

Abstract
Introduction
Previously, we demonstrated that inhibition of poly(ADP-ribose) polymerase (PARP) exerts protective effects against high-fat (HF) diet-induced atherogenesis, in part, by increasing tissue inhibitor of metalloproteinase (TIMP)-2 expression. Given that characteristics of dilated cardiomyopathy closely associate with atherosclerosis and are mediated by an imbalance between matrix metalloproteinases (MMPs) and TIMPs, we hypothesized that PARP-1 gene deletion may protect against HF-induced cardiac hypertrophy and dilatations by altering TIMP-2/MMPs balance in favor of a maintenance of tissue homeostasis.
Methods and Results
Hemodynamic parameters determined by echocardiography were similar in ApoE−/− mice and PARP-1-deficient ApoE−/− mice (DKO) fed a regular-diet (RD). However, histological analysis revealed that cardiomyocytes of ApoE−/− mice on RD were hypertrophied displaying an enlarged cell body and nucleus, traits that were absent in DKO animals. HF diet-fed ApoE−/− mice exhibited increased interventricular septum, left ventricular (LV) internal dimension, LV volume, and LV mass in addition to a separation of myocardial fibers suggestive of dilated cardiomyopathy. PARP-1 gene deletion protected against these degenerative changes. MMP activity was dramatically increased in hearts of ApoE−/− mice on HF diet and was accompanied by increased collagen degradation, mast cell degranulation and increased myocyte cell death. PARP-1 gene-knockout was associated with increased TIMP-2 expression antagonizing, as a result, the damaging effects of active MMPs.
Conclusions
The present study demonstrates that PARP-1 gene deletion exerts protective effects against HF diet-induced dilated cardiomyopathy by maintaining increased expression of TIMP-2. With additional protective effects against cell death and inflammation, PARP-1 deficiency preserves cardiac tissue homeostasis.
Keywords: PARP, echocardiography, matrix metalloproteinases, mast cell, cell death
Introduction
Cardiac hypertrophy starts as an adaptive response of the heart against hemodynamic overload induced by stress and/or other physiological factors such as exercise 1, 2. The deleterious progression of cardiac hypertrophy to dilatations and ultimately heart failure is very complex and involves activation of numerous secondary pathways including activation of matrix metalloproteinases (MMPs), TNF, signaling, and apoptotic pathways as well as activation of poly(ADP-ribose) polymerase (PARP) 3-6.
PARP-1 is a major member of the PARP protein family, whose function has been classically associated with sensing DNA lesions in response to DNA damaging agents 7. Oxidative and nitrosative stress triggers the activation of PARP, which contributes to the pathogenesis of various cardiovascular diseases 8-11. Because of its active participation in chronic inflammation, PARP has been suggested to play an active role in progression of atherosclerotic plaques 8-12. In previous studies, we reported that PARP inhibition provides stability to atherosclerotic plaques which was associated with increased expression of tissue inhibitor of MMPs (TIMP)-2 and decreased extracellular matrix (ECM) degradation 9.
An optimal balance between TIMPs and MMPs is required for the maintenance and tight regulation of the dynamic ECM environment. An alteration in such delicate balance may constitute a critical contributing factor to various cardiovascular disease states such as hypertension, atherosclerosis, aortic aneurysm and cardiac hypertrophy 13. Clinical as well as experimental studies have shown a strong correlation between MMP activity and manifestation of heart diseases 3, 14. In fact, myocardial matrix degradation and activation of MMPs in the failing heart have been suggested as viable therapeutic targets 3, 15. Cardiac hypertrophy is a potent risk factor for the development of cardiac arrhythmias, diastolic dysfunction, congestive heart failure, and death.
Since atherosclerosis and cardiac dysfunctions are so closely related, we hypothesized that PARP-1 gene deletion may protect against cardiac dysfunctions mediated by high fat (HF) diet-associated dyslipidemia and associated inflammation. Thus, we examined the effects of PARP-1 gene deletion on HF diet-induced cardiac functions and correlated the observed hemodynamic parameters with histopathological changes. We also investigated the protective effects of PARP-1 gene deletion on MMP activity, collagen degradation and tissue integrity (apoptotic cell death).
Materials and methods
Generation of ApoE−/−-PARP-1−/− double knockout mice (DKO)
C57BL/6 (wild type; WT), ApoE−/− (The Jackson Laboratory, Bar Harbor, ME), PARP-1−/−, and ApoE−/− :PARP-1−/− (DKO) mice were housed and bred in a pathogen-free animal care facility at LSUHSC, New Orleans, LA, and allowed full access to laboratory rodent chow and water. Experimental protocols were approved by the LSUHSC Animal Care and Use Committee. The generation of the C57BL/6 PARP-1−/− mice has been previously described 9. DKO mice were generated by breeding ApoE−/− into PARP-1−/− followed by cross breeding of the resulting heterozygous littermates8. All animals were genotyped by polymerase chain reaction (PCR). Six mice were used for each group unless stated otherwise. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Echocardiography
Echocardiograms were performed at various time points during the protocol. Mice were anesthetized with isoflurane (2% with 3 lpm O2). ECG electrodes were placed in a standard limb configuration to monitor heart rate. Ultrasound images were obtained using a Visualsonics VEVO 770 using a 30 MHz linear transducer. M-mode echocardiographic measurements of the interventricular septum (IS), posterior wall (PW) and LV diameter (LVD) were recorded in the parasternal short axis view at the level of the papillary muscles. LV systolic function was determined by fractional shortening (LVDdiastole-LVDsystole/LVDdiastole). All measurements were performed on 3 different cardiac cycles and the values averaged.
Histology, immunohistochemistry, TUNEL staining and assessment of lipid Profiles
Tissues were fixed in 10% formaldehyde/PBS overnight, dehydrated, and embedded in paraffin and sectioned. Hearts were cut in a cross section just below the level of the papillary muscle. The top half of the heart was formalin fixed and embedded in paraffin. Serial sections (5 μm) were prepared at 200 μm intervals. The sections were subjected to H&E staining for overall morphological examination, to Masson’s Trichrome staining for collagen detection, or to Giemsa staining visualization of mast cell degranulation. Terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling (TUNEL) staining was done according to the manufacturer’s instructions (Roche, Indianapolis, IN) or to immunohistochemistry with antibodies to poly(ADP-ribose) (Alexis, San Diego, CA), or to active caspase-3 (Cell Signaling, Carlsbad, CA) as described9, 11. For lipid profiles, animals were fasted for 4 h, anesthetized, and blood was drawn. The resulting sera samples were assessed for lipid contents by Cardiovascular Specialty Laboratories, Inc (Atlanta, GA) in a blinded fashion.
Zymography and Western Blot
Gelatinolytic activity of heart and aorta tissue homogenates were assessed by gelatin zymography using standard procedures. Briefly, immediately following heart and aorta removal from sacrificed animals, ventricular portion of the heart and thoracic aorta were frozen and crushed at liquid N2 temperature by mortar and pestle. The resulting powder was diluted 10 mM cacodylic acid, 150 mM NaCl, 20 mM CaCl2, 1 mM ZnCl2, 1.4 mM NaN3 and 0.01% Triton. This solution was then homogenized. The homogenate was centrifuged at 10,000g for 5min at 4 °C and the supernatant was kept on ice for immediate assay of MMP activities. Protein contents were measured by BCA method (Bio-Rad, Hercules, CA). Polyacrylamide gels (10%) copolymerized with gelatin (from Novex, Invitrogen, Carlsbad, USA) were used. Non-heated samples were diluted with water and sample buffer and a portion of the extracts (50μg) was loaded onto gels. After incubation with renaturing and developing buffers, the gels were stained with 0.05% Coomassie brilliant blue (G-250) and gelatinolytic activities were detected as transparent bands against a dark blue background. For western blot, hearts were used to assess expression of TIMP-2 in experimental mice after protein extraction by immunoblot analysis with antibodies to TIMP-2 (Chemicon. International Inc., Temecula, CA) or actin (Santa Cruz Biotechnology, Inc) as described 9. NIH/Scion imaging software was used for the quantitation of the immunoblot.
Cell Culture, Transfection, PCR and luciferase assay
Mouse embryonic fibroblasts (MEF) were transfected with a plasmid encoding luciferase under the control of the TIMP-2 promoter16, 17 using lipofectamine 2000 (Invitrogen Co., Carlsbad, USA) following the manufacturer’s instructions. Luciferase activity was assessed using the dual-glo Luciferase Assay kit (Promega, Madison, WI). All results were derived from four independent experiments.
Isolation of SMCs and fibroblasts from WT and PARP-1−/− mice were conducted as described 9. SMCs were used at passages 3-5. Human umbilical vein endothelial cells (HUVEC) cells were purchased from Cascade Biologies Inc. (Portland, OR, USA). HL-1, a cardiomycoyte cell line was, kindly provided by Dr. William Claycomb18. PARP inhibition in HUVECs and HL-1 cells was achieved pharmacologically by treatment with TIQ-A (450 nM).
RNA was extracted from heart, thoracic aortas, smooth muscle cells (SMCs), endothelial cells or MEFs and cDNA was generated by standard methods. Amplification, detection, and data analysis were performed with the iCycler real-time PCR system (Bio-Rad). Primer sets specific to actin, TIMP-2 or collagen type-1 have been described 9. Primers used for the detection of TIMP-3 message are: forward 5’-GGC-CTC-AAT-TAC-CGC-TAC-CA-3’; reverse 5’-CTG-ATA-GCC-AGG-GTA-CCC-AAA-A-3’.
Calculations and statistical analysis
Results are expressed as mean±SEM, from at least six mice per group unless stated otherwise. PRISM software (GraphPad, San Diego, CA, USA) was used to analyze the differences between experimental groups Significance of the differences between groups was determined by 1-repeated or 2-factor ANOVA, where appropriate. Differences were considered significant at P<0.05.
Results
PARP-1 gene deletion protected against cardiac dysfunction: hemodynamic parameters analysis
Cardiac hemodynamic parameters as measured by echocardiography are summarized in Table 1 and Table 2. There was no significant difference in the hemodynamic parameters in ApoE−/− mice and PARP-1-deficient ApoE−/− mice (DKO) on regular diet (RD). After 8 weeks on high-fat (HF) diet, there were evidences left ventricular (LV) hypertrophy and dilatations in ApoE vs DKO as suggested by parameters including interventricular septum (IVS;d;0.72±0.1 vs. 0.61 ± 0.03), left ventricular (LV) internal dimension LVID;d (4.57±0.35 vs. 3.68 ± 0.38), LVID;s (3.31±0.34 vs. 2.50 ± 0.44), LV vol;d (96.1±8.91 vs. 66.3 ± 8.5), LV vol;s (47.6±6.21 vs. 23.5 ± 3.84) and LV mass (126.9±17.0 vs. 91.9 ± 13.4). Ejection fraction (EF) and fractional shortening (FS) were not statistically different compared to DKO mice counterparts.
Table 1. Cardiac hemodynamic parameters suggest cardiac hypertrophy in ApoE−/− mice and protection by PARP-1 gene deletion.
| Parameters | ApoE−/− RD | DKO RD |
|---|---|---|
| IVS;d (mm) | 0.56 ± 0.06 | 0.58 ± 0.07 |
| LVID;d (mm) | 3.40 ± 0.14 | 3.52 ± 0.26 |
| LVPW;d (mm) | 0.66± 0.11 | 0.60 ± 0.13 |
| IVS;s (mm) | 1.01 ± 0.10 | 1.05 ± 0.16 |
| LVID;s mm | 2.33 ± 0.23 | 2.32 ± 0.22 |
| LVPW;s mm | 0.97± 0.14 | 0.96 ± 0.16 |
| LV Vol;d μl | 51.7 ± 6.11 | 52.3 ± 6.30 |
| LV Vol;s μl | 22.9 ± 6.67 | 20.1 ± 5.66 |
| %EF | 62.2 ± 5.33 | 64.0 ± 6.21 |
| % FS | 34.0 ± 3.32 | 34.6 ± 4.16 |
| LV Mass mg | 65.8 ± 11.1 | 63.2 ± 9.93 |
| LVM Corr. mg | 52.3 ± 8.86 | 50.6 ± 8.66 |
The data is expressed as mean ± SEM from at least 6 mice in each group for 3 different set of experiments. All measurements were performed on 3 different cardiac cycles and the values averaged. P value (<0.05)
Table 2. Cardiac hemodynamic parameters suggest cardiac hypertrophy in ApoE−/− mice and protection by PARP-1 gene deletion.
| Parameters | ApoE−/− HF 8wk | DKO HF 8wk | ApoE−/− HF 16wk | DKO HF 16wk |
|---|---|---|---|---|
| IVS;d mm | 0.72 ± 0.10* | 0.61 ± 0.03† | 0.75 ± 0.08 | 0.63 ± 0.13 |
| LVID;d mm | 4.57 ± 0.35* | 3.68 ± 0.38† | 4.31 ± 0.30 | 3.94 ± 0.17# |
| LVPW;d mm | 0.76 ± 0.10 | 0.58 ± 0.18 | 0.81 ± 0.11 | 0.67 ± 0.10 |
| IVS;s mm | 1.07 ± 0.12 | 0.95 ± 0.13 | 1.20 ± 0.14 | 1.11 ± 0.20 |
| LVID;s mm | 3.31 ± 0.34* | 2.50 ± 0.44† | 3.06 ± 0.24 | 2.11 ± 0.18# |
| LVPW;s mm | 1.08 ± 0.15 | 1.03 ± 0.25 | 1.23 ± 0.15 | 1.01 ± 0.13 |
| LV Vol;d ul | 96.1 ± 8.91* | 66.3 ± 8.5†‡ | 83.9 ± 11.2 | 61.3 ± 7.82# |
| LV Vol;s ul | 47.6± 6.21* | 23.5 ± 3.84† | 34.4 ± 6.39 | 26.8 ± 4.81# |
| %EF | 51.3 ± 6.87 | 59.4 ± 5.01 | 57.2 ± 6.27 | 65.9 ± 10.2 |
| % FS | 26.2 ± 4.30 | 27.8 ± 4.60 | 29.9 ± 4.21 | 36.5 ± 8.4 |
| LV Mass mg | 126.9 ± 17.0* | 91.9 ± 13.4† | 125.0 ± 12.0 | 87.3 ± 11.7# |
| LV Mass Corr.mg | 105.4 ± 11.2* | 73.5 ± 7.34† | 100.0 ± 9.57 | 69.8 ± 9.38# |
The data is expressed as mean ± SEM from at least 6 mice in each group for 3 different set of experiments. All measurements were performed on 3 different cardiac cycles and the values averaged. P value (<0.05)
compared to ApoE−/− RD
compared to ApoE−/− HF (8 weeks),
compared DKO-RD and
compared to ApoE−/− HF (16 weeks).
After feeding mice HF diet for an additional 8 weeks (total 16 weeks), IVS lost its significant difference; however, LVID (d&s), LV Vol (d&s) and LV mass remained higher in ApoE−/− mice compared to the DKO counterpart mice. The total body weight in these mice was not statistically different (data not reported). Overall these results suggest that HF diet may induce cardiac hypertrophy in ApoE−/− mice and that PARP-1 may play an important role in this process.
Histological examination substantiate cardiac hypertrophy in ApoE−/− mice: protective effects by PARP-1 gene deletion
Using echocardiography (Figure 1A), PARP-1 gene deletion was protective against HF diet induced cardiac hypertrophy in ApoE−/− mice.
Figure 1. Cardiac cross-sections revealed hypertrophy and dilatations in ApoE−/− mice; protective effects by PARP-1 gene deletion.

(A) Representative images from echocardiogram of different experimental group. (B) Histological cross-sections of heart from the different experimental groups were formalin-PBS perfused and were visualized with H&E staining. (C) HE sections showing “Box car like nuclei” characteristic of cardiac hypertrophy in ApoE−/− mice on regular diet (arrows). PARP-1 gene deletion prevented development of cardiac hypertrophy. HF diet fir 16 weeks caused severe dilatations in ApoE−/− mice which were protected by PARP-1 gene deletion. Scale bar: 50 μm (D) Shows that HF diet in ApoE−/− mice was associated with PARP activation (PAR immunoreactivity).
Histological analysis of cross-sections of the hearts of RD-fed ApoE−/− mice demonstrated the presence of large numbers of hypertrophied myocytes consisting of enlarged myocytes with boxcar-shaped nuclei and occasional binucleated myocytes as well as focal areas of myocyte disarray, suggestive of early and reversible stages of cardiac hypertrophy (Figure 1B and 1C). Interestingly, these histological changes were not revealed in the evaluated hemodynamic parameters. Hearts from DKO on the same RD regimen showed less hypertrophy with fewer hypertrophied myocytes and histology resembled the hearts of wild-type (WT) mice (data not shown), suggesting protective effects of PARP-1 gene deletion on early signs of cardiac hypertrophy. Feeding ApoE−/− mice HF diet for 16 weeks promoted clear signs of myocardial fibers separation accompanied by individual myocyte degeneration in cardiac tissue (Figure 1C). The latter features represent findings seen in dilated cardiomyopathy, in which hearts were usually increased in weight with dilatation of all four chambers. Again, PARP-1 gene deletion markedly prevented the manifestation of these cardiac defects. Consistent with the observed pathologies, HF diet was associated with a dramatic increase in PARP activation as determined by poly- (ADP-ribose) (PAR) immunoreactivity (Figure 1D). As expected, no PAR immunoreactivity was observed in the DKO counterpart mice eliciting the specificity of the PAR signal.
PARP-1 gene deletion lowers Atherogenic Index in ApoE−/− mice
Atherogenic index of plasma (AIP) has been suggested as a better indicator to predict cardiac dysfunctions 19. To determine the effect of PARP-1 gene deletion on AIP, we first conducted an assessment of lipid profiles of the different experimental groups. Figure 2A-C shows that PARP-1 gene deletion significantly increased total cholesterol and to a lesser extent LDL- cholesterol in ApoE−/− mice in the RD group. There was also a 2-3 fold increase in HDL-cholesterol in these mice compared to that detected in sera of ApoE−/− mice on a similar diet regimen. Feeding HF diet for 8 weeks dramatically promoted an increase in total and LDL-cholesterol with concomitantly decrease in HDL-cholesterol in both ApoE−/− and DKO mice compared to the respective counterparts on a RD (Figure 2A-C). However, while the increase in the total and LDL-cholesterol in DKO mice was significantly less than that in ApoE−/− mice, the promoted decrease in HDL was similar between the two mouse strains. AIP calculated using a formula suggested by Takasaki et al 20 was significantly lower in DKO mice on RD compared to that of ApoE−/− mice on similar diet (Figure 2D). PARP-1 gene deletion maintained a lower AIP on a HF diet regimen compared to the ApoE−/− counterparts. These results suggest the PARP-1 gene deletion may lower the risk of developing cardiovascular pathogenesis.
Figure 2. PARP-1 gene deletion decreases atherogenic index in plasma of ApoE−/− mice on HF diet.
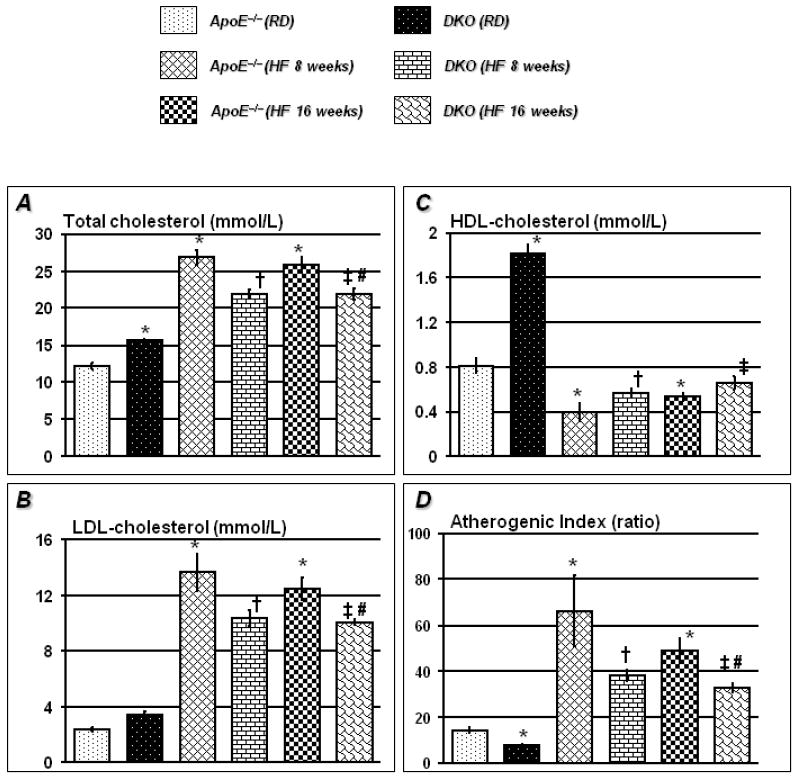
Lipid profile data of 4h starved mice shows that PARP-1 gene deletion significantly increased HDL-cholesterol and total cholesterol in ApoE−/− mice on regular diet (Figure 2A and 2C). After feeding HF diet for 8 weeks, total and LDL-cholesterol increased dramatically in ApoE−/− mice. The increase in total and LDL-cholesterol in DKO mice was significantly lesser than ApoE−/− mice on similar diet regimen (Figures 2A and 2B). Though HF diet significantly decreased HDL-cholesterol in DKO mice, atherogenic index was still significantly higher in ApoE−/− mice than DKO mice (Figure 2D). HF for further 8 weeks (total 16 weeks) did not alter the lipid profile further such that atherogenic index remained lower in sera of DKO mice than ApoE−/− mice on similar diet regimen. The data is expressed as mean ± SEM (mmol/L) from at least 6 mice in each group for 3 different set of experiments. P value (<0.05) *compared to ApoE−/− RD †compared to ApoE−/− HF (8 weeks), ‡compared DKO-RD and #compared to ApoE−/− HF (16 weeks).
PARP-1 gene deletion prevents HF diet-associated ECM degradation in ApoE−/− mice
The integrity of cardiac ECM of RD-fed ApoE−/− mice, as assessed by trichrome staining, did not seem to be influenced by PARP-1 gene deletion (Figure 3A and B). Upon a HF diet regimen for 16 weeks, a dramatic decrease in collagen content indicative of a loss of ECM integrity was observed in ApoE−/− mice. PARP-1 gene deletion was associated with a robust and significant preservation of ECM integrity (Figure 3A and B). The protective effect from PARP-1 gene deletion was not associated with an increase in collagen 1 synthesis in cardiac tissues as assessed by real-time PCR analysis (Figure 3C). Infact, collagen synthesis appeared to be significantly higher in HF diet-fed ApoE−/− mice than that in the DKO counterparts. Overall, these results suggest that the protective effect of PARP-1 gene deletion may be associated with prevention of ECM degradation rather than an increase in collagen synthesis. Further, the increase in collagen synthesis observed in HF diet-fed ApoE−/− mice may constitute a compensatory response to increased degradation of ECM in these mice.
Figure 3. HF diet promotes collagen degradation in hearts of ApoE−/− mice; protection by PARP-1 gene deletion.
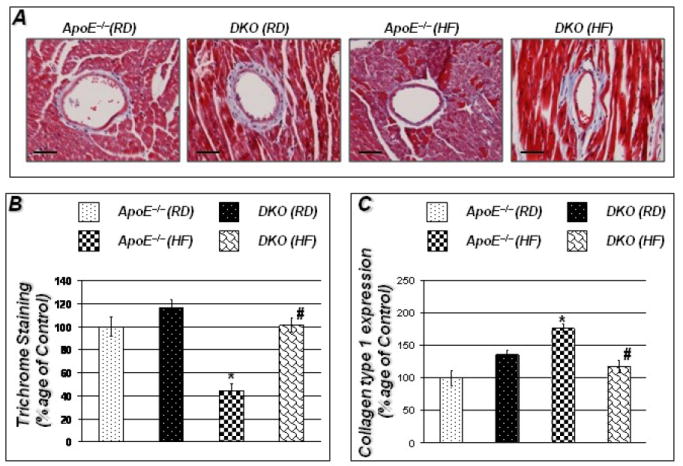
Figures 3A show trichrome staining for collagen around vessels in hearts of the experimental groups. Figure 3B represents quantitation of Trichrome staining conducted using Image-Pro Plus software and expressed percentage of control. Figure 3C shows that decreased collagen content in ApoE−/− mice was accompanied by increased collagen synthesis as determined by real time PCR for collagen type 1. cDNA was subjected to real-time PCR with primers specific to murine collagen type-1; β-actin was used as an internal control for normalization of expression values. Scale bar: 50μm P value (<0.05) *compared to ApoE−/− RD, #compared to ApoE−/− HF (16 weeks).
PARP-1 gene deletion exerts opposing effects on expression of MMP-9 and TIMP-2 in cardiac tissue
Since our results suggest that PARP-1 gene deletion may prevent degradation of ECM rather than an increase in collagen synthesis, we examined whether PARP-1 gene deletion influenced expression and/or activity of MMP-9 as well as expression of TIMP-2. Figure 4A shows that MMP-9/MMP-2 activity did not differ in cardiac tissue of ApoE−/−and DKO mice when fed a RD as assessed by zymography. However, upon a 16 weeks HF regimen, MMP-9 expression (data not shown) and activity dramatically increased in cardiac tissue of ApoE−/− mice, an effect that was largely absent in the DKO counterparts (Figure 4A). These results suggest a potential specificity between HF diet and induction of expression and activity of MMP-9 in cardiac tissue of ApoE−/− mice. The low levels of MMP-9 expression and activity in hearts of HF diet-fed DKO animals coincided with an increase in TIMP-2 expression as assessed by immunoblot analysis compared to that observed in the ApoE−/− counterparts (Figure 4B&C). Overall, these results suggest that PARP-1 gene deletion prevented ECM degradation by promoting a decrease in MMP-9 activity and an increase in endogenous production of TIMP-2 and, as a result, may explain the protective effect against ECM degradation and the ultimate prevention of HF diet-induced cardiac hypertrophy.
Figure 4. PARP-1 gene deletion maintains TIMP-2/MMP balance in response to HF diet in ApoE−/− mice on HF diet for 16 weeks.
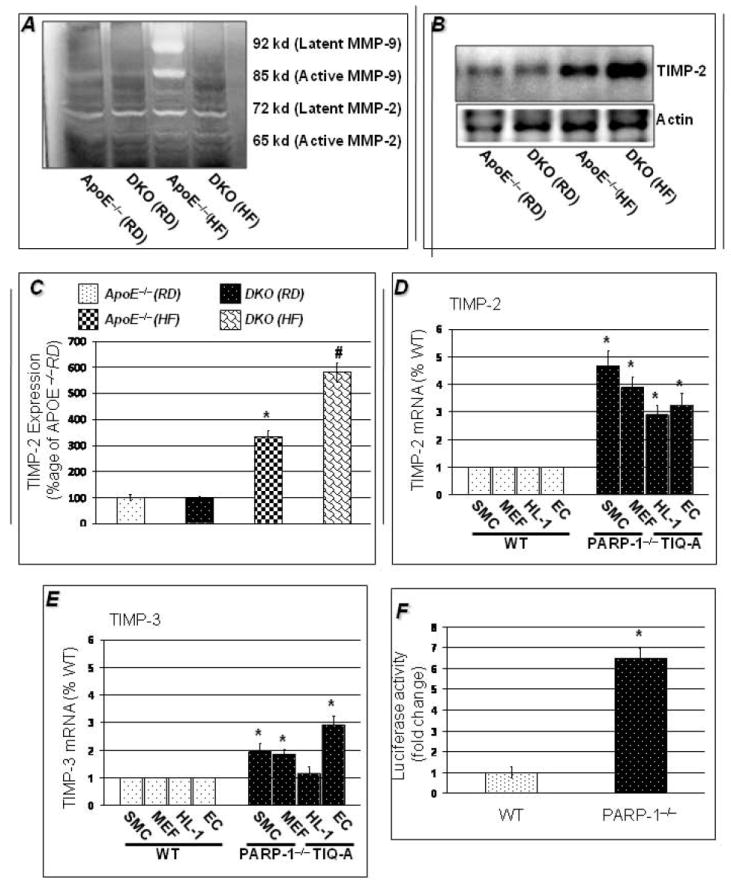
MMP activity as determined by gel zymography was comparable in ApoE−/− and DKO mice on regular diet, but HF diet induced marked increase in MMP activity in hearts of ApoE−/− mice only. PARP-1 gene deletion promoted a decrease in MMP-9 activity and an increase in endogenous production of TIMP-2 thus protecting against ECM degradation and dilatations (4B). Figure 4C shows the quantitation of TIMP-2 expression in heart normalized to actin expression. Figure 4D shows that PARP-1 gene deletion increases TIMP-2 expression by 3-5 folds in SMC and fibroblasts than wild-type cells and PARP inhibitor, TIQ-A increases TIMP_2 expression by 3-4 folds than wild-type counterparts. PARP-1 gene deletion or TIQ-A also increased TIMP-3 expression by2-3 folds (4D). Figure 4E shows increases luciferase activity of TIMP-2 promoter in MEFs by PARP-1 gene deletion. The data is expressed as mean ± SEM from at least 4 different set of experiments. P value (<0.05) *compared to wild-type cells.
PARP-1 regulates TIMP-2 expression at the promoter level
To determine whether there a direct relationship between PARP-1 and TIMP-2, we assessed the expression of TIMP-2 and TIMP-3 mRNA in cells of the vasculature (cardiomyocytes, endothelial cells, SMCs, and fibroblasts). Data in Figures 4D and E confirmed the suppressing effect of PARP-1 on gene expression of TIMPs as PARP-1 gene deletion coincided with 3-5 folds increase in TIMP-2 (4D) in the SMCs and fibroblasts as compared with the wild type counterparts. A potent PARP inhibitor TIQ-A also increased expression of TIMP-2 in cardiomyocytes (HL-1 cells) and endothelial (HUVEC) cell lines by about 3 folds. PARP-1 gene deletion or TIQ-A also increased expression of TIMP-3 albeit to lesser extent (4E). To further confirm the regulatory effect of PARP-1 on TIMP-2 expression, we examined the effect of PARP-1 gene deletion on the expression of luciferase under the control of the TIMP-2 promoter in primary fibroblasts. Figure 4E shows that PARP-1 gene deletion was associated with a significant increase (6-7 folds) in luciferase activity as assessed by a dual-glo-luciferase assay compared to activity observed in wild type fibroblasts. Overall, these results suggest that PARP-1 gene deletion prevented ECM degradation by promoting a specific increase in endogenous production of TIMP-2.
PARP-1 gene deletion significantly decreased mast cell degranulation and blocks induction of caspases-3-dependent apoptotic cell death in cardiac tissue of ApoE−/− mice fed a HF diet
In cardiac tissue of RD-fed ApoE−/− mice, mast cell number and degranulation were low and PARP-1 gene deletion did not exert any noticeable effect on these parameters (Figure 5A and B). Upon a 16 week HF diet regimen, the number of total mast cells exhibited a modest but significant increase (Figure 5B), an effect that was completely blocked in the DKO counterparts. The modest increase in mast cell number in cardiac tissue of HF diet-fed ApoE−/− mice coincided with a large increase in degranulation levels (Figure 5C). Although a slight but significant increase in mast cell degranulation was observed in cardiac tissue of HF diet-fed DKO, such an increase was substantially and significantly less than that observed in the ApoE−/− counterparts (Figure 5C). These results suggest that PARP-1 gene deletion may be preventing cardiac hypertrophy, in part, through a reduction in mast cell degranulation.
Figure 5. Increased MMP activity in ApoE−/− mice caused striking mast cell degranulation with modest increase in total number and reduction apoptotic cell death.
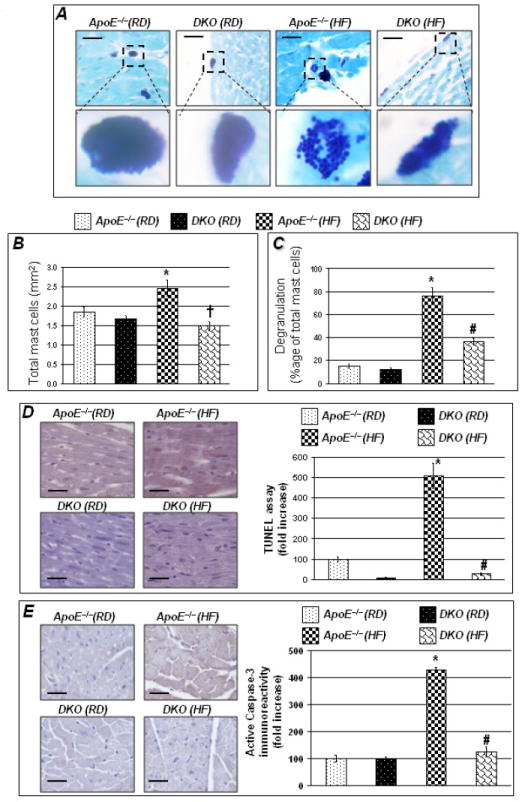
Noticeable increase in mast cell degranulation was observed in ApoE−/− mice fed HF diet for 16 weeks (Figure 5A top panel; bottom panel shows mast cells in magnification). Mast cells were stained by Giemsa stain. Figures 5B&C show quantitation of mast cell degranulation and total number. Hearts isolated from these experimental groups were collected and subjected to TUNEL assay (D) or immunohistochemistry with antibodies to active caspase-3 (E). Figure 5 D &E left panel represents quantitation of immunoreactivity was conducted using Image-Pro Plus software and expressed as immunoreactivity/mm2. Scale bar: 50 μm P value (<0.05) *compared to ApoE−/− RD, #compared to ApoE−/− HF (16 weeks).
In order to determine whether a potential correlation between HF diet-induced pathological events and induction of cell death exists, apoptosis was evaluated. Data in Figure 5D-E show that a RD diet regimen was not associated with any noticeable induction of cell death or activation of caspases-3 in either mouse strains as assessed by TUNEL and immunohistochemical analysis with antibodies to the active peptide of caspases-3, respectively. However, a 16 week HF diet regimen was associated with a significant increase in both TUNEL positivity and active caspases-3 immunoreactivity indicative of a caspases-3-dependent apoptosis in cardiac cells of ApoE−/− mice. PARP-1 gene deletion appeared to markedly reduce apoptotic cell death as evidenced by the reduced TUNEL positivity and active caspase-3 immunoreactivity in cardiac tissue of HF diet–fed DKO. Overall the data suggests that apoptotic cell death associated with cardiac hypertrophy is significantly prevented by PARP-1 gene deletion which is suggestive of a better preservation of cardiac tissue integrity. However, a direct relationship between the decrease in mast cell degranulation and the decrease in apoptosis in HF diet–fed DKO still remains to be elucidated.
Discussion
Cardiac hypertrophy is closely related to atherosclerosis and is considered a disease of ECM imbalance. We hypothesized a protective effect of PARP-1 gene deletion on cardiac hypertrophy in the mouse model of atherosclerosis. HF diet regimen caused cardiac hypertrophy and dilatation of the heart of ApoE−/− mice which was found to be associated with increased MMP-9 activity in heart, degranulation of mast cells and increased caspase-3-dependent apoptotic cell death. These traits are closely related to those induced by several inducers of cardiac hypertrophy including pressure overload, chronic hypoxia 21 or angiotensin 22, 23. PARP-1 gene deletions not only prevented the faltering of hemodynamic parameters associated with cardiac hypertrophy; but also, protected against progression of this condition to deleterious dilatations. At the molecular level, PARP-1 gene deletion was associated with a reduced expression of MMP-9 and an increased TIMP-2 expression in cardiac tissues. This observation was confirmed using an in vitro system encompassing several cells of vascular origin. The maintenance of TIMP-2/MMP balance favorable for maintenance of ECM integrity may be at the core of the reduced activation of mast cell degranulation and apoptotic cell death in the HF diet-fed DKO animals. Others have shown the development of cardiac hypertrophy in ApoE−/− mice albeit to a different extent depending upon the inducing agent and duration 24, 25. Some of these results are consistent with those recently reported on the effect of a PARP inhibitor in a rat model of chronic heart failure after isoproterenol-induced myocardial infarction 26, 27. Overall, the data suggest protective effects of PARP-1 gene deletion against cardiac hypertrophy and its progression to dilatations by a direct effect on TIMP-2 expression and a preservation of ECM integrity.
It is increasingly recognized that myocardial ECM is a complex microenvironment that contains a battery of matrix proteins, signaling molecules, protease and cell types that collectively may orchestrate the process of myocardial remodeling 28-30. Excessive production of MMPs and apoptosis has been established as the most potent stimulator of cardiac hypertrophy and its progression to dilatation which results in widespread matrix degradation. The proteolytic potential of MMPs is tightly regulated by endogenous physiological inhibitors (TIMPs), a family of four members 31, 32. Our data suggest that protective effects of PARP-1 gene deletion against cardiac hypertrophy and its progression to dilatations may be mediated mainly by increased TIMP-2 expression and decreased MMP activity. In ApoE−/− animals with active PARP, an aggravated dyslipidemia upon HF diet resulted in an activation of MMPs and increased degradation of collagen, which may result in the dissolution of the interstitial collagenous matrix of the myocardium. The latter effects may also be responsible for the activation of various inflammatory pathways that ultimately cause degranulation of mast cells, thus leading to dilated cardiomyopathy and ultimately heart failure.
TIMPs specifically inhibit both active and latent forms of MMPs, and the disturbance of this balance leads to pathological consequences 33. TIMP-2 is a unique member of the TIMP family in that it may directly suppress proliferation of endothelial cells, independently of its function to inhibit MMP activity 32. TIMP-2 and TIMP-3 have been reported to possess anti-apoptotic effects against angiogenesis in a manner independent of MMP inhibitors mediated functions 32, 34. In our studies, PARP-1 gene deletion increased expression of TIMP-2 by 4-5 folds (Figure 5) and TIMP-3 albeit to a lesser extent. Our preliminary data suggests that PARP-1 binds to the promoter region of TIMP-2 and regulates its basal expression. Further, these results were supported by the finding that re-establishment of PARP-1 expression by adenoviral vector partially decreased TIMP-2 expression (unpublished data).
Atherogenic index has been suggested as better index of dyslipidemia and an important tool to analyze the results considering the complexity of lipoproteins 19, 20. Our data suggests that though PARP-1 gene deletion moderately increased total cholesterol levels upon a HF diet regimen, an observation also reported by von Lukowicz et al 12, the atherogenic index was significantly decreased by PARP-1 gene deletion. PARP-1 gene deletion was associated with a doubling in HDL levels in the RD experimental group, an effect which was abolished by HF diet. Although the mechanism by which PARP-1 gene deletion may promote an elevation in HDL levels is unknown, an increase in HDL-cholesterol has been reported to closely correlate with a protection against left ventricular hypertrophy35.
Cardiac mast cells are increasingly being associated with the development of cardiac pathologies including hypertrophy given their potential role in activating MMPs, which, in turn, are responsible for ECM degradation 36. It is noteworthy that mast cells contribute to cardiac hypertrophy despite their rather small numbers within the tissue and constitute only 1-2 cells/mm2 37. Although latent mast cells show little to no MMP activity, in response to oxidative stress and free radical mast cells, they become activated and produce large amount of MMPs and other inflammatory factors including TNF and inteleukins 37, 38. Studies done by Chancey et al39 have reported that mast cell degranulation leads to an increase in MMP activity, collagen degradation and altered ventricular diastolic function. Therefore, PARP-1 gene deletion-associated decrease in MMP activity may be the cause for the reduced mast cell degranulation with a HF diet regimen seen in the present study. Such reduction in mast degranulation may further prevent an amplification of MMP expression and activation. While there is no direct evidence for a regulatory effect of PARP-1 on degranulation of mast cells, PARP activity has been associated with increased mast cell degranulation 40. In this regard, the observed increase in mast degranulation in cardiac tissue of HF diet-fed ApoE−/− mice was associated with a marked increase in PARP activity as assessed by immunohistochemistry. Activation and degranulation of mast cells as well as ECM degradation have been suggested to contribute to cardiomyocyte apoptosis 37, 38, 41.
The protective effect of PARP-1 gene deletion against HF diet-induced cardiac hypertrophy in a model of atherosclerosis is consistent with that recently reported by Gupta, et al. using a pharmacological inhibitor and an angiotensin-II-induced cardiac hypertrophy 5. These results are well in keeping with those very recently reported by our laboratory demonstrating that PARP-1 gene deletion is also protective against dyslipidemia-induced endothelial and vascular dysfunction8. The mechanism by which PARP-1 influences these factors is presently under intense investigation in our laboratory. In summary we provide evidence that PARP-1 gene deletion protects against cardiac hypertrophy and dyslipidemia induced dilatations by maintaining increased expression of TIMP-2 thus increasing the ratio of TIMP/MMP in a mouse model of atherosclerosis and is also beneficial in partially decreasing atherogenic index.
Over all the data suggests that, PARP-1 deficiency will afford cardioprotection by maintaining the optimum TIMP-2/MMP balance, reducing ECM degradation and protecting against cell death. These beneficial effects of PARP-1 deficiency on TIMP-2/MMP balance may open up new avenues of research in combating the deleterious effects of ECM degradation on cardiac function and present PARP-1 as an appropriate target for the modulation of cardiac remodeling, and provide a basis for the development of new class of drugs associated with PARP-1 that are specific to the treatment of cardiovascular diseases.
Acknowledgments
We thank Dr. Yves A. DeClerck (The Saban Research Institute, Division of Hematology/Oncology, Children’s Hospital of Los Angeles, CA) for providing us the TIMP-2 construct. We also thank Elizabeth McIlwain for echocardiography analysis and Dr. William Claycomb for providing the HL-1 cardiomyocytes.
Source of funding This work was supported by the National Heart Blood and Lung Institute at the National Institute of Health [(HL072889 and 1P20RR18766 (overall PI, D. Kapusta)]; the American Cancer Society (RSG-116608) to A.H.B, and the American Heart Association Postdoctoral fellowship (0825470E) to C.P.H.
Footnotes
Conflict of Interests: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carreno JE, Apablaza F, Ocaranza MP, Jalil JE. Cardiac hypertrophy: molecular and cellular events. Rev Esp Cardiol. 2006;59(5):473–486. [PubMed] [Google Scholar]
- 2.Gupta S, Das B, Sen S. Cardiac hypertrophy: mechanisms and therapeutic opportunities. Antioxid Redox Signal. 2007;9(6):623–652. doi: 10.1089/ars.2007.1474. [DOI] [PubMed] [Google Scholar]
- 3.Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. 2007;87(4):1285–1342. doi: 10.1152/physrev.00012.2007. [DOI] [PubMed] [Google Scholar]
- 4.Haudek SB, Taffet GE, Schneider MD, Mann DL. TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J Clin Invest. 2007;117(9):2692–2701. doi: 10.1172/JCI29134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pillai JB, Gupta M, Rajamohan SB, Lang R, Raman J, Gupta MP. Poly(ADP-ribose) polymerase-1-deficient mice are protected from angiotensin II-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2006;291(4):H1545–1553. doi: 10.1152/ajpheart.01124.2005. [DOI] [PubMed] [Google Scholar]
- 6.Balakumar P, Singh M. Possible role of poly(ADP-ribose) polymerase in pathological and physiological cardiac hypertrophy. Methods Find Exp Clin Pharmacol. 2006;28(10):683–689. doi: 10.1358/mf.2006.28.10.1037495. [DOI] [PubMed] [Google Scholar]
- 7.Chalmers AJ. Poly(ADP-ribose) polymerase-1 and ionizing radiation: sensor, signaller and therapeutic target. Clin Oncol (R Coll Radiol) 2004;16(1):29–39. doi: 10.1016/s0936-6555(03)00223-1. [DOI] [PubMed] [Google Scholar]
- 8.Hans CP, Feng Y, Naura AS, Zerfaoui M, Rezk BM, Xia H, Kaye AD, Matrougui K, Lazartigues E, Boulares AH. Protective effects of PARP-1 knockout on dyslipidemia-induced autonomic and vascular dysfunction in ApoE mice: effects on eNOS and oxidative stress. PLoS One. 2009;4(10):e7430. doi: 10.1371/journal.pone.0007430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oumouna-Benachour K, Hans CP, Suzuki Y, Naura A, Datta R, Belmadani S, Fallon K, Woods C, Boulares AH. Poly(ADP-ribose) polymerase inhibition reduces atherosclerotic plaque size and promotes factors of plaque stability in apolipoprotein E-deficient mice: effects on macrophage recruitment, nuclear factor-kappaB nuclear translocation, and foam cell death. Circulation. 2007;115(18):2442–2450. doi: 10.1161/CIRCULATIONAHA.106.668756. [DOI] [PubMed] [Google Scholar]
- 10.Pacher P, Szabo C. Role of Poly(ADP-ribose) polymerase 1 (PARP-1) in Cardiovascular Diseases: The Therapeutic Potential of PARP Inhibitors. Cardiovascular Drug Reviews. 2007;25(3):235–260. doi: 10.1111/j.1527-3466.2007.00018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hans CP, Zerfaoui M, Naura AS, Catling A, Boulares AH. Differential effects of PARP inhibition on vascular cell survival and ACAT-1 expression favouring atherosclerotic plaque stability. Cardiovasc Res. 2008;78(3):429–439. doi: 10.1093/cvr/cvn018. [DOI] [PubMed] [Google Scholar]
- 12.von Lukowicz T, Hassa PO, Lohmann C, Boren J, Braunersreuther V, Mach F, Odermatt B, Gersbach M, Camici GG, Stahli BE, Tanner FC, Hottiger MO, Luscher TF, Matter CM. PARP1 is required for adhesion molecule expression in atherogenesis. Cardiovasc Res. 2008;78(1):158–166. doi: 10.1093/cvr/cvm110. [DOI] [PubMed] [Google Scholar]
- 13.Zervoudaki A, Economou E, Stefanadis C, Pitsavos C, Tsioufis K, Aggeli C, Vasiliadou K, Toutouza M, Toutouzas P. Plasma levels of active extracellular matrix metalloproteinases 2 and 9 in patients with essential hypertension before and after antihypertensive treatment. J Hum Hypertens. 2003;17(2):119–124. doi: 10.1038/sj.jhh.1001518. [DOI] [PubMed] [Google Scholar]
- 14.Frantz S, Stork S, Michels K, Eigenthaler M, Ertl G, Bauersachs J, Angermann CE. Tissue inhibitor of metalloproteinases levels in patients with chronic heart failure: an independent predictor of mortality. Eur J Heart Fail. 2008;10(4):388–395. doi: 10.1016/j.ejheart.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 15.Newby AC. Dual Role of Matrix Metalloproteinases (Matrixins) in Intimal Thickening and Atherosclerotic Plaque Rupture. Physiol Rev. 2005;85(1):1–31. doi: 10.1152/physrev.00048.2003. [DOI] [PubMed] [Google Scholar]
- 16.Hammani K, Blakis A, Morsette D, Bowcock AM, Schmutte C, Henriet P, DeClerck YA. Structure and Characterization of the Human Tissue Inhibitor of Metalloproteinases-2 Gene. J Biol Chem. 1996;271(41):25498–25505. doi: 10.1074/jbc.271.41.25498. [DOI] [PubMed] [Google Scholar]
- 17.Zhong ZD, Hammani K, Bae WS, DeClerck YA. NF-Y and Sp1 cooperate for the transcriptional activation and cAMP response of human tissue inhibitor of metalloproteinases-2. J Biol Chem. 2000;275(24):18602–18610. doi: 10.1074/jbc.M001389200. [DOI] [PubMed] [Google Scholar]
- 18.Claycomb WC, Lanson NA, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(6):2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murakami S, Kondo-Ohta Y, Tomisawa K. Improvement in cholesterol metabolism in mice given chronic treatment of taurine and fed a high-fat diet. Life Sciences. 1998;64(1):83–91. doi: 10.1016/s0024-3205(98)00536-0. [DOI] [PubMed] [Google Scholar]
- 20.Takasaki Y. Serum Lipid Levels and Factors Affecting Atherogenic Index in Japanese Children. Journal of PHYSIOLOGICAL ANTHROPOLOGY and Applied Human Science. 2005;24(4):511–515. doi: 10.2114/jpa.24.511. [DOI] [PubMed] [Google Scholar]
- 21.Yamashita C, Hayashi T, Mori T, Tazawa N, Kwak CJ, Nakano D, Sohmiya K, Okada Y, Kitaura Y, Matsumura Y. Angiotensin II receptor blocker reduces oxidative stress and attenuates hypoxia-induced left ventricular remodeling in apolipoprotein E-knockout mice. Hypertens Res. 2007;30(12):1219–1230. doi: 10.1291/hypres.30.1219. [DOI] [PubMed] [Google Scholar]
- 22.Ainscough JF, Drinkhill MJ, Sedo A, Turner NA, Brooke DA, Balmforth AJ, Ball SG. Angiotensin II type-1 receptor activation in the adult heart causes blood pressure-independent hypertrophy and cardiac dysfunction. Cardiovasc Res. 2009;81(3):592–600. doi: 10.1093/cvr/cvn230. [DOI] [PubMed] [Google Scholar]
- 23.Polizio AH, Balestrasse KB, Yannarelli GG, Noriega GO, Gorzalczany S, Taira C, Tomaro ML. Angiotensin II regulates cardiac hypertrophy via oxidative stress but not antioxidant enzyme activities in experimental renovascular hypertension. Hypertens Res. 2008;31(2):325–334. doi: 10.1291/hypres.31.325. [DOI] [PubMed] [Google Scholar]
- 24.Hartley CJ, Reddy AK, Madala S, Martin-McNulty B, Vergona R, Sullivan ME, Halks-Miller M, Taffet GE, Michael LH, Entman ML, Wang YX. Hemodynamic changes in apolipoprotein E-knockout mice. Am J Physiol Heart Circ Physiol. 2000;279(5):H2326–2334. doi: 10.1152/ajpheart.2000.279.5.H2326. [DOI] [PubMed] [Google Scholar]
- 25.Wang YX. Cardiovascular functional phenotypes and pharmacological responses in apolipoprotein E deficient mice. Neurobiol Aging. 2005;26(3):309–316. doi: 10.1016/j.neurobiolaging.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 26.Bartha E, Kiss GN, Kalman E, Kulcsar G, Kalai T, Hideg K, Habon T, Sumegi B, Toth K, Halmosi R. Effect of L-2286, a poly(ADP-ribose)polymerase inhibitor and enalapril on myocardial remodeling and heart failure. J Cardiovasc Pharmacol. 2008;52(3):253–261. doi: 10.1097/FJC.0b013e3181855cef. [DOI] [PubMed] [Google Scholar]
- 27.Bartha E, Solti I, Kereskai L, Lantos J, Plozer E, Magyar K, Szabados E, Kalai T, Hideg K, Halmosi R, Sumegi B, Toth K. PARP inhibition delays transition of hypertensive cardiopathy to heart failure in spontaneously hypertensive rats. Cardiovasc Res. 2009 doi: 10.1093/cvr/cvp144. [DOI] [PubMed] [Google Scholar]
- 28.Hutchinson KR, Stewart JA, Jr, Lucchesi PA. Extracellular matrix remodeling during the progression of volume overload-induced heart failure. J Mol Cell Cardiol. 2009 doi: 10.1016/j.yjmcc.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahmed SH, Clark LL, Pennington WR, Webb CS, Bonnema DD, Leonardi AH, McClure CD, Spinale FG, Zile MR. Matrix metalloproteinases/tissue inhibitors of metalloproteinases: relationship between changes in proteolytic determinants of matrix composition and structural, functional, and clinical manifestations of hypertensive heart disease. Circulation. 2006;113(17):2089–2096. doi: 10.1161/CIRCULATIONAHA.105.573865. [DOI] [PubMed] [Google Scholar]
- 30.Malemud CJ. Matrix metalloproteinases (MMPs) in health and disease: an overview. Front Biosci. 2006;11:1696–1701. doi: 10.2741/1915. [DOI] [PubMed] [Google Scholar]
- 31.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovascular Research. 2006;69(3):562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 32.Stetler-Stevenson WG, Seo D-W. TIMP-2: an endogenous inhibitor of angiogenesis. Trends in Molecular Medicine. 2005;11(3):97–103. doi: 10.1016/j.molmed.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 33.Loftus IM, Naylor AR, Bell PR, Thompson MM. Matrix metalloproteinases and atherosclerotic plaque instability. Br J Surg. 2002;89(6):680–694. doi: 10.1046/j.1365-2168.2002.02099.x. [DOI] [PubMed] [Google Scholar]
- 34.Seo D-W, Li H, Guedez L, Wingfield PT, Diaz T, Salloum R, Wei B-y, Stetler-Stevenson WG. TIMP-2 Mediated Inhibition of Angiogenesis: An MMP-Independent Mechanism. Cell. 2003;114(2):171–180. doi: 10.1016/s0092-8674(03)00551-8. [DOI] [PubMed] [Google Scholar]
- 35.Anan F, Yonemochi H, Masaki T, Takahashi N, Fukunaga N, Teshima Y, Iwao T, Kaneda K, Eshima N, Saikawa T, Yoshimatsu H. High-Density Lipoprotein Cholesterol and Insulin Resistance Are Independent and Additive Markers of Left Ventricular Hypertrophy in Essential Hypertension. Hypertens Res. 2007;30(2):125–131. doi: 10.1291/hypres.30.125. [DOI] [PubMed] [Google Scholar]
- 36.Brower GL, Chancey AL, Thanigaraj S, Matsubara BB, Janicki JS. Cause and effect relationship between myocardial mast cell number and matrix metalloproteinase activity. Am J Physiol Heart Circ Physiol. 2002;283(2):H518–525. doi: 10.1152/ajpheart.00218.2000. [DOI] [PubMed] [Google Scholar]
- 37.Janicki JS, Brower GL, Gardner JD, Forman MF, Stewart JA, Jr, Murray DB, Chancey AL. Cardiac mast cell regulation of matrix metalloproteinase-related ventricular remodeling in chronic pressure or volume overload. Cardiovasc Res. 2006;69(3):657–665. doi: 10.1016/j.cardiores.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 38.Balakumar P, Singh AP, Ganti SS, Krishan P, Ramasamy S, Singh M. Resident cardiac mast cells: are they the major culprit in the pathogenesis of cardiac hypertrophy? Basic Clin Pharmacol Toxicol. 2008;102(1):5–9. doi: 10.1111/j.1742-7843.2007.00147.x. [DOI] [PubMed] [Google Scholar]
- 39.Chancey AL, Brower GL, Janicki JS. Cardiac mast cell-mediated activation of gelatinase and alteration of ventricular diastolic function. Am J Physiol Heart Circ Physiol. 2002;282(6):H2152–2158. doi: 10.1152/ajpheart.00777.2001. [DOI] [PubMed] [Google Scholar]
- 40.Cuzzocrea S. Shock, inflammation and PARP. Pharmacol Res. 2005;52(1):72–82. doi: 10.1016/j.phrs.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 41.Zhang QY, Ge JB, Chen JZ, Zhu JH, Zhang LH, Lau CP, Tse HF. Mast cell contributes to cardiomyocyte apoptosis after coronary microembolization. J Histochem Cytochem. 2006;54(5):515–523. doi: 10.1369/jhc.5A6804.2005. [DOI] [PubMed] [Google Scholar]