

Abstract
Objective
To assess the contribution of wild-type, mutant and loss of leucine-rich repeat kinase 2 (LRRK2; Lrrk2) on dendritic neuronal arborization.
Background
LRRK2 mutations are recognized as the major genetic determinant of susceptibility to Parkinson’s disease for which a cellular assay of Lrrk2 mutant function would facilitate the development of targeted molecular therapeutics
Methods
Dendritic neuronal arborization (neurite length, branching and the number of processes per cell) was quantified in primary hippocampal and midbrain cultures derived from five lines of recombinant LRRK2 mice, including human BAC wild-type and mutant overexpressors (Y1699C and G2019S), murine knock-out and G2019S knock-in animals
Results
Neuronal arborization in cultures from BAC Lrrk2 wild-type animals is comparable to non-transgenic littermate controls, despite high levels of human transgene expression. In contrast, primary neurons from both BAC mutant overexpressors presented with significantly reduced neuritic outgrowth and branching, although the total number of processes per cell remained comparable. The mutant-specific toxic gain-of-function observed in cultures from BAC mutant mice may be partially rescued by staurosporine treatment, a non-specific kinase inhibitor. In contrast, neuronal arborization is far more extensive in neuronal cultures derived from murine knock-out mice that lack endogenous Lrrk2 expression. In Lrrk2 G2019S knock-in mice, arguably the most physiologically relevant system, neuritic arborization is not impaired.
Conclusions
Impairment of neuritic arborization is an exaggerated, albeit mutant-specific, consequence of Lrrk2 over-expression in primary cultures. The phenotype and assay described provides a means to develop therapeutic agents that modulate the toxic gain-of-function conferred by mutant Lrrk2.
Keywords: LRRK2, Parkinson’s disease, PD, neuritic outgrowth, drug screening
Introduction
Autosomal-dominant mutations in leucine-rich repeat kinase-2 (LRRK2) have been identified as an important cause for familial and sporadic Parkinson’s disease (PD) [1]. LRRK2 encodes a 286KDa protein, previously classified as a member of the ROCO superfamily [2,3], characterized by the presence of tandem Ras-related GTPase (Roc), C-terminal of Roc (COR) and kinase domains. Additional domains include armadillo and ankyrin repeats at the N-terminus, a central leucine-rich repeat (LRR), and a WD40 domain towards the C-terminus.
Given the epidemiological importance of LRRK2 mutations [4-6] intensive efforts have been made to elucidate Lrrk2-specific signaling pathways and the mechanisms underlying PD. Rare pathogenic mutations focused initial studies on the enzymatic activity and regulation of Lrrk2 kinase and GTPase domains [7,8]. Several studies now report Lrrk2 is involved in the regulation of neurite maintenance and survival [9-11]. Cellular over-expression of the LRRK2 transgene (wild-type and mutant) has been achieved using viral transduction, transfection of cDNA constructs, and the ex-vivo culture of mice neurons expressing LRRK2 cDNA with heterologous promoters. Each of the published models represents a valuable tool to investigate Lrrk2 biology but nevertheless none recapitulate endogenous wild-type or mutant Lrrk2 expression in brain [12]. The changes in neuritic arborization observed in ex-vivo transfected cultures are non-physiologic, thus their relevance to age-associated human disease is not intuitive.
We have used a comparative approach to assess the role of Lrrk2 in bacterial artificial chromosome (BAC) wild-type (hWT) and mutant over-expressing [G2019S (GS) and Y1699C (YC)] mice as well as in murine LRRK2 knock-out (KO) and more physiologic G2019S knock-in animals (KI). Using primary neuronal cultures from our BAC models, we have assessed whether deficits in neuritic outgrowth can be recapitulated, and whether they are a consequence of high-levels of transgene expression, are mutant-specific or a combination. Subsequently, in primary cultures from KO mice we have examined whether regulation of neuritic outgrowth, morphology and cellular homeostasis is an inherent function of Lrrk2. Finally, using KI animals, we have assessed whether mutant Lrrk2 expressed at endogenous levels is sufficient to induce similar morphological changes.
Material and methods
Mouse models
hWT, GS and YC LRRK2 mice
A BAC (RP-11 568G5) containing the entire human wild-type LRRK2 gene and regulatory sequences was identified and recombination-based BAC mutagenesis performed to generate mutant G2019S BAC and Y1699C clones. Following confirmation of integrity, purified BAC DNA was injected into FVB/N (Taconic) fertilized oocytes and transplanted into pseudo-pregnant ICR (Harlan) female mice. Subsequent offspring were genotyped to identify founders. Transgenic founders were bred to FVB mice and transgenic F1 offspring analyzed for gene (RT-PCR) and protein expression (Western blot). Transgenic Lrrk2 expression levels in different founder lines were found to be ~20-fold for the hWT line, ~10-fold for G2019S line and ~11-fold for the Y1699C line (assuming the affinity of PAO362 antibody for human and mouse Lrrk2 is comparable).
LRRK2 KO mice
LRRK2 KO mice, generated by Ozgene PLC, were created utilizing a targeting construct designed to ablate LRRK2 exon 41 containing the kinase domain using standard gene targeting techniques. Resultant mice were bred to homozygosity. Routine genotyping was performed by a PCR-based strategy utilizing intronic primers that span exon 41. Deletion of exon 41 of LRRK2 was confirmed at the genomic, mRNA and protein level.
LRRK2 KI mice
Mice expressing endogenous LRRK2 G2019S were generated by Ozgene PLC. The ‘G’ nucleotide at cDNA position 6055 is conserved in mouse and human, however, mutagenesis of two nucleotides was required to change GGG to AGC in exon 41 of the targeting construct in the mouse. cDNA from resultant mice was re-sequenced to confirm the presence of the G201S mutations (Suppl. Figure 1a).
Experiments were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23, revised 1978). All efforts were made to minimize animal suffering, and to reduce the number of animals used for ex vivo experiments.
Cell culture
Pups were genotyped on postnatal day 1 (P1) by PCR. At postnatal day 2 (P2) brains were removed and transferred to a culture dish containing sterile ice-cold dissecting buffer A without Calcium (Hibernate A (Nalgene/ThermoFisher), B27 (Gibco/Invitrogen), 0.5 mM GMAX (Invitrogen). Brain hippocampi and midbrain sections were dissected under a microscope, collected in buffer A, cut to smaller pieces with a sterile razor blade and transferred to a conical tube containing buffer D (Hibernate A without Ca, 0.5mM GMAX, 2 mg/ml papain). For enzymatic digestion, the tissue/buffer suspension was kept shaking at 30°C for 30 minutes. After transfer to a conical tube containing buffer A, the tissue was mechanically digested by trituration. Cells were pelleted by centrifugation (300×g for 10 min), resuspended in Buffer B (Neurobasal A without Glutamate (Gibco/Invitrogen), B27, 0.5 mM GMAX, 50 μg/ml Gentamicin (Gibco/Invitrogen)) and seeded on poly-D-lysine (Sigma) coated coverslips or 6-well plates. After one hour buffer B was replaced by normal growth medium (Neurobasal A without Glutamate, B27, 0.5 mM GMAX, 5 ng/μl Fibroblast Growth Factor). For treatment with staurosporine (Sigma), an optimal concentration was determined in a pilot experiment testing several dilutions ranging from 50pM to 1μM; 200pM still induced neuritic outgrowth in hippocampal neurons derived from mutant LRRK2 transgenic animals while no sign of nuclear condensation or fragmentation could be detected by Hoechst staining. Cultures were maintained at 37°C in a humidified atmosphere of 95% air 5% CO2. One half of the medium was replaced with fresh growth medium (with or without staurosporine) every 48 hours.
Neurite outgrowth quantification
Cells were fixed at indicated time points and subsequently stained for microtubule-associated protein (MAP2, Sigma) as a dendritic marker for neurons, tyrosine hydroxylase (TH, Affinity Bioreagents) for the identification of catecholamine neurons, glial fibrillary acidic protein as a glial marker (GFAP, Sigma), and DAPI/Hoechst 33258 (Invitrogen).
For experiments evaluating axonal as opposed to dendritic outgrowth, tau (tau5 mouse mAB, LabVison/Thermo Scientific, Fremont, CA) was used as an axonal marker protein. For the quantification of neuritic outgrowth in hippocampal cultures at least 17 confocal images of MAP2 positive cells per condition were taken at 20× magnification (approximately 5-10 cells per image). Results from midbrain cultures are based on MAP2 staining of at least 15 TH positive neurons per condition. Using the MetaMorph Offline analysis software (Molecular Devices) each image was then evaluated for several parameters including mean process length, number of cell processes as well as general branching. Data was plotted (mean ± SEM) and statistically analyzed using unpaired t-test (Figures 2B & C, 3B & C, 4, Suppl.3B, C & D) or one way ANOVA comparisons (Figures 2A, 3A, Suppl.3A) in GraphPad Prism® software.
Figure 2. Neuritic outgrowth and branching in BAC primary hippocampal neurons.
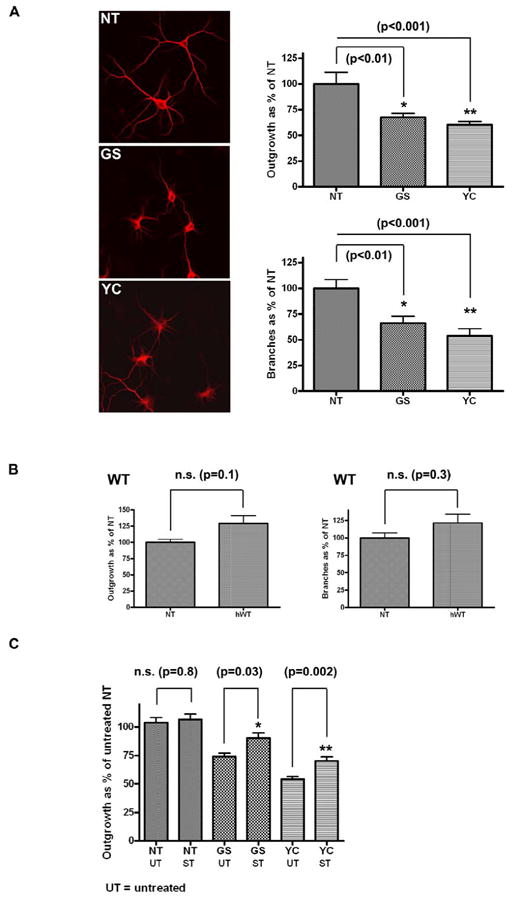
(A) Neurons from BAC mice overexpressing GS or YC mutant Lrrk2 display reduced neuritic outgrowth and branching compared with non transgenic (NT) littermates (one way ANOVA.) (B) in contrast, neurons from BAC mice overexpressing wild-type human Lrrk2 (hWT) show no deficit (unpaired t-test). (C) Treatment with 200pM staurosporine partially rescues the neurite outgrowth deficit observed in GS and YC lines while it does not have any affect on NT primary cultures (unpaired t-test). Error bars on all graphs represent standard errors of the mean.
Figure 3. Neuritic outgrowth and branching in KO primary hippocampal and midbrain cultures.
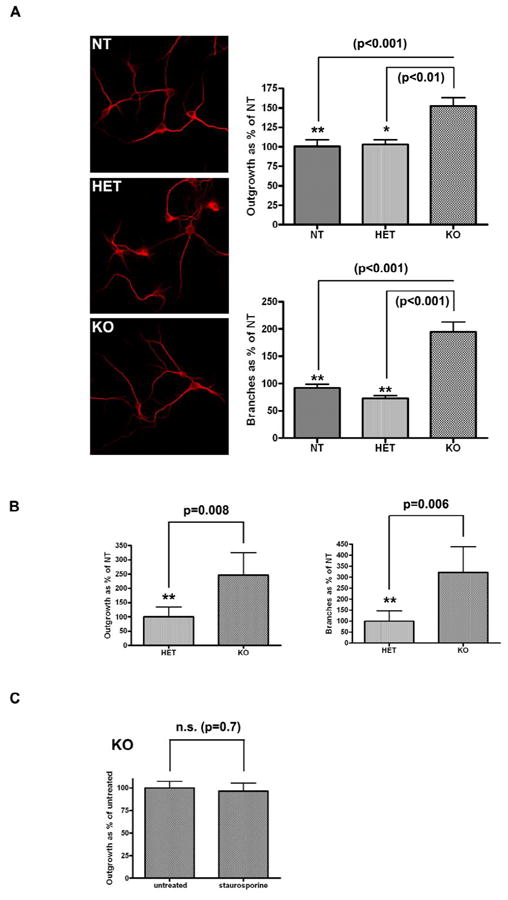
Homozygous knock-out (KO) but not heterozygous (HET) hippocampal (one way ANOVA) (A) and midbrain (unpaired t-test) (B) cultures show increased neuritic outgrowth and branching. (C) Treatment with 200pM staurosporine does not induce longer neuritic process length in KO primary hippocampal cultures (unpaired t-test).
Figure 4. Neuritic outgrowth in KI primary hippocampal and midbrain cultures.
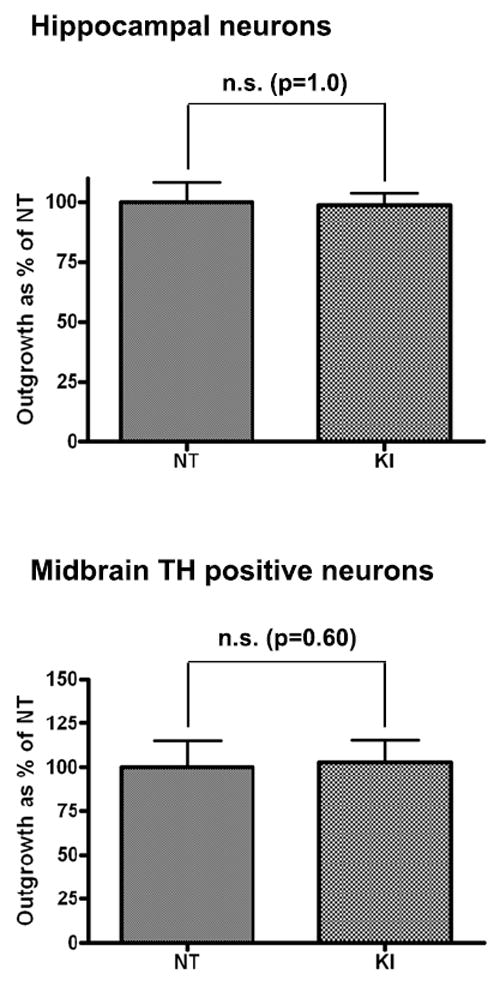
Deficits in neuritic outgrowth as indicated by the mean process length are not observed in homozygous hippocampal or midbrain TH positive neurons from KI mice (unpaired t-test). Error bars on all graphs represent standard errors of the mean.
To monitor long term survival, cells were fixed on day 24 in culture and stained for tau (tau5, see above; CP-13 gift from Dr. Peter Davies, Albert Einstein College of Medicine) and activated caspases 3, 8 and 9 (all Cell Signaling) using standard immunocytochemistry with fluorescent secondary antibodies (Alexa Fluor, Molecular Probes/Invitrogen).
Western Blot analysis
To test for Lrrk2 expression in P2 primary neurons, cells from a total of 4 six-well plates were combined and harvested using standard protocols on culture day 8. For analysis of regional LRRK2 transgene expression, adult mice were harvested by cervical dislocation and dissected into various brain regions. Crude lysates were sonicated, centrifuged at 13000 × g (4°C, 5 minutes) and separated into supernatant and pellet. 50μg of protein was loaded on 3-8% Tris-Acetate gels (Invitrogen), separated by SDS-PAGE and transferred to a PVDF membrane following standard protocols. Lrrk2 was detected using affinity purified rabbit polyclonal antibody PA0362 as previously described [14]. Due to low levels of endogenous Lrrk2 expression, membranes in supplementary Figure 1C were developed using undiluted fluorescence enhancer (SuperSignal, Pierce) with exposure times of up to 20 minutes. All blots were stripped and re-incubated with antibodies against the housekeeping protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Meridian Life Science).
Results
To explore whether we can recapitulate impaired neuritic outgrowth and branching in a LRRK2 over-expression model we prepared primary cultures from BAC LRRK2 transgenic animals and non-transgenic littermate controls. Human BAC LRRK2 transgenic animals express the transgene under the endogenous promoter, to mimic the temporal as well as regional expression patterns of the endogenous gene. Based on the expression pattern of full length hWT, GS and YC Lrrk2 proteins in various brain regions (Figure 1a) we decided to evaluate neuritic outgrowth in hippocampal cultures as the area of highest transgene expression. In the absence of reliable antibodies for immunocytochemistry, Western Blot analysis was applied to confirm Lrrk2 transgene expression in P2 primary cultures (Figure 1b). An initial time course experiment determined day 8 as optimal for harvesting (Suppl. Figure 2) as dendritic protein MAP2 expression was robust and uniform, serving as an ideal marker for neuritic process and branch quantification. In contrast, axonal marker proteins such as tau revealed morphological variability over the first days in culture and were difficult to quantify at later time points.
Figure 1. Lrrk2 protein expression in BAC transgenic mice and primary cultures.
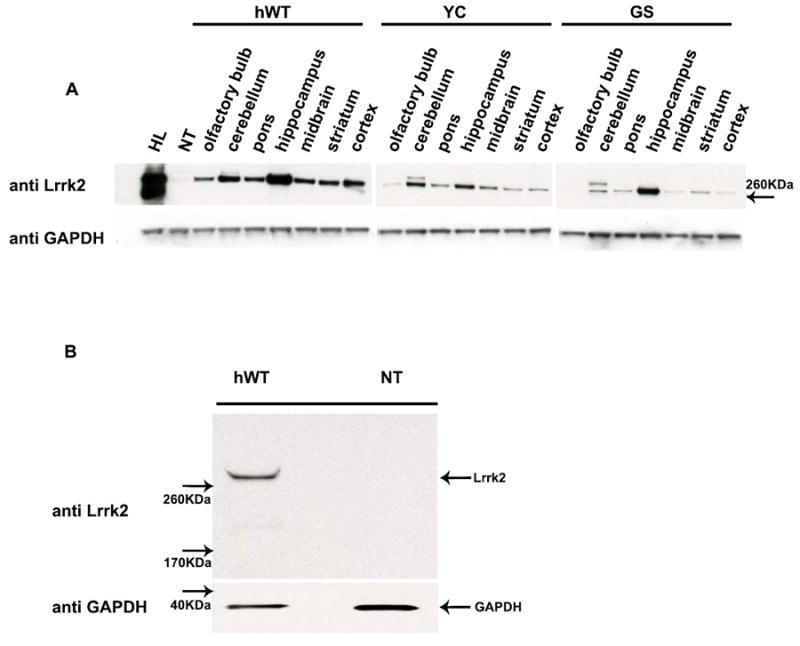
(A) Regional distribution of Lrrk2 (hWT, GS or YC) protein in adult transgenic mouse brain. Lysates of human lymphoblastoid cells (HL) and non-transgenic mouse brain (NT) served as controls. (B) P2 primary neurons (hWT) harvested on day 8 in-culture show robust Lrrk2 protein expression. GAPDH was used as a loading control.
To evaluate dendritic arborization, primary cultures of the various transgenic lines were analyzed for mean process length and the number of branches. Compared to non-transgenic littermate controls, primary neurons from both mutant lines (GS and YC) presented with significantly reduced neuritic outgrowth and branching (Figure 2a) while the number of processes per cell remained comparable (Suppl. Figure 3). We observed a similar outgrowth deficit in cells expressing YC or GS mutations, in contrast to previous reports based on viral transduction [11]. While over-expression of mutant Lrrk2 protein induced a significant deficit in neuritic outgrowth and branching, over-expression of Lrrk2 wild-type had no effect on these parameters (Figure 2b). Length, branching and the number of processes per cell were comparable between hippocampal neurons from hWT animals and littermate controls (NT) (Suppl. Figure 3).
To test the hypothesis that enhanced Lrrk2 kinase activity is responsible for the deficits in neuronal process morphology, cells over-expressing mutant Lrrk2 proteins (GS and YC) were treated with 200pM staurosporine (see material and methods sections for more details). This indolocarbazole derivate was recently confirmed as a Lrrk2 kinase inhibitor in-vitro, although its actions are promiscuous and may affect many cellular kinases [13,14]. Partial rescue of the neuritic outgrowth deficit was equivalent in both GS and YC cultures. The same concentration had no effect on non-transgenic littermate controls (Figure 2c).
A separate set of experiments was performed in LRRK2 KO models to investigate whether regulation of neuritic outgrowth and morphology is an inherent function of Lrrk2 (Suppl. Figure 1b,c). We have assessed homozygous KO, heterozygous KO and non-transgenic control littermates to evaluate the effect of zero, one and two endogenous Lrrk2 alleles. Hippocampal and midbrain primary cultures were included to compare results between regions of high (hippocampus) and low endogenous Lrrk2 expression (midbrain). TH-positive midbrain neurons were chosen because they are especially vulnerable to pathology and neuronal loss in PD.
We observed a comparable increase in mean process length and branching in both hippocampal and midbrain primary neurons derived from animals completely lacking endogenous LRRK2 (homozygous KO) in contrast to non-transgenic controls. Heterozygous KO animals, retaining one copy of the LRRK2 gene, revealed no change in either neuritic outgrowth or branching (Figure 3a and b).
As staurosporine is generally regarded as a relatively unspecific kinase inhibitor [14], we aimed to confirm that the partial rescue observed in BAC mutant LRRK2 cultures is due to Lrrk2 kinase inhibition. Hippocampal homozygous KO cultures were treated with 200pm staurosporine, as previously described. No significant difference in neuritic outgrowth was observed between treated and untreated cells (Figure 3c).
While all previous studies exploring the impact of mutant Lrrk2 on neuritic outgrowth [9-11] have focused on over-expression models, it remains unknown whether a comparable phenotype is reproducible in a system where mutant Lrrk2 is expressed at physiological levels and with the correct regional distribution. To evaluate whether Lrrk2 mutations expressed at endogenous levels induce a deficit in neuritic outgrowth and branching, primary hippocampal and midbrain cultures from homozygous G2019S knock-in (KI) animals (expressing mutant Lrrk2 endogenously) and littermate controls were generated. Endogenous LRRK2 mRNA is expressed in rodent brain during development and adulthood, beginning as early as embryonic day 15.5 [15,16]. Evaluation of mean process length (Figure 4), branching and the number of processes (data not shown) in either hippocampal or TH-positive midbrain neurons revealed no significant changes. Hence, endogenous expression of mutant Lrrk2 protein does not compromise neuritic outgrowth.
Discussion
Ex-vivo experiments in human BAC models revealed that over-expression of mutant but not wild-type Lrrk2 protein leads to pronounced deficits in mean process length and branching. These results compliment recent in-vivo studies where over-expression of human LRRK2 R1441G in mice lead to a marked diminution in the number of tyrosine hydroylase positive dendrites in the pars reticulata of the substantia nigra [17]. In both cases, toxic gain-of-function appears to be a mutant-specific phenotype. This is further underlined as Lrrk2 transgene and protein expression in BAC hWT animals is approximately twice that of GS or YC models (see materials and methods). Despite high Lrrk2 expression in primary cultures and within adult BAC mouse brain there is no evidence for apoptosis (nuclear condensation or fragmentation, or staining for caspases 3, 8 and 9; data not shown). A time course experiment of hippocampal cultures from mutant and hWT transgenic BAC mice demonstrates neuritic length and branching continues to increase up to our last harvesting time point at 21 days in culture. This is regardless of genotype and comparable to cultures derived from non-transgenic pups suggesting similar numbers of neurons survive. Our findings are in contrast to past results using acutely-induced LRRK2 cDNA expression, or mediated by viral or lipid transfection [18, 19]. Nevertheless in BAC models with constitutive (inherent) transgene expression there may be some developmental compensation.
Primary neurons from BAC GS and YC transgenic mice reveal similar deficits in neuritic outgrowth and branching. A previous study using viral transduction of Lrrk2 G2019S provided comparable results, but findings for the Y1699C mutation were not significant despite a similar trend [9]. Again, differences in how transgene expression was achieved may provide some explanation. Data from cultures over-expressing the Y1699C mutant, and other pathogenic mutations outside the kinase domain, may be insightful regarding the functional mechanism underlying the phenotypes observed.
Previously, neuronal polarity defined as the ratio of axons to dendrites was found to be unaffected by mutant Lrrk2 over-expression [9]. This finding supported a role for Lrrk2 in the regulation of lengthening and branching of existing neurites but not in the initial process formation and is reflected in our studies by a comparable number of total cellular processes for all conditions.
It is postulated that Lrrk2 kinase and Roc GTPase activities act in concert, possibly bridged by conformational changes in the Roc-COR-kinase structure, however there is currently no direct evidence supporting this hypothesis [20]. While Lrrk2 G2019S consistently enhances kinase activity in-vitro, data on the Y1699C mutation is equivocal and may be comparable to wild-type protein (reviewed by [21]). Staurosporine treatment partially rescues the neurite outgrowth deficit observed in both the GC and YC cultures, supporting enhanced kinase activity as the general mechanism. These results should be interpreted with caution since staurosporine is a relatively unspecific inhibitor that affects multiple cellular kinases [14]. Furthermore, staurosporine has previously been identified as a reagent that can induce neurite outgrowth in neuronal cells although the mechanism has yet to be resolved [22,23].Nevertheless, comparable dosing of staurosporine does not alter neuritic outgrowth in non-transgenic or KO lines, suggesting enhanced kinase activity in cells that express mutant Lrrk2 protein is responsible for the phenotype observed. Future studies focusing on more specific Lrrk2 inhibitors, and kinase and/or GTPase dead mutants, are required to define the role of Lrrk2 kinase activity in the regulation of neuritic outgrowth.
Results from the murine KO model corroborate an intrinsic role for Lrrk2 in the regulation of neuritic outgrowth and branching. Enhanced neuritic outgrowth was found in cultures derived from areas with both high (hippocampus) and low (midbrain) endogenous LRRK2 expression, emphasizing the importance of Lrrk2 in this process. In contrast, neurite outgrowth (process length and branching) in heterozygous animals, with only one functional endogenous copy of murine LRRK2, was comparable to non-transgenic littermates.
In nature, neither over-expression nor KO of Lrrk2 has been reported. Arguably, the most physiologically relevant model for studying Lrrk2-parkinsonism are KI mice, that express one or two alleles of the mutant protein [24]. Evaluation of neuritic outgrowth in hippocampal and midbrain primary cultures from G2019S KI mice suggests models with non-physiologic levels of Lrrk2 expression have an exaggerated response. Although deficits in neuritic length and branching were shown to be mutant specific, both features appear to be linked to high levels of transgene expression. In neurons from homo or heterozygous G2019S KI mice expressing endogenous levels of mutant Lrrk2 no deficits in neuritic outgrowth and branching were observed. As LRRK2 mutations are associated with late-onset PD, it is reasonable to expect over-expression of mutant protein, rather than physiological levels, are required to impair neuritic outgrowth and branching in primary cultures. Although compromised neuritic outgrowth may appear to have little in common with the pathologic changes observed in late-onset PD, many signaling pathways involved in neuritic outgrowth during development are reactivated during regeneration following toxic insults or may cause neurodegeneration if disregulated [25-28]. It is conceivable that mutant Lrrk2 renders neurons more susceptible to environmental stressors by compromising subsequent regenerative mechanisms. While over-expression of mutant Lrrk2 is sufficient to impair neuritic outgrowth on its own, endogenous levels of mutant protein might require the application of a second hit aimed to trigger Lrrk2-mediated deficits. Future studies that evaluate neuronal vulnerability in Lrrk2 over-expressing animals and KI models, and treated with a second hit are warranted. Results may better explain the susceptibility of LRRK2 mutation carriers for PD.
Supplementary Material
(A) Sequencing chromatogram confirming the presence of the G>A change at base 6055 and the G>C change at base 6057 (changing codon 2019 from GGG to AGC, and thus glycine to serine) in the murine cDNA isolated from Lrrk2 KI mice.
(B) Real time PCR with Taqman® gene expression probes to murine LRRK1 (Mm00713303_ml) and murine LRRK2 (Mm00481934_ml) in the mid-brain showing LRRK2 mRNA levels decrease in HET and KO mice compared with the NT mice, but LRRK1 mRNA levels remain similar. Murine GAPDH (Mm99999915_ml) was used a normalization control. Data presented as mean ± S.E.M.
(C) Immunoblots showing expression of Lrrk2 protein in NT, HET and KO mice utilizing Lrrk2 C-terminal antibody PA0362 (upper panel) and Lrrk2 N-terminal antibody Novus 267 (lower panel). GAPDH antibody was used as a loading control. Arrows denote the Lrrk2 band. 50μg of protein lysate isolated from one brain hemisphere was loaded per lane.
The graph reflects the increase in mean process length over time (arbitrary units). Cells were harvested after 2, 4, 6, 8, 14 and 20 days in culture. While an increase in mean process length was observed up to the last harvesting time point, the biggest changes were detected between day 6 and 8 in culture. Day 8 was selected as optimal for harvesting as potential differences may be most readily detectable. Error bars on all graphs represent standard errors of the mean.
Regardless of the respective genotype the mean numbers of processes are unchanged. The different graphs depict the number of processes in hippocampal primary cultures from BAC mutant (GS, YC), BAC hWT, KO and KI animals relative to cultures derived from non-transgenic littermate controls (A. one way ANOVA; B,C & D. unpaired t-test).
Acknowledgments
We would like to thank Gunnar P. Dietz and Karina Fog for their valuable advice and helpful discussions. Funding support was provided by the Mayo Clinic, NIH Grants NIA AG17216, NINDS NS40256, the Michael J Fox Foundation and H. Lundbeck A/S. Matt Farrer is a Canada Excellence Research Chairholder.
Abbreviations
- GS
G2019S
- HL
human lymphoblastoid cell line
- hWT
human wild-type
- KI
G2019S knock-in
- KO
mLRRK2 knock out
- NT
non-transgenic
- YC
Y1699C
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Farrer M, Ross O. LRRK2-Related Parkinson’s Disease. In: Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews. Seattle (WA): University of Washington, Seattle; 1993-2010. Internet; http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=lrrk2. [Google Scholar]
- 2.Bosgraaf L, Van Haastert PJ. Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta. 2003;1643:5–10. doi: 10.1016/j.bbamcr.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 3.Marin I, van Egmond WN, van Haastert PJ. The Roco protein family: a functional perspective. Faseb J. 2008;9:3103–10. doi: 10.1096/fj.08-111310. [DOI] [PubMed] [Google Scholar]
- 4.Farrer MJ, Stone JT, Lin CH, Dachsel JC, Hulihan MM, Haugarvoll K, et al. Lrrk2 G2385R is an ancestral risk factor for Parkinson’s disease in Asia. Parkinsonism Relat Disord. 2007;2:89–92. doi: 10.1016/j.parkreldis.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7:583–90. doi: 10.1016/S1474-4422(08)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hulihan MM, Ishihara-Paul L, Kachergus J, Warren L, Amouri R, Elango R, et al. LRRK2 Gly2019Ser penetrance in Arab-Berber patients from Tunisia: a case-control genetic study. Lancet Neurol. 2008;7:591–4. doi: 10.1016/S1474-4422(08)70116-9. [DOI] [PubMed] [Google Scholar]
- 7.Greggio E, Taymans JM, Zhen EY, Ryder J, Vancraenenbroeck R, Beilina A, et al. The Parkinson’s disease kinase LRRK2 autophosphorylates its GTPase domain at multiple sites. Biochem Biophys Res Commun. 2009;389(3):449–54. doi: 10.1016/j.bbrc.2009.08.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greggio E, Zambrano I, Kaganovich A, Beilina A, Taymans JM, Daniels V, et al. The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J Biol Chem. 2008;283(24):16906–14. doi: 10.1074/jbc.M708718200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52(4):587–93. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 10.Parisiadou L, Xie C, Cho HJ, Lin X, Gu XL, Long CX, et al. Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J Neurosci. 2009;29(44):13971–80. doi: 10.1523/JNEUROSCI.3799-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plowey ED, Cherra SJ, 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008;105(3):1048–56. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melrose HL, Kent CB, Taylor JP, Dachsel JC, Hinkle KM, Lincoln SJ, et al. A comparative analysis of leucine-rich repeat kinase 2 (Lrrk2) expression in mouse brain and Lewy body disease. Neuroscience. 2007;147(4):1047–58. doi: 10.1016/j.neuroscience.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 13.Covy JP, Giasson BI. Identification of compounds that inhibit the kinase activity of leucine-rich repeat kinase 2. Biochem Biophys Res Commun. 2009;378(3):473–7. doi: 10.1016/j.bbrc.2008.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nichols RJ, Dzamko N, Hutti JE, Cantley LC, Deak M, Moran J, et al. Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson’s disease. Biochem J. 2009;424(1):47–60. doi: 10.1042/BJ20091035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biskup S, Moore DJ, Rea A, Lorenz-Deperieux B, Coombes CE, Dawson VL, et al. Dynamic and redundant regulation of LRRK2 and LRRK1 expression. BMC Neurosci. 2007;8:102. doi: 10.1186/1471-2202-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Westerlund M, Belin AC, Anvret A, Bickford P, Olson L, Galter D. Developmental regulation of leucine-rich repeat kinase 1 and 2 expression in the brain and other rodent and human organs: Implications for Parkinson’s disease. Neuroscience. 2008;152(2):429–36. doi: 10.1016/j.neuroscience.2007.10.062. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Liu W, Oo TF, Wang L, Tang Y, Jackson-Lewis V, et al. Mutant LRRK2 R1441G BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat Neurosci. 2009;12(7):826–8. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9(10):1231–3. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- 19.Ho CC, Rideout HJ, Ribe E, Troy CM, Dauer WT. The Parkinson disease protein leucine-rich repeat kinase 2 transduces death signals via Fas-associated protein with death domain and caspase-8 in a cellular model of neurodegeneration. J Neurosci. 2009;29(4):1011–6. doi: 10.1523/JNEUROSCI.5175-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng J, Lewis PA, Greggio E, Sluch E, Beilina A, Cookson MR. Structure of the ROC domain from the Parkinson’s disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc Natl Acad Sci U S A. 2008;105(5):1499–504. doi: 10.1073/pnas.0709098105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greggio E, Cookson MR. Leucine-rich repeat kinase 2 mutations and Parkinson’s disease: three questions. ASN Neuro. 2009 April 14;1(1) doi: 10.1042/AN20090007. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li P, Matsunaga K, Yamakuni T, Ohizumi Y. Nardosinone, the first enhancer of neurite outgrowth-promoting activity of staurosporine and dibutyryl cyclic AMP in PC12D cells. Brain Res Dev Brain Res. 2003;145(2):177–83. doi: 10.1016/s0165-3806(03)00239-6. [DOI] [PubMed] [Google Scholar]
- 23.Min JY, Park MH, Park MK, Park KW, Lee NW, Kim T, et al. Staurosporin induces neurite outgrowth through ROS generation in HN33 hippocampal cell lines. J Neural Transm. 2006;113(11):1821–6. doi: 10.1007/s00702-006-0500-z. [DOI] [PubMed] [Google Scholar]
- 24.Ishihara L, Gibson RA, Warren L, Amouri R, Lyons K, Wielinski C, et al. Screening for Lrrk2 G2019S and clinical comparison of Tunisian and North American Caucasian Parkinson’s disease families. Mov Disord. 2007;22(1):55–61. doi: 10.1002/mds.21180. [DOI] [PubMed] [Google Scholar]
- 25.Haas MA, Vickers JC, Dickson TC. Rho kinase activates ezrin-radixin-moesin (ERM) proteins and mediates their function in cortical neuron growth, morphology and motility in vitro. J Neurosci Res. 2007;85(1):34–46. doi: 10.1002/jnr.21102. [DOI] [PubMed] [Google Scholar]
- 26.Iguchi Y, Katsuno M, Niwa J, Yamada S, Sone J, Waza M, et al. TDP-43 depletion induces neuronal cell damage through dysregulation of Rho family GTPases. J Biol Chem. 2009;284(33):22059–66. doi: 10.1074/jbc.M109.012195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacquier A, Buhler E, Schafer MK, Bohl D, Blanchard S, Beclin C, et al. Alsin/Rac1 signaling controls survival and growth of spinal motoneurons. Ann Neurol. 2006;60(1):105–17. doi: 10.1002/ana.20886. [DOI] [PubMed] [Google Scholar]
- 28.Temporin K, Tanaka H, Kuroda Y, Okada K, Yachi K, Moritomo H, et al. IL-1beta promotes neurite outgrowth by deactivating RhoA via p38 MAPK pathway. Biochem Biophys Res Commun. 2008;365(2):375–80. doi: 10.1016/j.bbrc.2007.10.198. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Sequencing chromatogram confirming the presence of the G>A change at base 6055 and the G>C change at base 6057 (changing codon 2019 from GGG to AGC, and thus glycine to serine) in the murine cDNA isolated from Lrrk2 KI mice.
(B) Real time PCR with Taqman® gene expression probes to murine LRRK1 (Mm00713303_ml) and murine LRRK2 (Mm00481934_ml) in the mid-brain showing LRRK2 mRNA levels decrease in HET and KO mice compared with the NT mice, but LRRK1 mRNA levels remain similar. Murine GAPDH (Mm99999915_ml) was used a normalization control. Data presented as mean ± S.E.M.
(C) Immunoblots showing expression of Lrrk2 protein in NT, HET and KO mice utilizing Lrrk2 C-terminal antibody PA0362 (upper panel) and Lrrk2 N-terminal antibody Novus 267 (lower panel). GAPDH antibody was used as a loading control. Arrows denote the Lrrk2 band. 50μg of protein lysate isolated from one brain hemisphere was loaded per lane.
The graph reflects the increase in mean process length over time (arbitrary units). Cells were harvested after 2, 4, 6, 8, 14 and 20 days in culture. While an increase in mean process length was observed up to the last harvesting time point, the biggest changes were detected between day 6 and 8 in culture. Day 8 was selected as optimal for harvesting as potential differences may be most readily detectable. Error bars on all graphs represent standard errors of the mean.
Regardless of the respective genotype the mean numbers of processes are unchanged. The different graphs depict the number of processes in hippocampal primary cultures from BAC mutant (GS, YC), BAC hWT, KO and KI animals relative to cultures derived from non-transgenic littermate controls (A. one way ANOVA; B,C & D. unpaired t-test).