled receptor subtypes named S1P1,2,3,4,5 (formerly Edg1,5,3,6,8 receptors).9–10 S1P1–5 members share approximately 40% sequence identity, with a conserved glutamic acid residue at the third transmenbrane domain responsible for the ligand specificity versus the closely related lysophosphatidic acid (LPA) Edg2,4,7 receptors family.11 The coupling of S1P1–5 to different signaling cascades along with their differential expression pattern explains the broad biological effects of S1P. S1P1 is expressed ubiquitously, particularly in brain, lung, spleen, heart, liver as well in a variety of immune system cells lineages.7 High affinity S1P1 agonists have been widely studied and emerged as major immunomodulatory molecules with therapeutic applications being tested in multiple sclerosis and allogenic transplantation.12 S1P3 is also widely distributed with highest levels in the heart where it is co-expressed with S1P1 and S1P2. S1P2 has been shown to be expressed in rat brain during embryogenesis as well as in most other developing tissues. S1P5 is highly present in adult rat brain, while in human and mouse high expression of the receptor is also found in the spleen. 13
S1P4 has been shown to bind S1P with lower affinity and have a narrower tissue distribution than the other family members. First isolated from human and mouse dendritic cells (DCs), S1P4 is highly expressed in lymphoid and hematopoietic tissue.13 S1P4 have been reported to couple to Gαi, Gαo and Gα12/13 proteins leading to the stimulation of MAPK/ERK signaling pathways, as well as PLC and Rho-Cdc42 activation.14–15 Molecules targeting S1P-metabolizing enzymes have been recently proposed as innovative potential therapeutics for viral diseases.1,12a,16 Consistent with these data, local S1P receptor modulation in the lung has been demonstrated to control immunopathological features of influenza virus infections by impairing the accumulation of DCs and cytokine release in the draining lymph nodes without altering the essential activity of virus-specific T-cells toward virus-infected cells.12a Therefore, regulation of pulmonary immune response by S1P receptor modulators may have therapeutic implications for alleviating excessive immune response responsible for exacerbating airway diseases. Based on the evidence that modulation of S1P1 alone did not inhibit DC-dependent T cell activation, and that the sphingosine analog used in the experiments did not bind to S1P2, it was hypothesized that either the single activation of S1P3, S1P4, S1P5 or the combined activity on S1P1,3,4,5 is responsible for the functional impairment of DCs.12a Reports showing that, in contrast to S1P5 and S1P2, S1P4 is highly expressed in DCs10 confirm that the S1P4 chemical activation in the airway may be effective at controlling the immunopathological response to viral infections, thus offering novel mechanism-based potential therapeutics for airway viral diseases.
Both in vitro and in vivo experiments have recently provided strong evidence that S1P4 is involved in the late stage of megakaryocyte differentiation. In S1P4–deficient mice the bone marrow is characterized by the presence of morphologically aberrant megakaryocytes, and platelet repopulation of the peripheral blood after thrombocytopenia is delayed. Indeed, S1P4 has been proposed as a suitable target either for increasing thrombocyte production in clinical conditions requiring increased platelets number, or for inhibiting a potentially detrimental reactive thrombocytosis.8
Despite the emerging therapeutic potential, aspects of the biological role of S1P4 remain unclear, partly due to the lack of ligands with high selectivity against the S1P1–3,5 subtypes. Herein we report on the synthesis, biological evaluation and structure-activity relationships (SAR) of the first class of selective S1P4 antagonists.
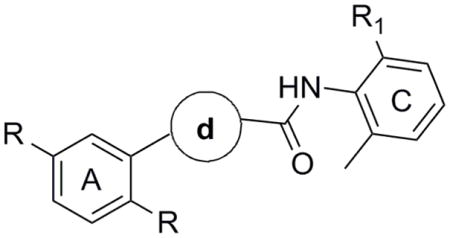
Recently, investigations from our laboratories have led to the discovery of the first class of potent and selective S1P4 antagonists.17 Synthesis and SAR analysis of various derivatives based on a 5-aryl furan-2-arylcarboxamide scaffold were carried out on regions A and C of the original hit 1a identified through a high-throughput screening campaign (Figure 1, Table 1). Similar biological properties were found for the 2,5-dimethylphenyl analog 1b (Figure 1). It was postulated that disubstitution on positions 2 and 6 of the phenyl ring C with small alkyl groups (e.g. methyl, ethyl) was essential to increase the potency. Remarkably, steric and electronic effects at position 4 of the phenyl ring C did not affect the functional activity to any appreciable extent, thus allowing the installation of solubility enhancing features such as alcohols and amines. However, safety concerns might arise from the presence of the furan ring given the number of furan-containing drug candidates demonstrating hepatotoxic and hepatocarcinogenic effects as a result of furan cytochrome P450-catalyzed oxidative metabolism and the covalent binding of the electrophilic metabolites to macromolecules.18 Thus, our chemistry efforts were successively focused on the SAR analysis of the central moiety B with the aim to acquire more insight into the receptor binding mode and identify new chemotypes to address potential metabolic and toxicity issues. For investigational purposes we fragmented the moiety B into aryl ring d and amide group e (Figure 1) while conserving regions A and C as in 1a and 1b.
Figure 1.

Novel S1P4 antagonists based on a 5-aryl furan-2-arylcarboxamide scaffold
Table 1.
S1P4 antagonist activity of synthesized molecules
 | ||||
|---|---|---|---|---|
| cpd | R | d | R1 | IC50 (nM)α |
| 1a | Cl |
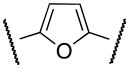
|
Me | 78 |
| 1b | Me |
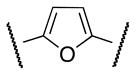
|
Me | 64 |
| 9 | Cl |
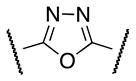
|
Et | NA |
| 14 | Me |
|
Me | 6300 |
| 19 | Me |
|
Me | 59 |
| 23 | Me |
|
Me | 233 |
| 28 | Me |
|
Me | 211 |
| 33 | Me |
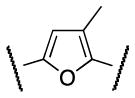
|
Me | 173 |
| 37 | Me |
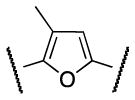
|
Me | 911 |
| 47 | Me |
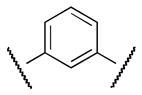
|
Me | 75 |
| 48 | Me |
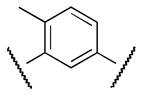
|
Me | 2400 |
| 49 | Cl |
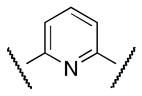
|
Me | NA |
Data are reported as mean for n = 3 determinations. NA = not active at concentrations up to 25 μM.
In our previous studies17 it was hypothesized that the hydrogen-bond donor capability of the amide group was important for the functional activity. Herein, the impact of the carbonyl group as hydrogen-bond acceptor was explored by replacing the amide functionality by a methyleneaniline as in 5. Suzuki coupling19 between bromide 2 and boronic acid 3 followed by reductive amination with 2,6-dimethylaniline 4 furnished the final compound 5 (Scheme 1).
Scheme 1. Synthesis of aniline derivative 5.

Reagents and conditions: (i) Pd(PPh3)4 (0.1 equiv), 3 (1.5 equiv), 2M aq. Na2C03 (2 equiv), 1,4-dioxane, 80 °C. 12 h, 65%; (ii) 4 (1.2 equiv), NaBH(OAc)3 (1.3 equiv), AcOH (1.5 equiv), DCE, rt, overnight, 75%.
Notably, 5 was found to be inactive in the S1P4 antagonist assay,20 indicating that the amide function is an essential molecular feature in the binding mode.
A systematic SAR analysis of the furan ring d was performed by the synthesis of molecules containing variously substituted aromatic rings as outlined in Schemes 2–9. Initially, the furan ring was replaced by 1,3,4-oxadiazole as a substructure with potentially improved toxicity profile18 (9, Scheme 2). Condensation of hydrazide 6 with ethyl oxalyl chloride 7 followed by cyclization using Burgess reagent afforded oxadiazole ethyl ester 8. Subsequent ester hydrolysis of 8 followed by amidation with 4 furnished the final compound 9.
Scheme 2. Synthesis of 1,3,4-oxadiazole derivative 9.

Reagents and conditions: (i) 7 (1.0 equiv), Et3N (2 equiv), CH2Cl2, 0 °C, 1 h; (ii) Burgess reagent (1.5 equiv), THF, 80 °C, 10 h, 40% (over 2 steps); (iii) 2N aq. NaOH, MeOH, rt, 2 h; (iv) 4 (1.2 equiv), EDCl (1.2 equiv), HOBt (1.2 equiv), DMF, rt, overnight, 70 % (over 2 steps).
Scheme 9. Synthesis of phenyl and pyridine analogs 47–49.

Reagents and conditions: (i) 3 or 16 (1.5 equiv), Pd(OAc)2 (0.05 equiv), Cy3P (0.1 equiv), K3PO4 (3 equiv), toluene/H2O (8:1), 100 °C, 12 h. 40–75%: (ii) LiOH (1.3 equiv), THF/MeOH/H2O (2: 2:1). rt, 3 h;(iii) (a) (COCl)2 (1.3 equiv), DMF, CH2Cl2, rt, 6 h; (b) Et3N (2 equiv), 4 (2 equiv), CH2Cl2, rt, overnight, 60–85% (over 3 steps).
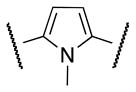
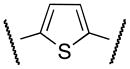
Aware of literature evidence of pyrrole and thiophene toxicity which mostly arises from hydroxylation at C2 and C5 positions,18 N-methyl pyrrole 14 and thiophene 19 analogs were prepared (Schemes 3, 4) primarily to elucidate the electronic effects of the oxygen atom on the functional activity. Suzuki coupling of phenyl iodide 10 with N-Boc-pyrrole boronic acid 11 furnished the N-deprotected pyrrole 12 as the major product.19 Alkylation of 12 with methyl iodide under strong basic conditions followed by hydrolysis of the methyl ester afforded carboxylic acid 13. Coupling the acyl chloride of 13 with 4 furnished compound 14.
Scheme 3. Synthesis of N-methyl pyrrole analog 14.

Reagents and conditions: (i) Pd(PPh3)4 (0.05 equiv), Na2CO3 (2 equiv), 1,4-dioxane/H2O (10:1), 100 °C. 4 h, 50%: (ii) (a) Mel (1.5 equiv), NaH (1.5 equiv), DMF, 0 °C to rt, overnight, 65%: (b) LiOH (1.3 equiv), THF/MeOH/H2O (2: 2: 1), rt, 2 h;(iii) (a) (COCl)2 (1.3 equiv), DMF, CH2Cl2, 0 °C to rt, 8 h; (b) Et3N (2 equiv), 4 (2 equiv), CH2Cl2, 0 °C, 2 h, 50% (over 3 steps).
Scheme 4. Synthesis of thiophene analog 19.

Reagents and conditions: (i) (a) conc. H2SO4, MeOH, reflux, 12 h; (b) Pd(OAc)2 (0.05 equiv), Cy3P (0.1 equiv), 16 (1.4 equiv), K3PO4 (3 equiv), toluene/H2O (8:1), 100 °C, 12 h, 50% (over 2 steps): (ii) 1M aq. LiOH (1 equiv), THF, 24 h, 25 °C; (iii) (a) (COCl)2 (1.3 equiv), DMF, CH2Cl2, rt, 8 h; (b) Et3N (2 equiv), 4 (1.2 equiv), CH2Cl2, rt, overnight, 75% (over 3 steps).
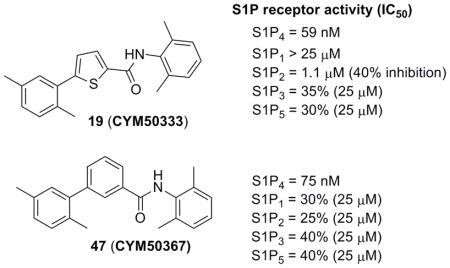
The synthesis of intermediate 17 commenced with the esterification of carboxylic acid 15 followed by Suzuki coupling with 2,5-dimethylphenyl boronic acid 16. Ester hydrolysis of 17, formation of the acyl chloride and coupling with 4 furnished thiophene derivative 19 (CYM50333) (Scheme 4).
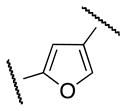
As regioisomers of 1b, 5-aryl furan-3-carboxamide 23 and 4-aryl furan-2-carboxamide 28 were synthesized in order to elucidate the electronic requirements of the 5-membered ring d, while keeping the pendant regions A and C geometrically similar to 1a and 1b. The synthesis of 23 is depicted in Scheme 5. Esterification of 20 followed by Suzuki coupling with boronic acid 16 afforded 22. Subsequent ester hydrolysis, formation of acyl chloride and coupling with 4 provided the desired compound 23.
Scheme 5. Synthesis of 5-aryl furan-3-carboxamide regioisomer 23.

Reagents and conditions: (i) conc. H2SO4, MeOH, reflux, 12h, 60%; (ii) 16 (1.5 equiv), Pd(OAc)2 (0.05 equiv). Cy3P (0.1 equiv), K3PO4 (3 equiv), toluene/H2O (8:1), 100 °C, 12 h, overnight, 78%; (iii) LiOH (1.2 equiv), THF/MeOH/H2O (2: 2:1), rt. 3 h; (iv) (a) (COCl)2. DMF, CH2Cl2, rt, 8 h; (b) 4 (1.2 equiv), Et3N(1.5 equiv), rt, overnight, 88% (over 3 steps).
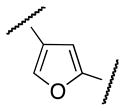
The 4-arylfuran-2-carboxamide 28 was synthesized as shown in Scheme 6. Selective debromination of 24 followed by esterification and Suzuki coupling afforded the 4-arylfuran-2-methyl ester 27. Synthesis of 28 was finally achieved by ester hydrolysis of 27, formation of acyl chloride and subsequent coupling with aniline 4.
Scheme 6. Synthesis of 4-aryl furan-2-carboxamide regioisomer 28.

Reagents and conditions: (i) aq. NH4OH, Zn (1 equiv), rt, 3 h, 80%; (ii) conc. H2SO4, MeOH, reflux, 12 h, 90%; (iii) 16 (1.4 equiv), Pd(OAc)2 (0.05 equiv), Cy3P (0.1 equiv), K3PO4 (3 equiv), toluene/H2O (8:1), 100 °C, 12 h, 60%; (iv) LiOH (1.3 equiv), THF/MeOH/H2O (2: 2:1), rt, 3 h;(v) (a) (COCl)2 (1.3 equiv), DMF, CH2Cl2, rt, overnight; (b) Et3N (2.0 equiv), 4 (1.4 equiv), CH2Cl2, rt, 2 h, 85% (over 3 steps).
Successively, a methyl group was inserted either on position 3 or 4 of the furan ring in order to evaluate the steric requirements of region B as well as conformational restrictions on the pendant regions A and C. Trisubstituted furans 33 and 37 were synthesized as illustrated in Scheme 7 and 8. Bromination of furan 29, followed by Suzuki coupling with boronic acid 16 afforded ester 31. Ester hydrolysis followed by acyl chloride preparation and amide coupling with 4 yielded the desired furan 33 (Scheme 7).
Scheme 7. Synthesis of 2,3,5-trisubstituted furan analog 33.

Reagents and conditions: (i) NBS (1.1 equiv), MeOH/THF, 0 °C to rt, 2 h, 60%; (ii) Pd(OAc)2 (0.05 equiv), Cy3P (0.01 equiv), 16 (1.4 equiv), K3PO4 (3 equiv), toluene/H2O (8:1), 100 °C, 12 h, 50%; (iii) LiOH (1.3 equiv), THF/MeOH/H2O (2: 2:1), rt, 3 h; (iv) (a) (COCl)2 (1.3 equiv), DMF, CH2Cl2, rt, overnight: (b) Et3N (2 equiv), 4 (1.5 equiv), CH2Cl2, rt, overnight, 80% ( over 3 steps).
Scheme 8. Synthesis of 2,4,5-trisubstituted furan analog 37.

Reagents and conditions: (i) Pd(OAc)2 (0.05 equiv), Cy3P (0.1 equiv), 16 (1.4 equiv), K3PO4 (3 equiv), toluene/H2O (8:1). 100 °C, 12 h. 40%; (ii) Pd[(Po-Tol)3]2Cl2 (0.05 equiv). Me4Sn (2 equiv), DMA, 90 °C, 6 h, 65%; (iii) LiOH (1.2 equiv), THF/MeOH/H2O (2: 2:1); (iv) (a) (COCl)2 (1.3 equiv), DMF, CH2Cl2, rt, 2 h; (b) Et3N (2 equiv), 4 (2 equiv), rt, overnight, 78% (over 3 steps).
Synthesis of the regioisomer 37 involved a selective Suzuki coupling between dibromo furan 34 and boronic acid 16 followed by methylation through Stille coupling,21 hydrolysis of ester 36, and coupling of the acyl chloride derivative with aniline 4 (Scheme 8).
Finally, new chemical space was explored by replacing the furan ring with phenyl and pyridine as potential 6-membered ring bioisosteres. The synthesis is depicted in Scheme 9. Suzuki coupling of phenylboronic acids (3 or 16) with the appropriate bromo phenyl (38 or 39) or pyridine 40 derivatives afforded the corresponding biaryl esters 41–43. Successively, ester hydrolysis, acyl chloride formation and amide coupling with aniline 4 furnished the desired products 47 (CYM50367), 48, 49.
The S1P4 functional antagonist activity of the aforementioned compounds is listed in Table 1. Surprisingly, the 2,5-disubstituted oxadiazole 9 was inactive. Similarly, the N-methyl-pyrrole 14 was 98-fold less potent than 1b. Conversely, the thiophene was found to be a suitable bioisoster of the furan ring, with the 2,5-disubstituted thiophene analog 19 displaying similar potency to the hit and 1b. Furthermore, both regioisomers 23 and 28 were 3-fold less potent than 1b. Trisubstituted furan regioisomers 33 and 37 were respectively 3- and 14-fold less potent than 1b; the loss of potency was attributed to unfavorable steric interactions of the methyl group with the receptor or resulting conformational modification of the molecule. Particularly, it was hypothesized that the methyl in 37 forces the biaryl C-C bond angle into a less active anti-coplanar conformation. Geometrically similar molecules 9, 14, 19, 23 and 28, having the amide group e and the ring A arranged in a 1,3 substitution pattern on the heteroaromatic central system d, displayed very different potencies suggesting that electronic effects are essential for the S1P4 antagonist activity. Interestingly, the 1,3-disubstituted phenyl derivative 47 showed similar potency to 19, 1a and 1b, whereas the pyridine analog 49 was inactive. The lack of activity of oxadiazole 9 and pyridine 49 suggested that heterocycles with basic lone pares at the ring d are detrimental to the potency. In agreement with the SAR trend within the furan derivatives, the methylated phenyl analog 48 was 32-fold less potent than the corresponding homolog 47, supporting the hypothesis that a methyl group in the ring d ortho to the biaryl C-C bond is detrimental to the activity.
The most active compounds 19 and 47 were selected for functional assays at S1P1–3,5 subtypes (Table 2). Notably, both compounds displayed an exquisite selectivity for the S1P4 receptor versus the other receptor family subtypes.
Table 2.
S1P selectivity counter screen of compounds 19, 47
| cpd | IC50 (nM)a | ||||
|---|---|---|---|---|---|
| S1P4 | S1P1 | S1P2 | S1P3 | S1P5 | |
| 19 (CYM50333) | 59 | NA | 1100 (40%)b | 35%c | 30%c |
| 47 (CYM50367) | 75 | 30%c | 25%c | 40%c | 40%c |
Data are reported as mean for n = 3 determinations.
Percentage of inhibition.
Percentage of inhibition at 25μM. NA = not active at concentrations up to 25 μM.
In summary, a systematic SAR analysis was carried out on region B of the original hit 1a and its analog 1b. Structural modifications of the central ring d showed that electronic effects are fundamental for the activity, and revealed that a methyl substitution ortho to the C-C biaryl bond has a negative impact on the activity, perhaps by forcing the biaryl system into an anti-coplanar conformation. Interestingly, both thiophene and phenyl rings were found to be bioisosteres of the furan moiety, thus expanding the molecular diversity within the hit-derived molecules and including new chemotypes which may address potential metabolic/toxicity issues. Remarkably, compounds 19 (CYM50333) and 47 (CYM50367), as novel highly selective S1P4 antagonists with low nanomolar activity, represent valuable small molecule tools to investigate the biological and pharmacological role of the target receptor in megakaryocyte differentiation and fundamental immune processes. Based on the acquired SAR of the explored regions (A, B, C),17 additional studies are currently ongoing in our research group to improve the physicochemical properties and further increase the S1P4 antagonist potency, while preserving the S1P1–3,5 selectivity profile. Our research progresses will be communicated in due course.
Acknowledgments
This work was supported by the National Institute of Health Molecular Library Probe Production Center grant U54 MH084512 (ER, HR), and by AI074564 (Michael Oldstone, PI). We thank Mark Southern for data management with Pub Chem.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Marsolais D, Hahm B, Walsh KB, Edelmann KH, McGavern D, Hatta Y, Kawaoka Y, Rosen H, Oldstone MB. Proc Natl Acad Sci USA. 2009;106(5):1560. doi: 10.1073/pnas.0812689106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marsolais D, Rosen H. Nat Rev Drug Discov. 2009;8:297. doi: 10.1038/nrd2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alfonso C, McHeyzer-Williams MG, Rosen H. Eur J Immunol. 2006;36:149. doi: 10.1002/eji.200535127. [DOI] [PubMed] [Google Scholar]
- 4.Jo E, Sanna MG, Gonzalez-Cabrera PJ, Thangada S, Tigyi G, Osborne DA, Hla T, Parrill AL, Rosen H. Chem Biol. 2005;12:703. doi: 10.1016/j.chembiol.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 5.Sanna G, Liao J, Jo E, Alfonso C, Ahn M, Peterson MS, Webb B, Lefebvre S, Chun J, Gray N, Rosen H. J Biol Chem. 2004;279:13839. doi: 10.1074/jbc.M311743200. [DOI] [PubMed] [Google Scholar]
- 6.Wei SH, Rosen H, Matheu MP, Sanna MG, Wang S, Jo E, Wong C, Parker I, Cahalan M. Nat Immunol. 2005;6:1228. doi: 10.1038/ni1269. [DOI] [PubMed] [Google Scholar]
- 7.López Almagro R, Tarrasón G, Godessart N. Structure, Signaling, And Physiology. Cambridge University Press; Cambridge: 2011. Protein-Coupled Receptors. [Google Scholar]
- 8.Golfier S, Kondo S, Schulze T, Takeuchi T, Vassileva G, Achtman AH, Gräler MH, Abbondanzo SJ, Wiekowski M, Kremmer E, Endo Y, Lira SA, Bacon KB, Lipp M. FASEB J. 2010;24:4701. doi: 10.1096/fj.09-141473. [DOI] [PubMed] [Google Scholar]
- 9.Chun J, Goetzl EJ, Hla T, Igarashi Y, Lynch KR, Moolenaar W, Pyne S, Tigyi G. Pharmacol Rev. 2002;54:265. doi: 10.1124/pr.54.2.265. [DOI] [PubMed] [Google Scholar]
- 10.Maeda Y, Matsuyuki H, Shimano K, Kataoka H, Sugahara K, Kenji Chiba K. J Immunol. 2007;178:3437. doi: 10.4049/jimmunol.178.6.3437. [DOI] [PubMed] [Google Scholar]
- 11.Holdsworth G, Osborne DA, Pham TCT, Fells JI, Hutchinson G, Milligan G, Parrill AL. BMC Biochemistry. 2004;5:12. doi: 10.1186/1471-2091-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Marsolais D, Hahm B, Edelmann KH, Walsh KB, Guerrero M, Hatta Y, Kawaoka Y, Roberts E, Oldstone MB, Rosen H. Mol Pharmacol. 2008;74:896. doi: 10.1124/mol.108.048769. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder GF, Aradhye PB. Nat Rev Drug Discov. 2010;9:883. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- 13.Candelore MR, Wright MJ, Tota LM, Milligan J, Shei GJ, Bergstrom JD, Mandala SM. Biochem Biophys Res Commun. 2002;297:600. doi: 10.1016/s0006-291x(02)02237-4. [DOI] [PubMed] [Google Scholar]
- 14.Toman RE, Spiegel S. Neurochem Res. 2002;27:619. doi: 10.1023/a:1020219915922. [DOI] [PubMed] [Google Scholar]
- 15.Kohno T, Matsuyuki H, Inagaki Y, Igarashi Y. Genes Cells. 2003;8:685. doi: 10.1046/j.1365-2443.2003.00667.x. [DOI] [PubMed] [Google Scholar]
- 16.Seo YJ, Blake C, Alexander S, Hahm B. J Virol. 2010;84:8124. doi: 10.1128/JVI.00510-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guerrero M, Urbano M, Velaparthi S, Zhao J, Schaeffer M-T, Brown S, Rosen H, Roberts E. Bioorg Med Chem Lett. 2011 doi: 10.1016/j.bmcl.2011.04.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dalvie DK, Kalguktar AS, Khojasteh-Bakht SC, Obach RS, O’Donnell JP. Chem Res Toxicol. 2002;15:269. doi: 10.1021/tx015574b. [DOI] [PubMed] [Google Scholar]
- 19.McAllister LA, Hixon MS, Kennedy KP, Dickerson TJ, Janda KD. J Am Chem Soc. 2006;128:4176. doi: 10.1021/ja057699z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The activity was measured by using Tango™ EDG6-bla U2OS cells which contain the human Endothelial Differentiation Gene 6 linked to a GAL4-VP16 transcription factor via a TEV protease site. The cells also express a beta-arrestin/TEV protease fusion protein and a beta-lactamase (BLA) reporter gene under the control of a UAS response element. BLA expression is monitored by measuring fluorescence resonance energy transfer (FRET) of a cleavable, fluorogenic, cell-permeable BLA substrate.
- 21.Bach T, Krüger L. Eur J Org Chem. 1999;1999:2045. [Google Scholar]