

Abstract
Visual impairment and blindness is widespread across the human population, and the development of therapies for ocular pathologies is of high priority. The zebrafish represents a valuable model organism for studying human ocular disease; it is utilized in eye research to understand underlying developmental processes, to identify potential causative genes for human disorders, and to develop therapies. Zebrafish eyes are similar in morphology, physiology, gene expression and function to human eyes. Furthermore, zebrafish are highly amenable to laboratory research. This review outlines the use of zebrafish as a model for human ocular diseases such as colobomas, glaucoma, cataracts, photoreceptor degeneration, as well as dystrophies of the cornea and retinal pigmented epithelium.
Keywords: zebrafish, eye, ocular, disease, retina, lens, RPE, cornea, glaucoma, vasculature
I. Introduction
A. Overview
Visual impairments affect over 160 million people world-wide, of whom 37 million are blind [1]. Major causes of blindness include cataracts, glaucoma, age-related macular degeneration, and diabetic retinopathy, and the development of effective therapies for these disorders is of high priority. Model organisms with similar physiology to humans are vital to understand underlying developmental processes, identify potential causative genes for human disorders, and develop therapies.
This review focuses on the zebrafish as a model organism for studying human eye diseases. The zebrafish is a remarkably amenable model organism for scientific research, and it combines advantages characteristic of invertebrate models with those inherent to vertebrates for modeling human physiology. As will be explained in the following sections, the zebrafish eye is similar in morphology, physiology, gene expression, and function to the human eye. Zebrafish researchers utilize mutant zebrafish recovered from forward genetic screens along with a number of reverse genetic techniques to model many of the major eye diseases which plague humans (Table 1).
Table 1.
Zebrafish mutants and morphants with ocular phenotypes relevant to human disorders and associated pathologies. Due to space limitations, not all relevant mutant and morphant zebrafish are included in this table.
| Gene | Mutant / Morpholino |
Ocular Phenotype | References | Associated Human Ocular Disease* |
OMIM |
|---|---|---|---|---|---|
| Coloboma | |||||
| adenomatous polyposis coli (apc) | Mutant | Coloboma; defects in optic vesicle patterning and optic fissure closure | [98] | Familial Adenomatous Polyposis | 175100 |
| bcl6 co-repressor (bcor) | Morpholino | Coloboma; microphthalmia | [246] | Oculofaciocardiodental and Lenz microphthalmia | 300485 |
| cadherin 2 neuronal (cdh2, glass onion) | Mutant | Coloboma; optic fissure closure defect | [247] | — | — |
| laminin,β1 (lamb1, lamb1hi1113bTg), laminin,γ1 (lamc1, lamc1hi3890Tg) | Mutant | Coloboma; basement membrane defects | [105] | — | — |
| paired box gene 2a (pax2a, no isthmus) | Mutant | Coloboma | [81] | Renal-coloboma Syndrome | 167409 |
| patched1 (ptc1, blowout) | Mutant | Coloboma; defects in optic stalk morphogenesis | [84, 85] | — | — |
| thioredoxin-related transmembrane prtein 3 (tmx3) | Morpholino | Microphthalmia; coloboma | [248] | Microphthalmia and coloboma | — |
| transcription factor ap2 alpha (tfap2a) | Morpholino | Coloboma | [249] | Branchio-Oculo-Facial Syndrome | 107580 |
| zinc family member 2a (zic2a) | Morpholino | Coloboma; defects in optic stalk morphogenesis and optic vesicle patterning | [88] | — | — |
| zinc finger proteins 703 and 503 (znf703,503, also known as nlz1 and nlz2) | Morpholino | Coloboma; defects in optic vesicle patterning and optic fissure closure | [79] | — | — |
| Photoreceptors | |||||
| cone transducin α (tcα, no optokinetic response f) | Mutant | Reduced sensitivity to light by cones | [250] | — | — |
| crumbs homolog 2 (crb2, oko meduzy) | Mutant | Photoreceptor defects | [119] | — | — |
| crumbs homolog 2, like (crb2l) | Morpholino | Photoreceptor defects | [119] | — | — |
| dihydrolipoamide S-acetyltransferase (pdhe2, no optokinetic response a) | Mutant | Blindness; photoreceptor synaptic transmission defects | [133, 136] | Pyruvate Dehydrogenase Deficiency | — |
| dynactin 1a (dctn1a, mikre oko) | Mutant | Retinal degeneration | [123, 251] | — | — |
| dynactin 2 (p50), (dctn2, ale oko) | Mutant | Retinal degeneration | [120] | — | — |
| erythrocyte membrane protein band 4.1-like 5 (epb41l5, mosaic eyes) | Mutant | Photoreceptor defects | [111, 112] | — | |
| fleer (flr) | Mutant | Rod outer-segment defects | [129, 252] | — | — |
| intraflagellar transport proteins, 57, 80, 88, and 172 (ift57hi3417Tg, oval(ift88), ift172hi2211Tg) | Mutants (ift57, 88, 172), Morpholino (ift80) | Outer segment defects; retinal degeneration | [104, 126, 127, 253] | — | — |
| membrane protein, palmitoylated 5a (mpp5a, nagie oko) | Mutant | Disrupted RPE; retinal laminaiton defects; photoreceptor defects | [254, 255] | — | — |
| Novel protein (partial optokinetic response b) | Mutant | Cone degeneration | [256, 257] | — | — |
| phosphodiesterase 6 alpha (pde6α, eclipse) | Mutant | Cone degeneration | [258] | — | — |
| protein kinase C iota (prkci, heart and soul) | Mutant | Photoreceptor morphogenesis defects | [259] | — | — |
| protocadherin 15b (pcdh15b) | Morpholino | Photoreceptor defects; visual function defects | [260] | Usher Syndrome | 605514 |
| TNF receptor-associated factor 3 interacting protein (traf3ip, elipsa) | Mutant | Photoreceptor defects; visual defects | [128, 129] | — | — |
| Unknown (brudas) | Mutant | Photoreceptor defects | [129] | — | — |
| Unknown (niezerka) | Mutant | Photoreceptor defects | [129, 261] | — | — |
| Unknown (nightblindness a,b,e,f,g) | Mutant | Visual function defects; retinal degeneration (nba, nbe, nbf) | [137, 138, 141] | — | — |
| Other Retinal Phenotypes | |||||
| patched2 (ptc2, leprechaun) | Mutant | Müller glial reactivity; vitreo-retinal abnormalities | [165] | Basal Cell Naevus Syndrome (BCNS) | 601309 |
| phosphatase and tensin homolog b (ptenb) | Mutant | Ocular tumors | [262] | — | — |
| RPE | |||||
| choroideremia (chm) | Mutant | Retinal degeneration; RPE defects | [145, 146] | Choroideremia | 300390 |
| protein kinase C iota (prkci, heart and soul) | Mutant | RPE morphogenesis defects | [259] | — | — |
| silver homolog a (silva, fading vision) | Mutant | RPE defects; photoreceptor defects | [158] | — | — |
| vacuolar protein sorting 18p (vps18, vps18phi2499aTg) | Mutant | Melanosome maturation defects; reduced visual function | [149] | — | — |
| vacuolar protein sorting 39 homolog (vps39, leberknodel) | Mutant | RPE vesicle traffic defects; PR defects | [148] | — | — |
| v-ATPase complex (multiple genes) | Mutants | Melanosome defects; photoreceptor outer segment defects; | [153] | — | — |
| Unknown (bleached) | Mutant | Pigmentation defect; blindness; retinal degeneration | [155] | — | — |
| Unknown (fade out) | Mutant | RPE defects; photoreceptor defects | [156] | — | — |
| Unknown (gantenbein) | Mutant | Cone dystrophy; RPE degeneration | [154] | — | — |
| Hyaloid Vasculature | |||||
| forkhead box C1a (foxc1a) + forkhead box C1b (foxc1b) | Morpholino (coinjection) | reduced hyaloid basement membrane integrity | [186] | Axenfield-Reiger Syndrome, glaucoma | 601090 |
| heparan sulfate 6-O-sulfotransferase 2 (hs6st2) | Morpholino | aberrant patterning of hyaloid vasculature | [185] | — | — |
| laminin, α1 (lama1) | Morpholino | hyaloid vasculature dysmorphogenesis | [209] | — | — |
| laminin, α1 (lama1, bashful) | Mutant | no hyaloid vasculature | [185] | — | — |
| mab-21-like 2 (mab21l2) | Morpholino | reduced hyaloid vasculature | [185] | — | — |
| microfibrillar-associated protein 2 (mfap2) | Morpholino | reduced hyaloid vasculature branching | [185] | — | — |
| plexin D1 (plxnd1, out of bounds) | Mutant | aberrant patterning of hyaloid vasculature | [185] | — | — |
| syndecan 2 (sdc2) | Morpholino | no vasculature on the lens | [185] | — | — |
| Unknown (fused eyes) | Mutant | no hyaloid vasculature | [185] | — | — |
| Unknown (margin affected) | Mutant | reduced, then absent hyaloid vasculature | [185] | — | — |
| Unknown (platinum) | Mutant | premature detachment of hyaloid vasculature from lens | [185] | — | — |
| Lens | |||||
| cadherin 4, retinal (cdh4) | Morpholino | Small opaque lens | [263] | — | — |
| CDP-diacylglycerol-inositol 3-phosphatidyltransferase (cdipt, lens opaque) | Mutant | Lens cell hyperproliferation; lens degeneration | [264–266] | — | — |
| choroideremia (chm) | Mutant | Small opaque lens | [217] | Choroideremia | 300390 |
| coatomer protein complex, subunit ζ1 (copz1, copz1hi528Tg) | Mutant | Cortical lens defects | [104] | — | — |
| connexin 48.5 (cx48.5) | Morpholino | Cataracts and small lens | [267] | Cataracts | 121015 |
| decapentaplegic and Vg-related 1 (dvr1) | Morpholino | Retention of nuclei in lens fibers | [268] | — | — |
| fibroblast growth factor 19 (fgf19) | Morpholino | Defective lens cell survival and differentiation | [240, 269] | — | — |
| forkhead box E3 (foxe3) | Morpholino | Lens dysmorphogenesis; epithelial cell hyperproliferation; defective fiber differentiation | [202, 203] | Congenital primary aphakia, Peter's anomaly, Cataracts | 601094 |
| growth differentiation factor 6a (gdf6a) | Morpholino | Cortical lens defects; defective lens gene expression | [270, 271] | Microphthalmia | 601147 |
| heat shock cognate 70-kd protein (hsp70) | Morpholino | Immature lens | [272] | — | — |
| heat shock transcription factor 1 (hsf1) | Morpholino | Small lens | [273] | — | — |
| integrator complex subunit 7 (ints7, ints7hi1548Tg, ints7hi3649Tg) | Mutant | Devere lens disorganization | [104] | — | — |
| laminin, α1 (lama1) | Morpholino | Lens degeneration | [210] | — | — |
| laminin, α1 (lama1, bashful) | Mutant | Lens degeneration, focal corneal dysplasia | [209, 215, 216, 219, 264] | — | — |
| laminin, β1 (lamb1, grumpy, lamb1hi1113bTg) | Mutant | Cortical lens defects; lens dysplasia | [105, 217, 219] | — | — |
| laminin, γ1 (lamc1, sleepy, lamc1hi3890Tg) | Mutant | Cortical lens defects; lens dysplasia and degeneration | [104, 105, 219] | — | — |
| lengsin, lens protein with glutamine synthetase domain (lgsn) | Morpholino | Lens dysmorphogenesis; lens fiber defects | [274] | — | — |
| mab-21-like 2 (mab21l2) | Morpholino | Lens cell death | [275] | — | — |
| paired box gene 2a (pax2a, no isthmus) | Mutant | Small opaque lens by 6 dpf | [217] | Coloboma | 167409 |
| paired box gene 6b (pax6b) | Morpholino | Variable phenotype: small lens to absent lens | [276] | Peter's anomaly, Aniridia | 607108 |
| paired box gene 6b (pax6b, sunrise) | Mutant | Small lens | [277, 278] | Peter's anomaly, Aniridia | 607108 |
| paired-like homeodomain transcription factor 3 (pitx3) | Morpholino | Lens dysmorphogenesis: disordered epithelial cells, defective fiber differentiation, fiber cell death | [200–202] | Cataracts, Anterior segment dysgenesis | 602669 |
| patched 1 (ptc1) | Morpholino | Small, dysplastic lens | [84] | — | — |
| retinal homeobox gene 3 (rx3, chokh) | Mutant | Small lens, no retina | [279] | Anophthalmia/ Microphthalmia | 601881 |
| RNA binding motif 42 (rbm42, rbm42hi2735ATg) | Mutant | Cortical lens defects | [104] | — | — |
| syndecan 2 (sdc2) | Morpholino | Small lens | [185] | — | — |
| ubiquitin-like, containing PHD and RING finger domains, 1 (uhrf1, uhrf1hi272Tg, uhrf1hi3020Tg) | Mutant | Lens disorganization and degeneration | [104] | — | — |
| WD repeat domain 36 (wdr36, wdr36hi3630aTg) | Mutant | Thickening of lens epithelium; cortical lens defects; lens degeneration | [280] | Primary open-angle glaucoma | 609669 |
| Unknown (bumper) | Mutant | Lens epithelial cell hyperproliferation, lens fiber degeneration | [135, 278, 281] | — | — |
| Unknown (chiorny) | Mutant | Small lens | [282] | — | — |
| Unknown (cloche) | Mutant | Opaque lens with retained nuclei | [205] | — | — |
| Unknown (disrupted lens) | Mutant | Disorganized lens fibers | [215, 264] | — | — |
| Unknown (dou yan) | Mutant | Small lens | [283] | — | — |
| Unknown (korinthe) | Mutant | Lens degeneration | [278] | — | — |
| Unknown (margin affected) | Mutant | Small lens | [185] | — | — |
| Unknown (platinum) | Mutant | Small lens | [185] | — | — |
| Unknown (rosine) | Mutant | Lens degeneration | [278] | — | — |
| Unknown (yol) | Mutant | Lens degeneration | [164] | — | — |
Online Mendelian Inheritance in Man (OMIM) entries are listed if the disrupted zebrafish gene is orthologous to a human gene associated with an ocular disease.
B. The Zebrafish as a Model Organism
The zebrafish, Danio rerio, is a common aquarium fish that originated in the Ganges region of India [2]. The rapid increase in its popularity for the study of vertebrate development and genetics is mainly due to its transparent embryos that develop ex-utero, making the visualization of developmental events possible. In addition, its large clutch sizes and ease of maintenance, features that are commonly associated with invertebrate model organisms such as Drosophila, make it an attractive alternative to model species such as the mouse. Zebrafish have a short generation time of 2–4 months and a single mating pair can produce around 200 offspring on a weekly basis [3]. These, combined with the low relative cost of raising zebrafish, make it an ideal model organism as evidenced by the rapid growth of the zebrafish research community.
Since its introduction as a model organism three decades ago, advances in available technologies pertaining to zebrafish research have quickly closed the gap with those available for other model organisms (see ‘Tools and Techniques’ section). Importantly, the Zebrafish Information Network (ZFIN) maintains a central comprehensive online database that provides researchers with access to information and services that include gene expression pattern databases, mutant and transgenic lines, and links to a fully sequenced genome (www.zfin.org).
C. Tools and Techniques
External development of zebrafish embryos provides the opportunity for various manipulations from the time of fertilization. These include the injection of tracer dyes or of mRNA (for transient expression of protein). Established fate-maps [4, 5] enable the transplantation of labeled blastula- or gastrula-stage donor cells into specific regions of host embryos. When the donor and host are of different genetic backgrounds, the resulting chimeric zebrafish can be used to determine whether a gene whose mutation is embryonic-lethal early in development has a role later in the life of the organism, or to establish the cell- or tissue-autonomy of a mutant phenotype [6]. Of particular relevance to this review, entire lenses can be transplanted between embryos to determine lens- versus retinal-contribution to an ocular phenotype [7].
1. Targeted Knockdown of Gene Expression
Homologous recombination strategies utilized in other model organisms to introduce specific mutations into the genome are not yet available in zebrafish. However, specific and heritable targeted mutagenesis has recently been effected with zinc-finger nucleases [8, 9]. Additionally, transient gene knock-down is commonly performed in zebrafish by injection of morpholinos to block gene expression [10, 11]. Morpholinos are effective and inexpensive oligomeric constructs designed to bind mRNA and prevent proper splicing or translation. Morpholinos are effective for several days, and their transient activity is sufficient for the embryonic timeframe of many zebrafish experiments. Morpholinos can also be introduced into specific tissues later in development by electroporation [12], and photoactivatable morpholinos (which can be activated in specific cells of transparent embryos by a laser) allow precise spatiotemporal control of gene knock-down [13]. Thus, zinc finger nucleases and morpholinos complement the catalog of mutant fish lines generated from mutagenesis screens.
2. Transgenic Techniques
Transient or stable introduction of transgenes in zebrafish is effected in a number of ways. Injection of DNA into a fertilized egg results in mosaic, transient expression in the embryo and low-efficiency integration into the genome [14–16]. Tol2 transposon-mediated strategies result in much higher efficiency transgene integration [17], and transgenic lines of zebrafish are routinely created by utilizing this technique. Specific spatiotemporal control of transgene expression is achieved by placing the transgene under the control of a ubiquitous promoter [18], heat-shock promoter [19], or tissue-specific enhancer/promoter elements [20]. In addition to spatiotemporal control over expression of genes of interest, these techniques may be utilized to express dominant negatives [21] or genetically-encoded reporter constructs such as GFP [18]. Genetically-encoded photoconvertible proteins, such as Kaede, may be utilized for lineage-tracing experiments [22]. Transgenes can also be used to ablate specific cells by expression of either diptheria toxin [23] or nitroreductase (in which case cell-autonomous ablation is not elicited until the application of a prodrug) [24].
Many of the transgenic tools originally implemented in mouse and Drosophila research are also utilized in zebrafish. The Gal4-UAS system for ectopic gene expression involves spatiotemporally-controlled transcription of gal4 under a promoter of choice. The Gal4 protein then binds to and activates expression of genes downstream of its upstream activation sequence (UAS) [20, 25, 26]. Cre-LoxP recombination [27], which allows for excision or inversion of a segment of transgene DNA upon activation of Cre, has also been utilized in zebrafish [28]. Cre-recombinase can be activated in zebrafish in many ways, including most recently the photo-uncaging of 4-OH-cyclofen for activation of a ligand-inducible Cre [29]. Therefore, the future of zebrafish transgenesis is extremely exciting.
D. Genetic Screens
Large-scale genetic screens in Drosophila and C. elegans have identified numerous genes required for embryonic development [30, 31]. Similar approaches were thought to not be feasible in vertebrates due to long generation times and small number of progeny of traditional vertebrate models such as the mouse and chick [3]. However, the pioneering work of George Steisinger nearly three decades ago established the zebrafish as a powerful genetic model organism for the identification of genes important for vertebrate development [32, 33]. Two large-scale genetic screens performed in Christiane Nusslein-Volhard and Wolfgang Driever’s labs followed fifteen years later and were published in a special issue of the journal Development, describing mutations affecting various aspects of vertebrate development [34, 35]. Since then, multiple large- and small-scale mutagenesis efforts have produced numerous mutant lines that have been studied to better understand vertebrate development, as well as disease.
Most genetic screens in zebrafish utilize the chemical mutagen ethylnitrosourea (ENU) due to its high mutagenic efficiency (Figure 1A) [36, 37]. Like ethyl methanesulphonate (EMS), which is commonly used in genetic screens in Drosophila, ENU is an alkylating agent that produces point mutations in the adult germline. While ENU mutagenesis has proved to be a powerful method for the induction of point mutations in the zebrafish, the identification of the mutated gene can be laborious. Positional cloning of a single mutation requires the work of one researcher for approximately six to twelve months [38]. A separate screen performed in Nancy Hopkins’ lab used retroviral insertional mutagenesis as an alternative (Figure 1B). Since the genetic sequence of the insertion is known, the location of the retroviral insertion can be assayed using standard PCR techniques, quickly and reliably revealing the affected gene [39, 40]. While the efficiency of mutagenesis using retroviral insertion is estimated to be about one-seventh that of ENU mutagenesis, the cloning of one mutation requires only three to four weeks to perform [38]. The main disadvantage of insertional mutagenesis is that, due to low efficiency, larger screens need to be performed as compared to ENU-based screens in order to identify equal numbers of mutants. As a result, the need for larger facilities make these screens impractical for most academic labs.
Figure 1. Chemical versus insertional mutagenesis screens in zebrafish.
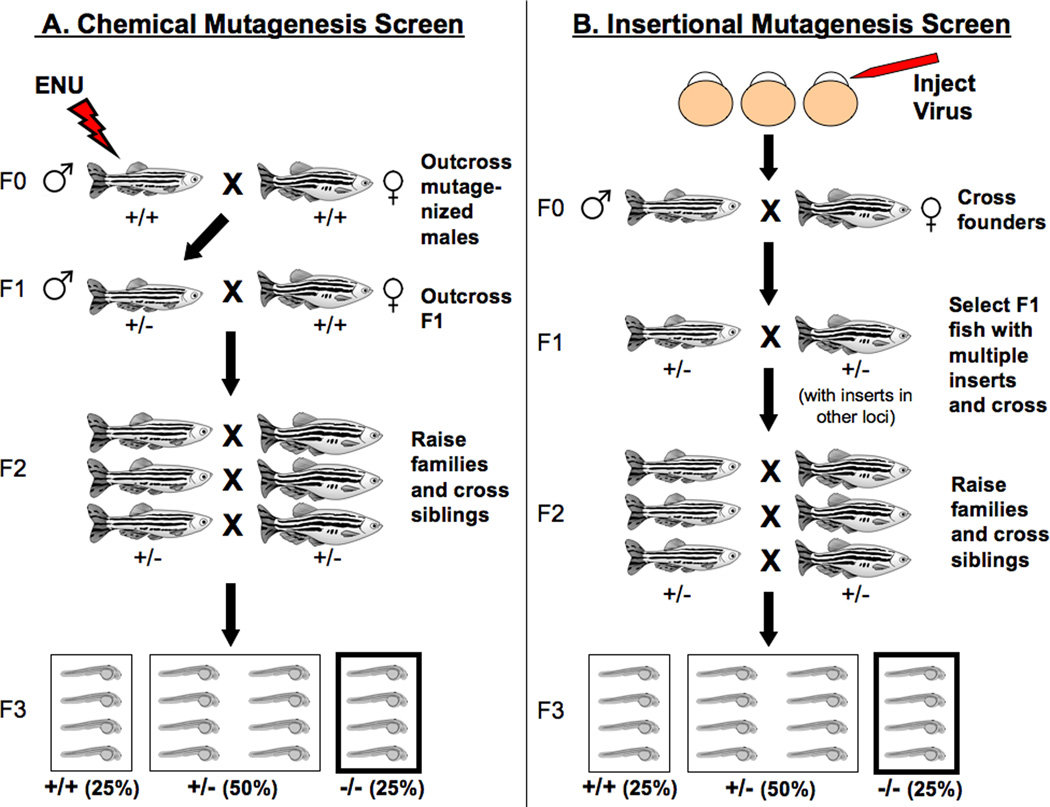
In chemical screens (A), the mutagen ethynitrosourea (ENU) is used to induce mutations in the adult male germline. The mutagenized males (F0 generation) are then out-crossed to generate a heterozygous F1 population. F1 progeny are crossed again to wild-type fish, giving rise to F2 families that carry a specific mutation. F2 siblings are bred to each other in order to generate an F3 generation out of which 25% will be homozygous for the mutation [284, 285]. In an insertional screen (B), a retrovirus is injected into fertilized eggs to generate transgenic adults (F0) that will be subsequently bred to generate an F1 generation with multiple viral inserts. PCR and Southern blot analysis is then performed in order to identify F1 fish that carry multiple inserts. As in a chemical screen, F1 fish are bred to generate F2 families; those will be subsequently bred to siblings to give rise to F3 progeny that will be screened for physiological and/or behavioral phenotypes [40, 285].
The identification of ENU-induced point mutations can be simplified using a technique known as TILLING (Targeted Induced Local Lesions in Genomes). Unlike traditional screens in which mutants are isolated in the F3 generation and then positionaly cloned, with TILLING mutations can be identified in the F1 generation. Mutagenized males are bred to generate large populations of F1 offspring whose DNA is analyzed by PCR in order to identify mutations in a gene of interest. A mutation of interest is then isolated by out-crossing a single identified carrier [41]. This approach has generated over 150 loss-of-function alleles yet to be published [42]. In addition, a consortium has been recently established to consolidate TILLING efforts, greatly improving the likelihood of finding mutations of interest in the zebrafish genome. Researchers can now place requests for mutations in specific genes online (www.sanger.ac.uk/projects/D_rerio/mutres).
Mutants isolated from genetic screens can further be used to screen for small molecules that might suppress the mutant phenotype. These compounds can then be further studied as potential therapeutic agents for a particular defect or disease [43]. Small molecule screens, like genetic screens, utilize the already mentioned advantages of zebrafish: large clutch size, rapid development, and transparency of the embryos allowing for the rapid screening of compounds that affect certain aspects of development. The dosage, as well as the time during development of administration can be controlled and the effects on embryonic development assayed by various methods such as in-situ hybridization, vital dyes and transgenics to visualize effects on specific tissues, as well as behavioral assays [44–47]. Since the first large-scale small molecule screen was published ten years ago [48], multiple screening efforts have identified compounds that affect various biological processes including cell cycle and cancer, control of stem cell populations, and the formation of retinal vasculature [45, 46, 49].
E. Eye Development and Anatomy
The zebrafish has long been recognized as a useful model for the study of human ocular development and disease [50–53]. Detailed characterization of the embryonic development of the posterior segment of the eye, which includes the neural retina [54] and the RPE [55] and the anterior segment (which includes the lens, cornea, ciliary body, and the various tissues of the iridocorneal angle [56–68]), has not only shed light on the sequence of events in vertebrate eye development, but has also highlighted the similarities in the architecture of the zebrafish eye to that of the human eye.
In zebrafish, eye development is rapid. The optic vesicle, which will ultimately give rise to the neural retina and the retinal pigment epithelium, evaginates from the forebrain at around 12 hours post fertilization (hpf) and remains attached to and continuous with the forebrain through a transient structure called the optic stalk (Figure 2). The optic vesicle then gives rise to the optic cup through a series of morphogenetic events that occur from about 16 hpf to 20 hpf [68]. Morphogenesis of the optic cup continues as the optic fissure forms ventrally by 24 hpf and subsequently closes by 48 hpf. Neurogenesis begins at 28 hpf and by as early as 72 hpf zebrafish embryos exhibit visual function [67].
Figure 2. Development and morphogenesis of the zebrafish eye.
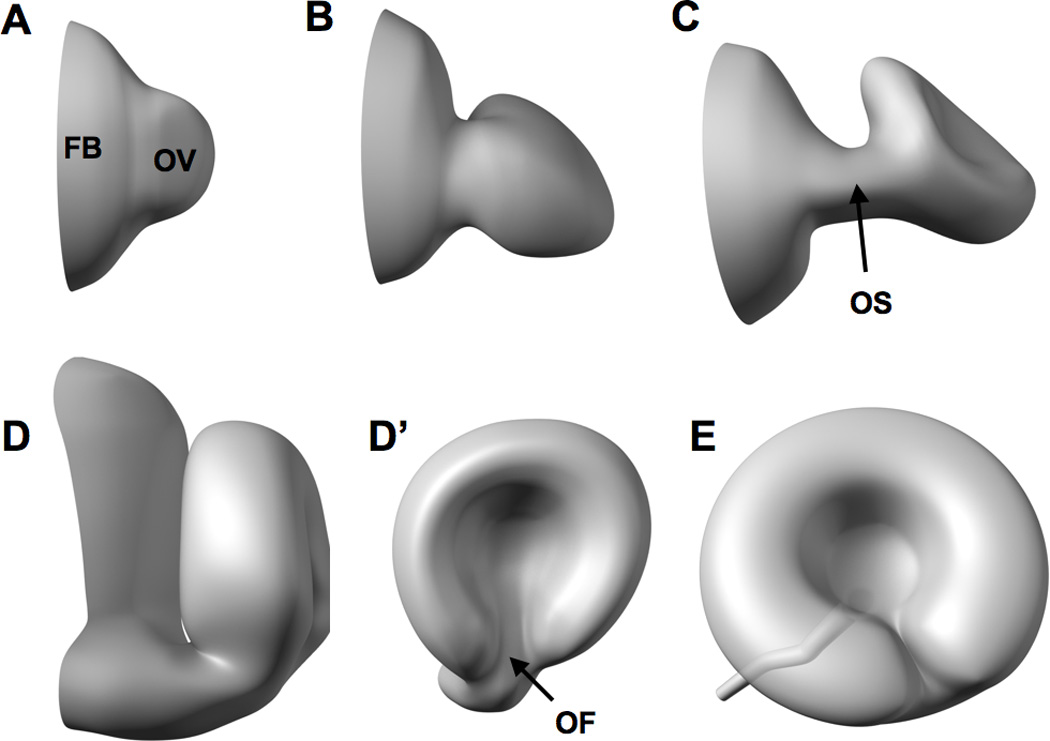
Eye development commences around 12 hpf as the optic vesicle (OV) evaginates from the forebrain (FB) (A). The optic vesicle then elongates into a flattened wing-like structure at around 16 hpf (B) that is attached to the forebrain through a transient structure called the optic stalk (OS in C). The eye subsequently rotates and invaginates (C) to form the ‘optic cup’ at around 24 hpf as depicted in D (anterior view) and D’ (lateral view). Morphogenesis of the embryonic eye is mostly complete by 48 hpf as the optic fissure (OF in D’) is closed and neurogenesis of the retina is underway [54, 68, 286].
The anterior segment of the embryonic eye develops concurrently with the events mentioned thus far. At 16 hpf, surface ectoderm cells overlying the optic cup thicken to form the lens placode ([57], Figure 3), the lens mass delaminates from the surface ectoderm at approximately 24 hpf, and fully detaches by 26 hpf [57, 59, 60, 67, 68]. The surface ectoderm overlying the lens becomes the corneal epithelium, which is two cell layers thick by 30 hpf [62]. Migratory periocular mesenchymal cells (which first enter the enter the anterior chamber of the eye at 24 hpf) coalesce to form the corneal endothelium between 30 and 36 hpf [60, 62, 67, 68].
Figure 3. Early lens development in zebrafish and mouse.

After [58]. In zebrafish (A–E) and mouse (A’–E’), the surface ectoderm overlying the optic cup (A, A’) thickens to form the lens placode (B, B’). In zebrafish, the lens mass delaminates from the surface ectoderm (C, D), while in mouse the lens placode evaginates to form the lens vesicle (C’, D’). Primary lens fibers in the zebrafish elongate within the lens mass in a circular fashion (E), whereas the primary lens fibers in the mouse elongate to the anterior to fill the lens vesicle space (E’). The remaining surface ectoderm becomes the corneal epithelium in both zebrafish and mouse. [57, 59, 67, 191]. Anterior is to the left.
Humans are a diurnal species, and day-time vision is predominantly mediated by cone photoreceptors in the retina. In contrast to nocturnal mice and rats, whose retinas contain few cones, larval zebrafish vision is mediated almost entirely by cone photoreceptors [69]. As in humans, the mature zebrafish retina is composed of three nuclear layers separated by two plexiform layers. Zebrafish possess four types of cones (blue, UV, and red/green double cones) and one rod cell type [70]. Rod and cone cell bodies reside in the outer nuclear layer (ONL), while the inner nuclear layer (INL) is occupied by amacrine, horizontal, bipolar cells, and Müller glia. Visual signals originating in the photoreceptors are transmitted through the retina to the ganglion cells, which make up the ganglion cell layer (GCL); their axons then relay the signal to the brain [54, 71].
II. Posterior Segment
A. Coloboma
Early during vertebrate eye development, the optic vesicle evaginates from the forebrain and ultimately forms the optic cup (see introduction). As a result of a series of morphogenetic processes that follow, a transient gap called the optic fissure forms ventrally and subsequently closes by the fusion of the surrounding tissue [68, 72]. Failure of optic fissure closure is the underlying cause of colobomas, which ultimately affect one or more parts of the eye including retina, choroid, iris cornea, lens, ciliary body, and optic nerve [73]. In humans, the incidence of coloboma is estimated at around 1 per 10,000 births [74–76]; severe colobomas may cause as much as 10% of childhood blindness [77]. The clinical presentations of colobomas are varied, as numerous genetic loci have been associated with this diverse class of ocular pathologies [78]. While the causes underlying most human coloboma conditions remain unknown, studies in zebrafish have contributed to our understanding of the developmental bases of optic fissure closure.
A gene expession profiling study was conducted recently by Brown et al to identify genes whose transcript levels are enriched in the optic fissure of the mouse. Two related genes, Nlz1 and Nlz2, were shown to be expressed in the optic fissure; subsequent morpholino knockdown of both in zebrafish resulted in coloboma [79]. Brown, et al demonstrated that nlz1 and nlz2 may function in the optic fissure by directly regulating the expression of the transcription factor pax2a. In nlz1/2-deficient embryos, pax2a expression was drastically reduced, and both the nlz1 and nlz2 proteins bound the pax2 promoter in vitro, suggesting direct transcriptional regulation. In humans, mutation in the PAX2 gene result in coloboma as part of renal-coloboma syndrome (OMIM 120330) [80], and the human PAX2 and zebrafish pax2a genes are expressed in the developing optic stalk and optic fissure [81, 82]. Interestingly, the zebrafish No-isthmus (noi) mutant line, which carries a non-sense mutation in the pax2a gene, exhibit optic fissure closure defects, resulting in coloboma [81].
Additional studies in zebrafish have shown that pax2a is transcriptionally regulated by Sonic hedgehog (Shh), a well-studied developmental morphogen. In the zebrafish mutant cyclops, in which a mutation affecting the nodal pathway results in a deficiency in Shh signaling, pax2a transcript levels are highly reduced in the optic stalk [83]. In blowout (blw) mutants, which possess a loss-of-function mutation in the patched1 gene, the expression domain of pax2a is increased, resulting in an enlarged optic stalk and colobomas [84, 85]. The Patched1 protein is the receptor and negative regulator of the Hedgehog pathway; mutations in patched genes are known to result in an overactive Hedgehog pathway [86]. Interestingly, colobomas have been described in a family carrying specific deletions in the human SHH gene [87], suggesting that the genetic interaction between Shh and pax2a in zebrafish might be conserved in humans. This interaction was recently shown in zebrafish to be mediated by zic2a, a transcription factor whose activity is controlled by Shh. zic2a restricts pax2a expression to the optic stalk, and loss of zic2a activity results in the expansion of pax2a expression into the retina, resulting in coloboma [88]. While human ZIC2 mutations have not yet been associated with the formation of coloboma, they have been linked to holoprosencephaly, a forebrain defect frequently associated with colobomas [88, 89]. Studies in zebrafish have therefore identified ZIC2 as an additional candidate gene in human colobomas.
The Hedgehog pathway has also been shown to control the expression of vax genes (vax1 and vax2), whose function has also been shown to be important for the proper development and specification of the optic stalk and ventral retina, tissues in which they are normally expressed [90, 91]. Morpholino knockdown of both vax1 and vax2 in zebrafish resulted in the loss of optic stalk and ventral retinal tissue identity, and the presence of colobomas. Over-expression of Shh results in the expansion of vax gene expression domains, while blocking the Hh pathway reduces vax gene expression. While Shh appears to control the expression of both pax2a and vax genes, these likely act in parallel to specify optic stalk identity, as the experimental loss of pax2a does not lead to transcriptional loss of vax genes, and vice versa [91]. Since both pax2a and vax genes encode transcription factors, it remains to be seen what factors might act downstream of pax2a and to directly control the proper development of the optic stalk and ventral retina.
While the Hedgehog pathway seems to play a central role in dictating optic stalk identity and proper optic fissure closure, other developmental pathways have been implicated in proper morphogenesis of the eye. The requirement of retinoic acid (RA) during ocular development, for example, is well established. RA is derived from vitamin A, which has been linked to ocular pathologies in humans, including coloboma [92]. In zebrafish, exposure to exogenous RA early during eye development results in an enlarged optic stalk and, depending on the dose of RA, duplication of the ventral retina [93, 94]. Conversely, embryonic exposure to citral, which blocks RA synthesis, results in optic stalk and ventral retinal loss and ocular coloboma [95]. In addition, morpholino knockdown of bcox, an enzyme critical for the synthesis of RA, results in numerous developmental phenotypes, including coloboma [96].
A possible relationship between RA synthesis and ocular colobomas has emerged through the characterization of the zebrafish adenomatous polyposis coli (apc) mutant. Human mutations in APC, a well studied tumor suppressor, underlie familial adenomatous polyposis syndrome [97]. These patients suffer from failure in the development of the ventral retina and coloboma, phenotypes that are also found in zebrafish apc mutants. Through elegant gain- and loss-of-function experiments, Nadauld et al demonstrated that Apc regulates RA synthesis indirectly through the regulation of Rdh5, an enzyme responsible for RA synthesis in the ventral retina. These findings suggest that a deficiency of retinoic acid, a vitamin A derivative, might underly ocular phenotypes in familial adenomatous polyposis syndrome [98].
The expression of RA synthesizing enzymes in the optic vesicle and optic cup in zebrafish, chick, and mouse suggests that RA is required autonomously in the eye for proper ocular development [99–101]. However, surprisingly, studies in mouse have demonstrated that RA activity in the neural crest-derived periocular mesenchyme (POM) is responsible for early eye morphogenesis [102]. Consistent with these findings, migration defects and subsequent apoptosis of POM in the zebrafish lmx1b morphant lead to morphogenetic defects in the optic fissure and colobomas. Loss of lmx1b results in a transient up-regulation of Fgf signaling, which in turn at least partially affects proper patterning and morphogenesis of the embryonic eye [103].
The developmental pathways discusses thus far most likely regulate optic fissure closure indirectly, through the regulation of other factors. Studies in zebrafish have also suggested that the molecular mechanisms underlying optic fissure closure are likely to involve interactions between the cells of the retina/optic stalk with the extracellular matrix (ECM). Mutations in laminin β1 and γ1 (lamβ1 and lamγ1), for example, result in disruptions in basement membrane integrity and the presence of a number of ocular phenotypes including coloboma [104, 105]. Laminins are major components of the ECM, which serves as a scaffold for tissue morphogenesis, as well as a site for the deposition of growth factors such as Fgf and Hedgehog ligands. In the future, it would be interesting to determine whether ocular phenotypes in laminin mutants might be a result of misregulation of these signaling pathways and/or a consequence of morphogenetic defects due to perturbed cell-ECM interactions.
B. Photoreceptors
Photoreceptor degenerations are the most common form of blindness in the Western world and involve the loss of vision due to dystrophy and/or death of retinal photoreceptors. These pathologies can be roughly divided into those conditions that initially affect rod photoreceptors, such as retinitis pigmentosa (RP), and those that initially affect cone photoreceptors, such as macular degeneration [53]. While photoreceptor degeneration can also be caused indirectly by primary defects in the RPE, which serves an important function in photoreceptor health and homeostasis, this topic will be discussed in a later section (see ‘RPE’).
Photoreceptors are highly polarized sensory cells that consist of an inner segment (IS) that is connected to an outer segment (OS) by a highly modified cilium (Figure 5) [106]. In mature photoreceptors, proteins that are required for growth and maintenance of photoreceptor OS’s, as well as for phototransduction, are transported along polarized microtubules from the basal IS to the apical OS. Photoreceptors are similar to epithelial cells in that their surfaces are divided into apical and basolateral domains by cell junctions. Their centrosomes are located at the apical surface, while their nuclei reside basally [107]. Proper development and subsequent maintenance of vertebrate photoreceptors relies on the establishment of proper apical-basal polarity, as well as the function of the transport machinery. It is therefore not surprising that of the more than 100 genetic loci known to cause photoreceptor degenerations in vertebrates, most affect the structure and function of the OS [108].
Figure 5. Structure of rod and cone photoreceptors.
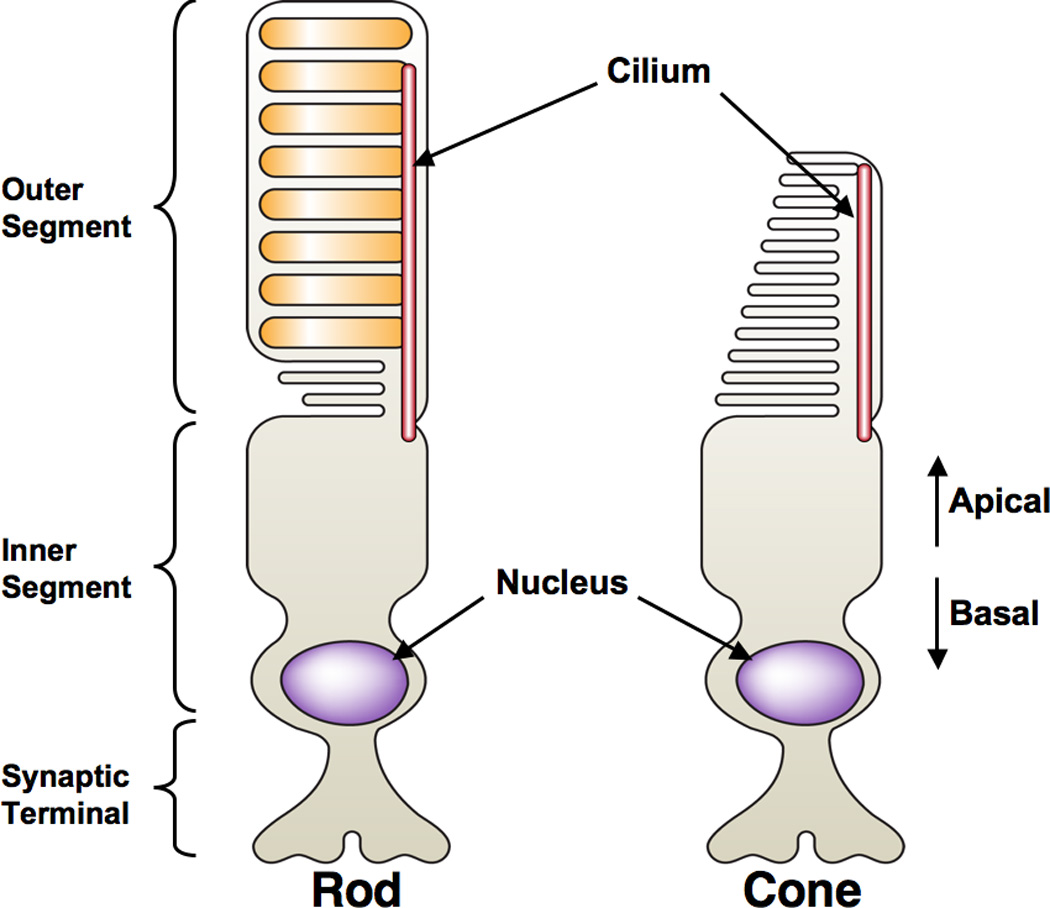
Photoreceptor cells are composed of an outer segment that contains visual pigment-bound membrane disks. The inner segment is the polarized cell body of the photoreceptor, where the nucleus resides basally and the cilium originates apically and extends into the OS. The synaptic terminal forms synaptic connections to the interneurons of the retina, relaying visual input [71, 287].
Genetic screens in zebrafish have shed light on the molecular bases of these cellular functions by the isolation of mutants in which photoreceptor development and/or maintenance are defective [104, 109, 110]. One such mutant, mosaic eyes (moe), was found to contain expanded apical features in photoreceptors [111, 112]. The moe locus encodes a FERM-domain containing protein that forms a complex with Crumbs proteins, which in turn have been shown to be critical for apical-basal polarity in Drosophila, as well as vertebrates [113–115]. Interestingly, mutations in the human orthologue of Crumbs, CRB1, are associated with retinal photoreceptor dystrophies such as retinitis pigmentosa 12 [116] and Leber’s congenital anamurosis (LCA1, [117, 118]). Other zebrafish mutants, such as nagie oko (nok) and heart and soul (has) do not only phenocopy the moe mutant phenotype, but the proteins that they encode (Pals1 and PKCi, respectively) also interact biochemically with Moe [111]. Moreover, morpholino knockdown of crb2b, a zebrafish Crumbs paralog, results in the reduction of IS size [119]. These studies have therefore begun to uncover the role of the Crumbs complex in vertebrate photoreceptor development and disease.
The formation of correct apical-basal polarity depends not only on the proper function of cell polarity determinants, but also on their transport and localization. Photoreceptors in the ale oko (ako) mutant retina accumulate the apical determinants Pals1 and PKCλ in their cell bodies and exhibit extensive photoreceptor death late in development [120]. The ako locus encodes the p50 subunit of the dynactin complex, which serves an important role in the transport of cargo along microtubules as part of the dynein motor complex [121]. In another mutant, mikre oko (mok), which possesses a mutation in dynactin-1, retinal degeneration is at least partially due to mislocalization of photoreceptor nuclei. Surprisingly, unlike ako mutants, apical determinants such as Crumbs and aPKCλ are not mislocalized, suggesting that degeneration in these mutants is not due to loss of cell polarity. Experimental mislocalization of the nucleus by the overexpression of the dynein motor resulted in photoreceptor cell death, supporting the role of nuclear position in photoreceptor survival [122]. While nuclear positioning defects in mok photoreceptors seems to have a cell-autonomous role, other non-cell autonomous components seem to play a role in mok degeneration. Mosaic analyses have revealed that mutant photoreceptors display over a 2.5 fold increase in survival when placed in a wild-type environment [123]. This finding suggests that photoreceptor health and survival depends on environmental cues from surrounding cells. These findings are consistent with defects associated with some human degenerations, such as retinitis pigmentosa, where an initial defect in the rod specific rhodopsin ultimately results in the loss of both rods and cones [124].
Transport from the cell body to the OS is also important to replace OS components that are lost due to the continual phagocytosis of photoreceptor OS’s by the overlying RPE [125]. In photoreceptors, as in cilia, transport occurs by a process known as Intraflagellar Transport (IFT) and mutations in IFT component genes such ift57, ift80, ift88, and ift172 result in OS atrophy and/or complete loss of OS’s in zebrafish [104, 126, 127]. Mutations in the elipsa gene, which encodes a protein that is thought to facilitate IFT [128], result in early photoreceptor loss [129], while morpholino knockdown of multiple subunits of the retrograde IFT motor dynein-2 has highlighted the importance of IFT in proper OS elongation and visual function [130]. Photoreceptor degenerations relating to defective IFT are some of the many cilia related phenotypes associated with Bardet-Biedl Syndrome (BBS) [131], and the further study of the function of IFT components in zebrafish might prove to be instrumental in gaining a better understanding of the molecular causes underlying BBS-related ocular pathologies.
While the mutants described thus far in this section were isolated in genetic screens utilizing mainly histological methods to detect photoreceptor defects, others were isolated in behavioral screens [132–135]. One such screen took advantage of the ability of zebrafish to exhibit vision-dependent behavior as early as 3dpf. Brockerhoff et al first used the optokinetic response assay (OKR) on mutagenized embryos to detect defects in visual function. A second assay involved the use of electroretinogram (ERG) recordings to identify whether isolated mutations affect outer retinal function [133]. From this screen, 18 mutants were isolated that were determined to have reduced visual function. One, no optokinetic response a (noa), possessed no gross photoreceptor abnormalities at 5dpf, but was found to be blind, as well as lethargic, and died prematurely. Analysis of the noa mutation revealed a deficiency in a subunit of the pyruvate dehydrogenase (PDH) complex, which regulates energy production in cells. The noa mutant line has been used as a model for the study of PDH deficiency (OMIM 245348), a human disorder that, like the zebrafish model, results in blindness, neurological defects, and early death [136]. Since current treatments for PDH deficiency in humans have resulted in limited success, Taylor et al utilized noa mutants to test a ketogenic diet which has shown some success in alleviating PDH deficiency symptoms in a limited number of human patients. Administration of this special diet restored normal behavior in noa mutants, highlighting the potential of studying PDH deficiency therapies in the noa mutant line.
Another mutant, no optokinetic response f (nof) was found to possess a mutation in the α subunit of cone transducin (Tcα), a G-protein required for phototransduction. In human patients suffering from a condition known as achromatopsia (OMIM 139340), mutations in Tcα underlie loss of color vision. In nof mutants, cone development occurs normally; they are, however, up to 1000x less sensitive to light, as detected by recordings of single photoreceptors. Extensive analysis of cone light responses revealed that residual phototransduction is light-dependent, but transducin independent. Ca+2 influx, which is important for photoreceptor light adaptation and was previously thought to be controlled by transducin, was still detectable in nof mutants. This study therefore revealed that some Ca+2 influx in cone photoreceptors might be transducin independent.
In a separate screen, dominant mutations that cause photoreceptor degeneration in adult zebrafish was performed in order to isolate genetic mutants that could be later used to study human inherited night blindness such as retinitis pigmentosa (RP) [137, 138]. In the case of RP, many genetic loci underlie this group of disorders. However, only about half of the cases of dominant RP have been linked to specific mutations at the time the screen was performed [138]. Li et al therefore screened mutagenized adult zebrafish by utilizing a known escape response exhibited by the fish. A lack of such response to a threatening cue was interpreted as a loss of vision, which was later confirmed using ERG recordings. In all, seven heterozygous mutants were isolated (nightblindness a,b,c,d,e, and f), out of which six exhibited photoreceptor degeneration [138–141]. Four of the six were found to be embryonic lethal as homozygotes, suggesting that the mutated genes underlying the photoreceptor phenotypes have other critical functions during embryonic development. This finding highlights the importance of such screens for the identification of dominant mutations that might not be easily isolated in traditional screening due to early embryonic lethality.
C. RPE
The RPE is a monolayer of pigmented cells that serves in the protection and maintenance of photoreceptors, and is therefore essential for visual function. In addition to its role in absorbing excess light entering the eye, the RPE transports essential nutrients from the blood to photoreceptors, while removing ions, water, and metabolic end products from the retina to the blood. Vertebrate phototransduction depends on the conversion of all-trans-retinal to 11-cis-retinal through a series of biochemical reactions termed the retinoid cycle, and most of these essential reactions are carried out by the RPE rather than photoreceptors [142]. Finally, the RPE supports photoreceptor renewal by the daily phagocytosis of about 10% of OS volume, while the photoreceptor itself regenerates roughly the same volume each day to maintain proper OS length [143]. Failure of any of these functions often results in retinal degeneration and loss of vision ([144]).
An important question in the study of many types of retinal degenerations is whether photoreceptor degeneration occurs due to a primary defect in photoreceptors themselves or as a secondary consequence of RPE degeneration. Since in many cases defects in both photoreceptor and RPE are present, a cause-and-effect relationship cannot be easily established between these cell types. Krock et al studied this relationship in the zebrafish rep1/chm mutant line, which exhibits retinal phenotypes consistent with those found in humans suffering from choroideremia, a form of hereditary retinal degeneration associated with mutations in the human REP1 gene [145]. Rep proteins are involved in the posttranslational modification of Rab protein, and are therefore critical for vesicle trafficking. Since both photoreceptors and the RPE depend on vesicle trafficking for transport of material (such as opsin) to the OS and degradation of phagocytosed OS material, respectively, the initial defect due to the rep1 mutation could potentially lay in either tissue. Through the use of mosaic analysis, Krock et al showed a loss of rep1 in the RPE is sufficient to cause photoreceptor degeneration and results in the localized accumulation of OS material. These findings, together with the observation that opsin was not mislocalized in rep1 photoreceptors suggest that defective cellular processes within the RPE are the primary cause of photoreceptor degeneration in choroideremia patients. In a separate study however, Moosajee et al showed that zebrafish do not possess an orthologue of a second human REP gene, REP2. REP2 plays an essential role in the pathogenesis of choroideremia as its function is thought to compensate for the loss of REP1. The authors of the study therefore concluded that the rep1 mutant line may not be a suitable model for the study of choroideremia in its present form [146].
Defects in vesicle trafficking, formation, and/or fusion are known to be the underlying cause of many human ocular diseases such Chediak-Higashi syndrome (CHS, OMIM 214500), Hermansky-Pudlak syndrome (HPS, OMIM 203300), and Griscelli syndromes (GS1-3, OMIM 214450, 607624, and 609227). The pathologies of these human syndromes include hypo-pigmentation of the RPE, as well as loss of visual function. The RPE contains lysosome-related organelles known as melanosomes that synthesize and store melanin, the main pigment present in the melanosomes of the RPE [147]. Proper fusion of lysosome-related organelles depends on the homotypic fusion and vacuolar protein sorting (HOPS) complex, as it is required for SNARE complex assembly. A model for the study of human conditions such as CHS and HPS has emerged recently in the zebrafish leberknödel (lbk) mutant line. lbk mutants possess a mutation in the vam6/vps39 gene, which encodes a component of the HOPS complex. Hypopigmentation of the lbk RPE, a common characteristic of vesicle traffic defects, results from a defect in melanosome maturation, and the RPE contains vesicles containing undigested photoreceptor OS’s. As a result, OS’s are shortened and lbk mutants display reduced visual function [148]. A mutant in another HOPS complex component, vps18, also possesses immature melanocytes and hypopigmentation, and can serve as an additional model for the study of the underlying cellular and molecular basis of ocular defects in hypopigmentation-related disorders [149].
In addition to their role in melanin synthesis, melanosomes are also thought to play a role in the degradation and detoxification of phagocytosed photoreceptor outer segments by the RPE [150]. In human patients suffering from age-related macular degeneration (ARMD), phagocytosed OS’s are not properly digested by the RPE. This defect results in the accumulation of the lipofuscin component A2E, which is thought to further inhibit OS component degradation, contributing to the progression of ARMD [151]. It is thought that A2E might inhibit the activity of vacuolar ATPases (v-ATPase), whose known role is the acidification of lysosomes and lysosome-related organelles, such as melanosomes [152]. Acidification of these organelles is, in turn, important for the activity of proteases and therefore for proper degradation of accumulated materials. Consistent with this model, a recent analysis of five separate zebrafish mutant lines defective in different subunits of the v-ATPase complex revealed accumulations of undigested OS material in the RPE [153]. While no mutations in v-ATPase have been linked to blindness in humans, these results suggest that defects in v-ATPase might directly or indirectly cause disorders that affect RPE function.
Some of the other mutants that have been shown to possess RPE defects coupled to retinal degeneration are gantenbein (gnn), bleached (blc), fading vision (fdv), and fade out (fad) [154–156]. Of those, only the mutation underlying the fdv phenotype has been cloned and identified. The fdv mutant was originally isolated during a mutagenesis screen due to reduced pigmentation in the eye as well as the rest its body [157]. Like the lbk, vps18, and v-ATPase mutants, the fdv RPE contains vesicular inclusions and their OS’s are reduced. However, the visual defects identified in fdv mutants are thought to be due to disrupted recycling of visual pigment, a defect that can be attributed to primary defects in melanosome biogenesis [158]. Positional cloning identified a mutation in the silver a/pmel17 gene, whose orthologue has been originally shown to be important for pigmentation in mice [159]. While silver orthologues in various species have been described and its overall function in melanosome biogenesis established, the molecular mechanisms underlying its roles in pigmentation in general and RPE function specifically are still controversial [160]. Further study of existing mutants, such as fdv, as well as the identification and cloning of additional pigmentation mutants, are therefore critical for our understanding of role of the RPE in ocular development and disease.
D. Müller Glia in Regeneration and Disease
The study of stem cells has been of major interest due to the growing potential of their use in therapeutic and regenerative medicine. The ability of zebrafish to regenerate various tissues after injury has attracted major attention from researchers seeking to identify the cellular and molecular mechanisms that enable this regenerative capacity, which is often absent in mammals [161–163]. In the vertebrate retina, two populations of stem/progenitor cells have been identified: the Müller glia of the central retina, and the ciliary marginal zone (CMZ) at the most peripheral edge of the retina. Both of these stem cell populations add new neurons and glia to the zebrafish retina throughout the lifetime of the animal. Müller glia give rise to rod photoreceptor precursors that migrate along Müller glial processes to the photoreceptor layer. All other retinal cell types are continually produced in the post-embryonic CMZ. While the study of the stem/progenitor populations of the CMZ in zebrafish has seen much progress in the past few years, including the identification of mutants with CMZ-specific phenotypes [104, 164–168], the remainder of this section will focus on recent findings regarding Müller glia.
Neuroprotection, maintenance of retinal homeostasis, and the establishment of proper retinal lamination are some of the many functions attributed to Müller glia in the healthy retina ([169]). In response to retinal disease or damage, Müller glia can become ‘reactive’, as characterized by changes in gene expression that are often followed by de-differentiation and proliferation [170, 171]. Virtually every human retinal disease is associated to some degree with Müller glial reactivity [169]. In some cases, such as diabetic and proliferative retinopathies, and retinal detachment, Müller glia are thought to become reactive in response to primary detects arising in another cell type. In others, Müller glia are thought to be the primary cell type affected. Still, in many human eye diseases, the role of Müller glia is the onset of the disease is unclear. In humans suffering from Basal Cell Naevus Syndrome (BCNS), epiretinal membranes (ERMs) - proliferative and structural abnormalites at the boundary between the retina and the vitreous, have been shown to contain a major glial component. Studies in Ptch mutant mice have shown that Müller glial reactivity is locally associated with BCNS-related ocular pathologies [172]. However, it is yet unclear whether ocular defects in BCNS patients are a result of a primary defects at the level of Müller glia, or conversely, whether Müller glia are reactive in response to another ocular defect. Recently, a zebrafish model for the study of BCNS-related ocular pathologies has emerged in the leprechaun (lep) mutant line, which possesses a loss-of-function mutation in the patched2 gene, the zebrafish orthologue of PTCH. lep mutants display retinal abnormalities that are similar to those observed in human BCNS patients, including disruptions at the vitreo-retinal interface [165]. Further study of ptc2 mutants is required to determine the role of Müller glia in BCNS-related ocular pathologies.
Müller glia have long been known to possess neurogenic potential, having an intrinsic ability to give rise to newborn neurons. In the adult retina of lower vertebrates, differentiated Müller glia continue to express molecular markers that are characteristic of early retinal progenitors, such as pax6 and rx1 [173]. Clusters of proliferating rod progenitors are closely associated with Müller glia and appear to migrate along their processes, which span the apical-basal width of the adult retina, to their final location in the photoreceptor layer [174, 175]. A recent study utilizing transgenic zebrafish that express GFP under the control of a Müller glia-specific promoter (Tg(gfap:GFP)) as a lineage tracer has shown that rod progenitors retain low levels of GFP, suggesting that Müller glia indeed give rise to rod photoreceptor precursors in the adult retina [176]. These findings support the neurogenic potential of Müller glia and raise the intriguing possibility of using Müller glia as a source for cell replacement therapies for humans suffering from age-related photoreceptor loss.
While in the uninjured adult retina Müller glia give rise strictly to progenitors destined to the rod lineage, Müller glia are also able to dedifferentiate, proliferate, and replenish all neuronal cell types in response to retinal damage [171, 176]. While conclusive lineage tracing to show Müller glia as the sole source of regenerated neurons have not yet been published, the use of zebrafish transgenic lines together with extensive immuno-histochemical analysis has strongly supported the role of Müller glia as the stem cell population responsible for the regenerative response in the central retina [176, 177]. Since mutant analysis in adults is labor intensive and time consuming, multiple labs have also performed microarray analyses after retinal damage in order identify genes that play a role in these processes. Consistent with the idea that regenerative processes often re-capitulate embryonic development, these studies identified known developmental pathways such as BMP, Notch, Wnt and Hedgehog as being transcriptionally regulated in reactive Müller glia [178, 179]. It remains to be seen what exact roles of each of these pathways in retinal regeneration, and whether the genetic and/or chemical manipulation of these pathways might have therapeutic value in humans.
E. Intraocular Vasculature
The vertebrate eye is highly metabolically active, and the intraocular vasculature provides oxygen and other nutrients [180, 181]. Many human diseases present with defects in intraocular vascularization, including diabetic retinopathy, retinopathy of prematurity, and age-related macular degeneration [181]. Similarities between zebrafish and human intraocular vasculature make the zebrafish an exciting new model organism in this field. For instance, Alvarez and colleagues [182] recently showed that a zebrafish model of hyperglycemia [183] recapitulates some aspects of non-proliferative diabetic retinopathy; zebrafish may therefore be a useful model for this very common human disease.
In humans and in zebrafish, the hyaloid vessel network surrounds the posterior lens during development [184, 185]. In humans, the hyaloid vasculature regresses completely by birth, during which time the retinal vasculature (which lines the inner limiting membrane) forms [184]. In contrast, hyaloid regression does not take place in zebrafish; the hyaloid vasculature instead remodels into the retinal vasculature [185]. In humans and in zebrafish, the choroid vasculature covers the outer surface of the retina [46, 181].
One study utilized a transgenic zebrafish line in which cells of the vasculature express GFP (Tg(fli1a:EGFP)y1) to screen various mutant and morphant zebrafish for altered formation of the intraocular vasculature [185]. Utilizing this strategy, the authors demonstrated that several genes are required for hyaloid and retinal vasculature development and maintenance, including syndecan 2 (morphants had no vasculature on the lens), and mab21l2 (morphants displayed disrupted hyaloid vessel patterning). Another study which investigated the effects of foxc1 depletion (by morpholino) on hyaloid vessel ultrastructure found that the integrity of the basement membrane of hyaloid vascular cells was disrupted [186].
Also utilizing transgenic zebrafish lines which express GFP in the vasculature (Tg(fli1a:EGFP)y1 and Tg(kdrl:GFP)la116), Kitambi and colleagues [46] performed a small molecule screen to search for compounds which altered retinal vasculature without significant alteration in trunk vasculature. The authors reported two classes of compounds: one which induced degeneration of vessels, and one which increased vessel diameter [46]. This study provides an example of the powerful role that zebrafish small molecule screens can play in therapeutics research.
III. Anterior Segment
A. Lens
The lens is a specialized transparent tissue which, in conjunction with the cornea, focuses incoming light onto the retina [187]. Lens opacity, or cataract, is the leading cause of human visual impairment world-wide [1]. Most cases of cataracts are age-related, and likely have both genetic and environmental causes [188]. In addition, mutations in multiple genes are known to underlie congenital cataracts (which arise in children during the first year of life) [189]. However, there are many cases of cataract and other lens disorders for which no causative gene is yet known [190].
Development and adult morphology of the lens is similar in humans and in zebrafish, but there are important differences as well. During the early stages of lens development in humans and most other vertebrates, cells of the surface ectoderm invaginate to form the lens vesicle [191, 192]. In zebrafish lens development, surface ectoderm cells within the lens placode delaminate as a solid cluster of cells rather than a vesicle (Figure 3) [57, 59, 67]. Despite this difference, cell fate analysis demonstrates that the relationship between zebrafish lens placode cell position and cell fate within the mature lens is consistent with the mammalian model [58], which indicates that a common genetic program may exist for lens development in vertebrates.
In zebrafish and humans, the mature lens is made up of tightly-packed lens fibers which are surrounded at the anterior periphery by a proliferative monolayer of lens epithelial cells (Figure 4C). Throughout the life of the organism, epithelial cells near the transition zone exit the cell cycle, migrate, elongate, and degrade their light-scattering organelles to become new secondary fibers [57, 59, 60, 193–195]. In zebrafish, this mature lens state is reached by 72 hpf [58], by which time the lens appropriately focuses an image onto the plane of the retinal photoreceptors [67]. As lens opacity results from many defective cellular processes [189], the early maturity of the zebrafish lens provides a convenient readout for gene requirement by the lens in mutants and knock-down experiments.
Figure 4. Structure of the zebrafish eye.

(A) Transverse histological section of a wild-type zebrafish eye at 5 dpf. (B) Illustration of the major neuronal and glial cell types in the zebrafish retina. Rods and cones relay sensory input to retinal interneurons (Horizontal and Bipolar Cells). Following synaptic interactions with Amacrine Cells, the information is passed to the output neurons, the Ganglion Cells. Müller glia perform multiple functions in the retina, including maintaining retinal health and structure (see ‘Müller Glia’ section in text). C, Cones. R, Rods. HC, Horizontal Cells. BP, Bipolar. AC, Amacrine Cells. RGC, Ganglion Cells. MG, Müller Glia. (C) Diagram of the zebrafish lens at 5 dpf. Lens epithelial cells surround the anterior periphery; proliferation of these cells is mainly restricted to the lateral proliferative zone. Epithelial cells at the transition zone exit the cell cycle, migrate, elongate, and degrade their light-scattering organelles to become new secondary fibers. Tightly-packed lens fibers which extend from the posterior suture to the anterior suture make up the bulk of the lens. [57–60, 193–195]. ECs, lens epithelial cells. LPZ, lateral proliferative zone. TZ, transition zone. Fs, lens fibers. AS, anterior suture. PS, posterior suture.
There are subtle differences in the mature structure of the lens between zebrafish and humans. The epithelial cell layer on the surface of the mature zebrafish lens extends further toward the posterior suture; in mammals, epithelial cells end near the lens equator [59]. The shape of the mature zebrafish lens is also more spherical than the human lens, and it is responsible for nearly all refraction to focus light onto the retina, unlike terrestrial vertebrates in which much of the light refraction is performed by the cornea [56, 196, 197].
When genes whose mutation causes human congenital cataracts are knocked down in zebrafish embryos, cataracts or other lens abnormalities often result (Table 1). Zebrafish therefore provide a useful model to characterize the molecular mechanism of cataract formation. Human mutations in transcription factors PITX3 and FOXE3 cause various anterior segment disorders, including cataracts [198, 199]. Expression of both of these genes has been knocked down in zebrafish via antisense morpholino [200–203], and for both genes this results in lens dysmorphogenesis. Further experiments utilizing these morpholino-injected embryos demonstrated that that foxe3 is genetically downstream of pitx3 in the zebrafish lens [202].
Zebrafish with lens defects identified in large-scale mutagenesis screens can also provide valuable information. Proteins known as crystallins contribute to the transparency and refractive power of the lens and cornea [187], and mutations in many lens crystallins are associated with cataracts in human patients [204]. The zebrafish mutant cloche (the genetic basis of which is unknown) presents with cataracts as well as severe vascular defects [205, 206]. In these mutants, both αA-crystallin mRNA and protein expression is decreased, and the γ-Crystallin present within the lens is predominantly insoluble. Both the cataract phenotype and γ-Crystallin insolubility were rescued in cloche mutants by overexpression of αA-crystallin by mRNA injection, likely due to the protein chaperone activity of αA-crystallin [205]. This study demonstrated the requirement for αA-crystallin for proper lens development, as well as providing an exciting example of cataract prevention.
The lens capsule is a transparent basement membrane which completely encloses the lens. Laminin is a component of the lens capsule, and the expression of specific Laminin subunits is conserved between zebrafish and humans [207]. One component of zebrafish and human lens capsules is Laminin-111, a heterotrimeric protein made up of Lama1, Lamb1, and Lamc1 subunits [105, 208–212]. None of these subunit genes have so far been associated with an ocular disease in humans, although humans with mutations in LAMB2 have lens defects including abnormal shape and cataract [213, 214]. However, zebrafish mutants or morphants in lama1, lamb1, and lamc1 all present with defective lens phenotypes [104, 105, 135, 209, 210, 215–219].
In lama1 morpholino-injected embryos, the lens degenerates completely by 48 hpf [210], and in various homozygous alleles of lama1 mutants, the lens is dysmorphic and eventually degenerates completely [209, 219]. Homozygous mutants in lamb1 or lamc1 present with very similar lens phenotypes characterized by lens dysplasia at 5 dpf [105], and the lens degenerates completely by 6–9 dpf [105, 219]. Zebrafish deficient in subunits of Laminin-111 provide a useful model system to elucidate the roles for laminin-containing extracellular matrix in the lens. For instance, one study found that reduced Focal Adhesion Kinase signaling likely underlies the lens phenotype in lama1 mutants [209].
B. Cornea
As the most anterior structure within the eye, the transparent cornea provides protection and, in terrestrial vertebrates, refraction of incoming light [56, 196, 197]. Major causes of corneal blindness include the infectious disease Trachoma, vitamin A deficiency, and injury [1]. Human genetic corneal dystrophies, though less common, are another cause of blindness due to corneal opacity [220].
In humans and in zebrafish, the mature cornea contains five major layers: the stratified corneal epithelium at the anterior, Bowman’s Layer, the stroma, Descemet’s membrane, and the corneal endothelium [60, 62, 63, 66, 221]. At 72 hpf in zebrafish, a rudimentary cornea consisting of a two-cell-layer epithelium, an acellular stroma, and the endothelium is in place [60, 62]. Bowman’s layer is formed by 5 dpf, and the zebrafish cornea (which is much thinner than the human cornea) is fully mature at 2 months post fertilization [62].
Many shared proteins are expressed in both the human and zebrafish cornea [62], although many of the highly-expressed corneal crystallins are taxon-specific [222]. In most mammals, including humans, the most abundant corneal crystallin is Aldehyde Dehydrogenase 3a1 [223]. In contrast, 50% of soluble protein in zebrafish adult cornea is made up of Scinderin-Like Protein A (also known as C/L-Gelsolin and Gelsolin-Like 1) [224].
There are several human corneal dystrophies which lead to opacity of the cornea. For many, the responsible gene is known, but few knockout or transgenic mouse mutants in the particular defective gene have been reported [220]. Some of these genes, including pip5k3 (Fleck Corneal Dystrophy, [225, 226]) and keratocan (Cornea Plana, [227, 228]) are expressed in the zebrafish cornea. Using blastula-stage transplants (see introduction), it is possible to study the function of genes whose mutation would otherwise be embryonic lethal in the mature zebrafish cornea [209]. Semina and colleagues [209] utilized this technique to show that loss of functional lama1 in eye tissues leads to focal corneal dysplasia in adult zebrafish. Utilizing early-stage lens ablation, the authors further showed that corneal and other eye defects in lama1 mutants are not a secondary consequence of lens degeneration.
C. Glaucoma
Second in prevalence only to cataracts in cases of world-wide human blindness [1], glaucoma encompasses a group of related progressive diseases characterized by death of retinal ganglion cells and subsequent degeneration of the optic nerve which leads to irreversible blindness [229]. Although many genes are associated with human cases of glaucoma, incomplete penetrance within families indicates that the various causes of glaucoma are multigenic [230].
Elevated intraocular pressure (IOP) is a major risk factor associated with glaucoma [231, 232]. IOP results from the balance between aqueous humor secretion (from the ciliary epithelium) and outflow, with defective outflow being the most common cause of elevated IOP [233, 234]. In humans, aqueous humor provides nutrients and removes waste products, and its exit from the anterior segment is through the trabecular meshwork outflow pathways [235].
Although one group has advised caution in using zebrafish as a model for glaucoma research due to the ultrastructural dissimilarity between the mammalian trabecular meshwork and the zebrafish annular ligament [65], a second group has more recently utilized aqueous humor tracing experiments to meticulously detail the outflow pathway in the zebrafish eye [61]. Gray and colleagues found that the appropriate zebrafish analog to the mammalian outflow pathway is in fact the ventral canalicular network, and not the annular ligament [61]. Further, the authors found that the ultrastructure of this region was overall quite similar in zebrafish and humans, although the aqueous humor flow pathway in zebrafish is vectorial (flowing from dorsal to ventral) rather than circumferential as in humans. Despite this difference, the overall similarities in aqueous humor outflow tissue structure make the zebrafish a potentially valuable model organism for human glaucomas.
The zebrafish is an ideal model system in which to study multigenic traits [236]. In addition, methodology for measuring zebrafish IOP has been developed, and average intraocular pressure ranges are similar in mammals and zebrafish [64]. Utilizing this technique, the zebrafish mutant brass (genetic basis unknown) was shown to have elevated IOP along with anterior segment defects [64]. Although brass zebrafish do not develop glaucoma, their genetic background could be used to uncover glaucoma modifier loci [64].
In humans, mutation of the gene FOXC1 is associated with Axenfeld-Reiger Syndrome, which includes glaucoma in some individuals [237–239]. Zebrafish foxc1 is expressed in the anterior segment and periocular mesenchyme [236]. By utilizing morpholinos to knock down foxc1 expression in zebrafish embryos along with other techniques, one study has provided evidence that FoxC1 directly activates expression of fgf19 within the periocular mesenchyme [240]. Fgf19 goes on to activate Fgfr4 receptors, and this interaction is required for appropriate anterior segment development [240]. Reduced foxc1 expression during zebrafish eye development also results in reduced expression of the cellular homeostasis and apoptosis regulator foxo1, and in increased apoptosis within the zebrafish eye [241].
Mutation of the gene lmx1b in humans is associated with Nail-Patella syndrome and an increased susceptibility to glaucoma [242]. Similar to the expression pattern in mice [243], zebrafish orthologues of this gene are expressed in cells of the periocular mesenchyme [103]. Reduction of lmx1b expression in zebrafish with morpholinos resulted in early eye morphological defects, enhanced ocular FGF activity, and in altered expression of two genes also implicated in glaucoma: foxc1 and pitx2 [103, 239, 244, 245]. Results from the study further suggest that lmx1b-expressing cells in the zebrafish eye have migration defects in the absence of lmx1b expression [103].
IV. Concluding Remarks
The zebrafish has emerged as a powerful model system for the study of the cellular and molecular underpinnings of human ocular pathologies. Large-scale genetic screens have identified genes whose study is relevant for understanding both eye development and disease, while the continual development of tools for the manipulation of gene function in vivo has enabled researchers to study their functions during eye development and maintenance. Current and future research efforts utilizing the zebrafish system promise to continue to provide important insights into human ocular conditions and contribute to the discovery and development of relevant therapeutics.
Acknowledgements
We apologize for the omission of other relevant zebrafish eye mutants and morphants due to space constraints, and the responsibility for this is borne entirely by the senior author. Work in the Gross lab has been supported by grants from the American Health Assistance Foundation Macular Degeneration Research Program, the Knights Templar Eye Foundation, the Retina Research Foundation, the Karl Kirchgessner Foundation, the E. Matilda Ziegler Foundation for the Blind, a CAREER Award from the National Science Foundation (IOS-0745782) and RO1-EY18005 from the National Institutes of Health. We are grateful to Marianna Grenadier for generating artwork, and to Robin Williamson, Edward Spaghetti and Simon Posford for editorial assistance.
Contributor Information
Jonathan Bibliowicz, Email: yoni@mail.utexas.edu.
Rachel K. Tittle, Email: rtittle@mail.utexas.edu.
Jeffrey M. Gross, Email: jmgross@mail.utexas.edu.
References
- 1.Resnikoff S, Pascolini D, Etya'ale D, Kocur I, Pararajasegaram R, Pokharel GP, Mariotti SP. Global data on visual impairment in the year 2002. Bull World Health Organ. 2004;82:844–851. [PMC free article] [PubMed] [Google Scholar]
- 2.Spence R, Gerlach G, Lawrence C, Smith C. The behaviour and ecology of the zebrafish, Danio rerio. Biol Rev Camb Philos Soc. 2008;83:13–34. doi: 10.1111/j.1469-185X.2007.00030.x. [DOI] [PubMed] [Google Scholar]
- 3.Haffter P, Nusslein-Volhard C. Large scale genetics in a small vertebrate, the zebrafish. Int J Dev Biol. 1996;40:221–227. [PubMed] [Google Scholar]
- 4.Kimmel CB, Warga RM, Schilling TF. Origin and organization of the zebrafish fate map. Development. 1990;108:581–594. doi: 10.1242/dev.108.4.581. [DOI] [PubMed] [Google Scholar]
- 5.Woo K, Shih J, Fraser SE. Fate maps of the zebrafish embryo. Curr Opin Genet Dev. 1995;5:439–443. doi: 10.1016/0959-437x(95)90046-j. [DOI] [PubMed] [Google Scholar]
- 6.Carmany-Rampey A, Moens CB. Modern mosaic analysis in the zebrafish. Methods. 2006;39:228–238. doi: 10.1016/j.ymeth.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto Y, Jeffery WR. Probing teleost eye development by lens transplantation. Methods. 2002;28:420–426. doi: 10.1016/s1046-2023(02)00261-x. [DOI] [PubMed] [Google Scholar]
- 8.Meng X, Noyes MB, Zhu LJ, Lawson ND, Wolfe SA. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol. 2008;26:695–701. doi: 10.1038/nbt1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doyon Y, McCammon JM, Miller JC, Faraji F, Ngo C, Katibah GE, Amora R, Hocking TD, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Amacher SL. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol. 2008;26:702–708. doi: 10.1038/nbt1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nasevicius A, Ekker SC. Effective targeted gene 'knockdown' in zebrafish. Nat Genet. 2000;26:216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- 11.Draper BW, Morcos PA, Kimmel CB. Inhibition of zebrafish fgf8 pre-mRNA splicing with morpholino oligos: a quantifiable method for gene knockdown. Genesis. 2001;30:154–156. doi: 10.1002/gene.1053. [DOI] [PubMed] [Google Scholar]
- 12.Thummel R, Bai S, Sarras MP, Jr, Song P, McDermott J, Brewer J, Perry M, Zhang X, Hyde DR, Godwin AR. Inhibition of zebrafish fin regeneration using in vivo electroporation of morpholinos against fgfr1 and msxb. Dev Dyn. 2006;235:336–346. doi: 10.1002/dvdy.20630. [DOI] [PubMed] [Google Scholar]
- 13.Shestopalov IA, Sinha S, Chen JK. Light-controlled gene silencing in zebrafish embryos. Nat Chem Biol. 2007;3:650–651. doi: 10.1038/nchembio.2007.30. [DOI] [PubMed] [Google Scholar]
- 14.Stuart GW, McMurray JV, Westerfield M. Replication, integration and stable germ-line transmission of foreign sequences injected into early zebrafish embryos. Development. 1988;103:403–412. doi: 10.1242/dev.103.2.403. [DOI] [PubMed] [Google Scholar]
- 15.Stuart GW, Vielkind JR, McMurray JV, Westerfield M. Stable lines of transgenic zebrafish exhibit reproducible patterns of transgene expression. Development. 1990;109:577–584. doi: 10.1242/dev.109.3.577. [DOI] [PubMed] [Google Scholar]
- 16.Culp P, Nusslein-Volhard C, Hopkins N. High-frequency germ-line transmission of plasmid DNA sequences injected into fertilized zebrafish eggs. Proc Natl Acad Sci U S A. 1991;88:7953–7957. doi: 10.1073/pnas.88.18.7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawakami K. Transgenesis and gene trap methods in zebrafish by using the Tol2 transposable element. Methods Cell Biol. 2004;77:201–222. doi: 10.1016/s0091-679x(04)77011-9. [DOI] [PubMed] [Google Scholar]
- 18.Amsterdam A, Lin S, Hopkins N. The Aequorea victoria green fluorescent protein can be used as a reporter in live zebrafish embryos. Dev Biol. 1995;171:123–129. doi: 10.1006/dbio.1995.1265. [DOI] [PubMed] [Google Scholar]
- 19.Halloran MC, Sato-Maeda M, Warren JT, Su F, Lele Z, Krone PH, Kuwada JY, Shoji W. Laser-induced gene expression in specific cells of transgenic zebrafish. Development. 2000;127:1953–1960. doi: 10.1242/dev.127.9.1953. [DOI] [PubMed] [Google Scholar]
- 20.Scheer N, Campos-Ortega JA. Use of the Gal4-UAS technique for targeted gene expression in the zebrafish. Mech Dev. 1999;80:153–158. doi: 10.1016/s0925-4773(98)00209-3. [DOI] [PubMed] [Google Scholar]
- 21.Hua JY, Smear MC, Baier H, Smith SJ. Regulation of axon growth in vivo by activity-based competition. Nature. 2005;434:1022–1026. doi: 10.1038/nature03409. [DOI] [PubMed] [Google Scholar]
- 22.Hatta K, Tsujii H, Omura T. Cell tracking using a photoconvertible fluorescent protein. Nat Protoc. 2006;1:960–967. doi: 10.1038/nprot.2006.96. [DOI] [PubMed] [Google Scholar]
- 23.Kurita R, Sagara H, Aoki Y, Link BA, Arai K, Watanabe S. Suppression of lens growth by alphaA-crystallin promoter-driven expression of diphtheria toxin results in disruption of retinal cell organization in zebrafish. Dev Biol. 2003;255:113–127. doi: 10.1016/s0012-1606(02)00079-9. [DOI] [PubMed] [Google Scholar]
- 24.Pisharath H, Rhee JM, Swanson MA, Leach SD, Parsons MJ. Targeted ablation of beta cells in the embryonic zebrafish pancreas using E. coli nitroreductase. Mech Dev. 2007;124:218–229. doi: 10.1016/j.mod.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 26.Halpern ME, Rhee J, Goll MG, Akitake CM, Parsons M, Leach SD. Gal4/UAS transgenic tools and their application to zebrafish. Zebrafish. 2008;5:97–110. doi: 10.1089/zeb.2008.0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sauer B. Functional expression of the cre-lox site-specific recombination system in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1987;7:2087–2096. doi: 10.1128/mcb.7.6.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langenau DM, Feng H, Berghmans S, Kanki JP, Kutok JL, Look AT. Cre/lox-regulated transgenic zebrafish model with conditional myc-induced T cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2005;102:6068–6073. doi: 10.1073/pnas.0408708102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sinha DK, Neveu P, Gagey N, Aujard I, Le Saux T, Rampon C, Gauron C, Kawakami K, Leucht C, Bally-Cuif L, Volovitch M, Bensimon D, Jullien L, Vriz S. Photoactivation of the CreER T2 recombinase for conditional site-specific recombination with high spatiotemporal resolution. Zebrafish. 2010;7:199–204. doi: 10.1089/zeb.2009.0632. [DOI] [PubMed] [Google Scholar]
- 30.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 32.Walker C, Streisinger G. Induction of Mutations by gamma-Rays in Pregonial Germ Cells of Zebrafish Embryos. Genetics. 1983;103:125–136. doi: 10.1093/genetics/103.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Streisinger G, Walker C, Dower N, Knauber D, Singer F. Production of clones of homozygous diploid zebra fish (Brachydanio rerio) Nature. 1981;291:293–296. doi: 10.1038/291293a0. [DOI] [PubMed] [Google Scholar]
- 34.Haffter P, Granato M, Brand M, Mullins MC, Hammerschmidt M, Kane DA, Odenthal J, van Eeden FJ, Jiang YJ, Heisenberg CP, Kelsh RN, Furutani-Seiki M, Vogelsang E, Beuchle D, Schach U, Fabian C, Nusslein-Volhard C. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development. 1996;123:1–36. doi: 10.1242/dev.123.1.1. [DOI] [PubMed] [Google Scholar]
- 35.Driever W, Solnica-Krezel L, Schier AF, Neuhauss SC, Malicki J, Stemple DL, Stainier DY, Zwartkruis F, Abdelilah S, Rangini Z, Belak J, Boggs C. A genetic screen for mutations affecting embryogenesis in zebrafish. Development. 1996;123:37–46. doi: 10.1242/dev.123.1.37. [DOI] [PubMed] [Google Scholar]
- 36.Grunwald DJ, Streisinger G. Induction of recessive lethal and specific locus mutations in the zebrafish with ethyl nitrosourea. Genet Res. 1992;59:103–116. doi: 10.1017/s0016672300030317. [DOI] [PubMed] [Google Scholar]
- 37.Solnica-Krezel L, Schier AF, Driever W. Efficient recovery of ENU-induced mutations from the zebrafish germline. Genetics. 1994;136:1401–1420. doi: 10.1093/genetics/136.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amsterdam A, Hopkins N. Mutagenesis strategies in zebrafish for identifying genes involved in development and disease. Trends Genet. 2006;22:473–478. doi: 10.1016/j.tig.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 39.Golling G, Amsterdam A, Sun Z, Antonelli M, Maldonado E, Chen W, Burgess S, Haldi M, Artzt K, Farrington S, Lin SY, Nissen RM, Hopkins N. Insertional mutagenesis in zebrafish rapidly identifies genes essential for early vertebrate development. Nat Genet. 2002;31:135–140. doi: 10.1038/ng896. [DOI] [PubMed] [Google Scholar]
- 40.Amsterdam A, Burgess S, Golling G, Chen W, Sun Z, Townsend K, Farrington S, Haldi M, Hopkins N. A large-scale insertional mutagenesis screen in zebrafish. Genes Dev. 1999;13:2713–2724. doi: 10.1101/gad.13.20.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wienholds E, van Eeden F, Kosters M, Mudde J, Plasterk RH, Cuppen E. Efficient target-selected mutagenesis in zebrafish. Genome Res. 2003;13:2700–2707. doi: 10.1101/gr.1725103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moens CB, Donn TM, Wolf-Saxon ER, Ma TP. Reverse genetics in zebrafish by TILLING. Brief Funct Genomic Proteomic. 2008;7:454–459. doi: 10.1093/bfgp/eln046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor KL, Grant NJ, Temperley ND, Patton EE. Small molecule screening in zebrafish: an in vivo approach to identifying new chemical tools and drug leads. Cell Commun Signal. 2010;8:11. doi: 10.1186/1478-811X-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Owens KN, Santos F, Roberts B, Linbo T, Coffin AB, Knisely AJ, Simon JA, Rubel EW, Raible DW. Identification of genetic and chemical modulators of zebrafish mechanosensory hair cell death. PLoS Genet. 2008;4:e1000020. doi: 10.1371/journal.pgen.1000020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trompouki E, Zon LI. Small molecule screen in zebrafish and HSC expansion. Methods Mol Biol. 2010;636:301–316. doi: 10.1007/978-1-60761-691-7_19. [DOI] [PubMed] [Google Scholar]
- 46.Kitambi SS, McCulloch KJ, Peterson RT, Malicki JJ. Small molecule screen for compounds that affect vascular development in the zebrafish retina. Mech Dev. 2009;126:464–477. doi: 10.1016/j.mod.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kokel D, Bryan J, Laggner C, White R, Cheung CY, Mateus R, Healey D, Kim S, Werdich AA, Haggarty SJ, Macrae CA, Shoichet B, Peterson RT. Rapid behavior-based identification of neuroactive small molecules in the zebrafish. Nat Chem Biol. 6:231–237. doi: 10.1038/nchembio.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peterson RT, Link BA, Dowling JE, Schreiber SL. Small molecule developmental screens reveal the logic and timing of vertebrate development. Proc Natl Acad Sci U S A. 2000;97:12965–12969. doi: 10.1073/pnas.97.24.12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murphey RD, Stern HM, Straub CT, Zon LI. A chemical genetic screen for cell cycle inhibitors in zebrafish embryos. Chem Biol Drug Des. 2006;68:213–219. doi: 10.1111/j.1747-0285.2006.00439.x. [DOI] [PubMed] [Google Scholar]
- 50.Gross JM, Perkins BD. Zebrafish mutants as models for congenital ocular disorders in humans. Mol Reprod Dev. 2008;75:547–555. doi: 10.1002/mrd.20831. [DOI] [PubMed] [Google Scholar]
- 51.Bilotta J, Saszik S. The zebrafish as a model visual system. Int J Dev Neurosci. 2001;19:621–629. doi: 10.1016/s0736-5748(01)00050-8. [DOI] [PubMed] [Google Scholar]
- 52.Fadool JM, Dowling JE. Zebrafish: a model system for the study of eye genetics. Prog Retin Eye Res. 2008;27:89–110. doi: 10.1016/j.preteyeres.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goldsmith P, Harris WA. The zebrafish as a tool for understanding the biology of visual disorders. Semin Cell Dev Biol. 2003;14:11–18. doi: 10.1016/s1084-9521(02)00167-2. [DOI] [PubMed] [Google Scholar]
- 54.Schmitt EA, Dowling JE. Early retinal development in the zebrafish, Danio rerio: light and electron microscopic analyses. J Comp Neurol. 1999;404:515–536. [PubMed] [Google Scholar]
- 55.Lister JA. Development of pigment cells in the zebrafish embryo. Microsc Res Tech. 2002;58:435–441. doi: 10.1002/jemt.10161. [DOI] [PubMed] [Google Scholar]
- 56.Greiling TM, Clark JI. The transparent lens and cornea in the mouse and zebra fish eye. Semin Cell Dev Biol. 2008;19:94–99. doi: 10.1016/j.semcdb.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Greiling TM, Clark JI. Early lens development in the zebrafish: a three-dimensional time-lapse analysis. Dev Dyn. 2009;238:2254–2265. doi: 10.1002/dvdy.21997. [DOI] [PubMed] [Google Scholar]
- 58.Greiling TM, Aose M, Clark JI. Cell fate and differentiation of the developing ocular lens. Invest Ophthalmol Vis Sci. 2010;51:1540–1546. doi: 10.1167/iovs.09-4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dahm R, Schonthaler HB, Soehn AS, van Marle J, Vrensen GF. Development and adult morphology of the eye lens in the zebrafish. Exp Eye Res. 2007;85:74–89. doi: 10.1016/j.exer.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 60.Soules KA, Link BA. Morphogenesis of the anterior segment in the zebrafish eye. BMC Dev Biol. 2005;5:12. doi: 10.1186/1471-213X-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gray MP, Smith RS, Soules KA, John SW, Link BA. The aqueous humor outflow pathway of zebrafish. Invest Ophthalmol Vis Sci. 2009;50:1515–1521. doi: 10.1167/iovs.08-3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao XC, Yee RW, Norcom E, Burgess H, Avanesov AS, Barrish JP, Malicki J. The zebrafish cornea: structure and development. Invest Ophthalmol Vis Sci. 2006;47:4341–4348. doi: 10.1167/iovs.05-1611. [DOI] [PubMed] [Google Scholar]
- 63.Akhtar S, Schonthaler HB, Bron AJ, Dahm R. Formation of stromal collagen fibrils and proteoglycans in the developing zebrafish cornea. Acta Ophthalmol. 2008;86:655–665. doi: 10.1111/j.1600-0420.2007.01135.x. [DOI] [PubMed] [Google Scholar]
- 64.Link BA, Gray MP, Smith RS, John SW. Intraocular pressure in zebrafish: comparison of inbred strains and identification of a reduced melanin mutant with raised IOP. Invest Ophthalmol Vis Sci. 2004;45:4415–4422. doi: 10.1167/iovs.04-0557. [DOI] [PubMed] [Google Scholar]
- 65.Chen CC, Yeh LK, Liu CY, Kao WW, Samples JR, Lin SJ, Hu FR, Wang IJ. Morphological differences between the trabecular meshworks of zebrafish and mammals. Curr Eye Res. 2008;33:59–72. doi: 10.1080/02713680701795026. [DOI] [PubMed] [Google Scholar]
- 66.Swamynathan SK, Crawford MA, Robison WG, Jr, Kanungo J, Piatigorsky J. Adaptive differences in the structure and macromolecular compositions of the air and water corneas of the "four-eyed" fish (Anableps anableps) FASEB J. 2003;17:1996–2005. doi: 10.1096/fj.03-0122com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Easter SS, Jr, Nicola GN. The development of vision in the zebrafish (Danio rerio) Dev Biol. 1996;180:646–663. doi: 10.1006/dbio.1996.0335. [DOI] [PubMed] [Google Scholar]
- 68.Schmitt EA, Dowling JE. Early eye morphogenesis in the zebrafish, Brachydanio rerio. J Comp Neurol. 1994;344:532–542. doi: 10.1002/cne.903440404. [DOI] [PubMed] [Google Scholar]
- 69.Bilotta J, Saszik S, Sutherland SE. Rod contributions to the electroretinogram of the dark-adapted developing zebrafish. Dev Dyn. 2001;222:564–570. doi: 10.1002/dvdy.1188. [DOI] [PubMed] [Google Scholar]
- 70.Robinson J, Schmitt EA, Harosi FI, Reece RJ, Dowling JE. Zebrafish ultraviolet visual pigment: absorption spectrum, sequence, and localization. Proc Natl Acad Sci U S A. 1993;90:6009–6012. doi: 10.1073/pnas.90.13.6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dowling JE. The Retina. Cambridge, MA: Harvard University Press; 1987. [Google Scholar]
- 72.Fitzpatrick DR, van Heyningen V. Developmental eye disorders. Curr Opin Genet Dev. 2005;15:348–353. doi: 10.1016/j.gde.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 73.Gregory-Evans CY, Williams MJ, Halford S, Gregory-Evans K. Ocular coloboma: a reassessment in the age of molecular neuroscience. J Med Genet. 2004;41:881–891. doi: 10.1136/jmg.2004.025494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Porges Y, Gershoni-Baruch R, Leibu R, Goldscher D, Zonis S, Shapira I, Miller B. Hereditary microphthalmia with colobomatous cyst. Am J Ophthalmol. 1992;114:30–34. doi: 10.1016/s0002-9394(14)77409-4. [DOI] [PubMed] [Google Scholar]
- 75.Bermejo E, Martinez-Frias ML. Congenital eye malformations: clinical-epidemiological analysis of 1,124,654 consecutive births in Spain. Am J Med Genet. 1998;75:497–504. [PubMed] [Google Scholar]
- 76.Stoll C, Alembik Y, Dott B, Roth MP. Congenital eye malformations in 212,479 consecutive births. Ann Genet. 1997;40:122–128. [PubMed] [Google Scholar]
- 77.Maumenee IH, Mitchell TN. Colobomatous malformations of the eye. Trans Am Ophthalmol Soc. 1990;88:123–132. discussion 133–125. [PMC free article] [PubMed] [Google Scholar]
- 78.Chang L, Blain D, Bertuzzi S, Brooks BP. Uveal coloboma: clinical and basic science update. Curr Opin Ophthalmol. 2006;17:447–470. doi: 10.1097/01.icu.0000243020.82380.f6. [DOI] [PubMed] [Google Scholar]
- 79.Brown JD, Dutta S, Bharti K, Bonner RF, Munson PJ, Dawid IB, Akhtar AL, Onojafe IF, Alur RP, Gross JM, Hejtmancik JF, Jiao X, Chan WY, Brooks BP. Expression profiling during ocular development identifies 2 Nlz genes with a critical role in optic fissure closure. Proc Natl Acad Sci U S A. 2009;106:1462–1467. doi: 10.1073/pnas.0812017106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sanyanusin P, Schimmenti LA, McNoe LA, Ward TA, Pierpont ME, Sullivan MJ, Dobyns WB, Eccles MR. Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat Genet. 1995;9:358–364. doi: 10.1038/ng0495-358. [DOI] [PubMed] [Google Scholar]
- 81.Macdonald R, Scholes J, Strahle U, Brennan C, Holder N, Brand M, Wilson SW. The Pax protein Noi is required for commissural axon pathway formation in the rostral forebrain. Development. 1997;124:2397–2408. doi: 10.1242/dev.124.12.2397. [DOI] [PubMed] [Google Scholar]
- 82.Terzic J, Muller C, Gajovic S, Saraga-Babic M. Expression of PAX2 gene during human development. Int J Dev Biol. 1998;42:701–707. [PubMed] [Google Scholar]
- 83.Macdonald R, Barth KA, Xu Q, Holder N, Mikkola I, Wilson SW. Midline signalling is required for Pax gene regulation and patterning of the eyes. Development. 1995;121:3267–3278. doi: 10.1242/dev.121.10.3267. [DOI] [PubMed] [Google Scholar]
- 84.Lee J, Willer JR, Willer GB, Smith K, Gregg RG, Gross JM. Zebrafish blowout provides genetic evidence for Patched1-mediated negative regulation of Hedgehog signaling within the proximal optic vesicle of the vertebrate eye. Dev Biol. 2008;319:10–22. doi: 10.1016/j.ydbio.2008.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koudijs MJ, den Broeder MJ, Groot E, van Eeden FJ. Genetic analysis of the two zebrafish patched homologues identifies novel roles for the hedgehog signaling pathway. BMC Dev Biol. 2008;8:15. doi: 10.1186/1471-213X-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 87.Schimmenti LA, de la Cruz J, Lewis RA, Karkera JD, Manligas GS, Roessler E, Muenke M. Novel mutation in sonic hedgehog in non-syndromic colobomatous microphthalmia. Am J Med Genet A. 2003;116A:215–221. doi: 10.1002/ajmg.a.10884. [DOI] [PubMed] [Google Scholar]
- 88.Sanek NA, Taylor AA, Nyholm MK, Grinblat Y. Zebrafish zic2a patterns the forebrain through modulation of Hedgehog-activated gene expression. Development. 2009;136:3791–3800. doi: 10.1242/dev.037820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brown SA, Warburton D, Brown LY, Yu CY, Roeder ER, Stengel-Rutkowski S, Hennekam RC, Muenke M. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet. 1998;20:180–183. doi: 10.1038/2484. [DOI] [PubMed] [Google Scholar]
- 90.Ohsaki K, Morimitsu T, Ishida Y, Kominami R, Takahashi N. Expression of the Vax family homeobox genes suggests multiple roles in eye development. Genes Cells. 1999;4:267–276. doi: 10.1046/j.1365-2443.1999.00257.x. [DOI] [PubMed] [Google Scholar]
- 91.Take-uchi M, Clarke JD, Wilson SW. Hedgehog signalling maintains the optic stalk-retinal interface through the regulation of Vax gene activity. Development. 2003;130:955–968. doi: 10.1242/dev.00305. [DOI] [PubMed] [Google Scholar]
- 92.Hornby SJ, Ward SJ, Gilbert CE. Eye birth defects in humans may be caused by a recessively-inherited genetic predisposition to the effects of maternal vitamin A deficiency during pregnancy. Med Sci Monit. 2003;9:HY23–HY26. [PubMed] [Google Scholar]
- 93.Hyatt GA, Schmitt EA, Marsh-Armstrong NR, Dowling JE. Retinoic acid-induced duplication of the zebrafish retina. Proc Natl Acad Sci U S A. 1992;89:8293–8297. doi: 10.1073/pnas.89.17.8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hyatt GA, Schmitt EA, Marsh-Armstrong N, McCaffery P, Drager UC, Dowling JE. Retinoic acid establishes ventral retinal characteristics. Development. 1996;122:195–204. doi: 10.1242/dev.122.1.195. [DOI] [PubMed] [Google Scholar]
- 95.Marsh-Armstrong N, McCaffery P, Gilbert W, Dowling JE, Drager UC. Retinoic acid is necessary for development of the ventral retina in zebrafish. Proc Natl Acad Sci U S A. 1994;91:7286–7290. doi: 10.1073/pnas.91.15.7286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lampert JM, Holzschuh J, Hessel S, Driever W, Vogt K, von Lintig J. Provitamin A conversion to retinal via the beta,beta-carotene-15,15'-oxygenase (bcox) is essential for pattern formation and differentiation during zebrafish embryogenesis. Development. 2003;130:2173–2186. doi: 10.1242/dev.00437. [DOI] [PubMed] [Google Scholar]
- 97.Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001;1:55–67. doi: 10.1038/35094067. [DOI] [PubMed] [Google Scholar]
- 98.Nadauld LD, Chidester S, Shelton DN, Rai K, Broadbent T, Sandoval IT, Peterson PW, Manos EJ, Ireland CM, Yost HJ, Jones DA. Dual roles for adenomatous polyposis coli in regulating retinoic acid biosynthesis and Wnt during ocular development. Proc Natl Acad Sci U S A. 2006;103:13409–13414. doi: 10.1073/pnas.0601634103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pittlik S, Domingues S, Meyer A, Begemann G. Expression of zebrafish aldh1a3 (raldh3) and absence of aldh1a1 in teleosts. Gene Expr Patterns. 2008;8:141–147. doi: 10.1016/j.gep.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 100.McCaffery P, Wagner E, O'Neil J, Petkovich M, Drager UC. Dorsal and ventral retinal territories defined by retinoic acid synthesis, break-down and nuclear receptor expression. Mech Dev. 1999;82:119–130. doi: 10.1016/s0925-4773(99)00022-2. [DOI] [PubMed] [Google Scholar]
- 101.Fischer AJ, Wallman J, Mertz JR, Stell WK. Localization of retinoid binding proteins, retinoid receptors, and retinaldehyde dehydrogenase in the chick eye. J Neurocytol. 1999;28:597–609. doi: 10.1023/a:1007071406746. [DOI] [PubMed] [Google Scholar]
- 102.Matt N, Ghyselinck NB, Pellerin I, Dupe V. Impairing retinoic acid signalling in the neural crest cells is sufficient to alter entire eye morphogenesis. Dev Biol. 2008;320:140–148. doi: 10.1016/j.ydbio.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 103.McMahon C, Gestri G, Wilson SW, Link BA. Lmx1b is essential for survival of periocular mesenchymal cells and influences Fgf-mediated retinal patterning in zebrafish. Dev Biol. 2009;332:287–298. doi: 10.1016/j.ydbio.2009.05.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gross JM, Perkins BD, Amsterdam A, Egana A, Darland T, Matsui JI, Sciascia S, Hopkins N, Dowling JE. Identification of zebrafish insertional mutants with defects in visual system development and function. Genetics. 2005;170:245–261. doi: 10.1534/genetics.104.039727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee J, Gross JM. Laminin beta1 and gamma1 containing laminins are essential for basement membrane integrity in the zebrafish eye. Invest Ophthalmol Vis Sci. 2007;48:2483–2490. doi: 10.1167/iovs.06-1211. [DOI] [PubMed] [Google Scholar]
- 106.Rohlich P. The sensory cilium of retinal rods is analogous to the transitional zone of motile cilia. Cell Tissue Res. 1975;161:421–430. doi: 10.1007/BF00220009. [DOI] [PubMed] [Google Scholar]
- 107.Badano JL, Teslovich TM, Katsanis N. The centrosome in human genetic disease. Nat Rev Genet. 2005;6:194–205. doi: 10.1038/nrg1557. [DOI] [PubMed] [Google Scholar]
- 108.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 109.Fadool JM, Brockerhoff SE, Hyatt GA, Dowling JE. Mutations affecting eye morphology in the developing zebrafish (Danio rerio) Dev Genet. 1997;20:288–295. doi: 10.1002/(SICI)1520-6408(1997)20:3<288::AID-DVG11>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 110.Malicki J, Neuhauss SC, Schier AF, Solnica-Krezel L, Stemple DL, Stainier DY, Abdelilah S, Zwartkruis F, Rangini Z, Driever W. Mutations affecting development of the zebrafish retina. Development. 1996;123:263–273. doi: 10.1242/dev.123.1.263. [DOI] [PubMed] [Google Scholar]
- 111.Hsu YC, Willoughby JJ, Christensen AK, Jensen AM. Mosaic Eyes is a novel component of the Crumbs complex and negatively regulates photoreceptor apical size. Development. 2006;133:4849–4859. doi: 10.1242/dev.02685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jensen AM, Walker C, Westerfield M. mosaic eyes: a zebrafish gene required in pigmented epithelium for apical localization of retinal cell division and lamination. Development. 2001;128:95–105. doi: 10.1242/dev.128.1.95. [DOI] [PubMed] [Google Scholar]
- 113.Tepass U, Theres C, Knust E. crumbs encodes an EGF-like protein expressed on apical membranes of Drosophila epithelial cells and required for organization of epithelia. Cell. 1990;61:787–799. doi: 10.1016/0092-8674(90)90189-l. [DOI] [PubMed] [Google Scholar]
- 114.Wodarz A, Hinz U, Engelbert M, Knust E. Expression of crumbs confers apical character on plasma membrane domains of ectodermal epithelia of Drosophila. Cell. 1995;82:67–76. doi: 10.1016/0092-8674(95)90053-5. [DOI] [PubMed] [Google Scholar]
- 115.Roh MH, Fan S, Liu CJ, Margolis B. The Crumbs3-Pals1 complex participates in the establishment of polarity in mammalian epithelial cells. J Cell Sci. 2003;116:2895–2906. doi: 10.1242/jcs.00500. [DOI] [PubMed] [Google Scholar]
- 116.den Hollander AI, ten Brink JB, de Kok YJ, van Soest S, van den Born LI, van Driel MA, van de Pol DJ, Payne AM, Bhattacharya SS, Kellner U, Hoyng CB, Westerveld A, Brunner HG, Bleeker-Wagemakers EM, Deutman AF, Heckenlively JR, Cremers FP, Bergen AA. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12) Nat Genet. 1999;23:217–221. doi: 10.1038/13848. [DOI] [PubMed] [Google Scholar]
- 117.Lotery AJ, Jacobson SG, Fishman GA, Weleber RG, Fulton AB, Namperumalsamy P, Heon E, Levin AV, Grover S, Rosenow JR, Kopp KK, Sheffield VC, Stone EM. Mutations in the CRB1 gene cause Leber congenital amaurosis. Arch Ophthalmol. 2001;119:415–420. doi: 10.1001/archopht.119.3.415. [DOI] [PubMed] [Google Scholar]
- 118.Gerber S, Perrault I, Hanein S, Shalev S, Zlotogora J, Barbet F, Ducroq D, Dufier J, Munnich A, Rozet J, Kaplan J. A novel mutation disrupting the cytoplasmic domain of CRB1 in a large consanguineous family of Palestinian origin affected with Leber congenital amaurosis. Ophthalmic Genet. 2002;23:225–235. doi: 10.1076/opge.23.4.225.13879. [DOI] [PubMed] [Google Scholar]
- 119.Omori Y, Malicki J. oko meduzy and related crumbs genes are determinants of apical cell features in the vertebrate embryo. Curr Biol. 2006;16:945–957. doi: 10.1016/j.cub.2006.03.058. [DOI] [PubMed] [Google Scholar]
- 120.Jing X, Malicki J. Zebrafish ale oko, an essential determinant of sensory neuron survival and the polarity of retinal radial glia, encodes the p50 subunit of dynactin. Development. 2009;136:2955–2964. doi: 10.1242/dev.037739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schroer TA. Dynactin. Annu Rev Cell Dev Biol. 2004;20:759–779. doi: 10.1146/annurev.cellbio.20.012103.094623. [DOI] [PubMed] [Google Scholar]
- 122.Tsujikawa M, Omori Y, Biyanwila J, Malicki J. Mechanism of positioning the cell nucleus in vertebrate photoreceptors. Proc Natl Acad Sci U S A. 2007;104:14819–14824. doi: 10.1073/pnas.0700178104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Doerre G, Malicki J. A mutation of early photoreceptor development, mikre oko, reveals cell-cell interactions involved in the survival and differentiation of zebrafish photoreceptors. J Neurosci. 2001;21:6745–6757. doi: 10.1523/JNEUROSCI.21-17-06745.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dryja TP, Li T. Molecular genetics of retinitis pigmentosa. Hum Mol Genet. 1995;4(Spec No):1739–1743. doi: 10.1093/hmg/4.suppl_1.1739. [DOI] [PubMed] [Google Scholar]
- 125.Young RW. The renewal of photoreceptor cell outer segments. J Cell Biol. 1967;33:61–72. doi: 10.1083/jcb.33.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sukumaran S, Perkins BD. Early defects in photoreceptor outer segment morphogenesis in zebrafish ift57, ift88 and ift172 Intraflagellar Transport mutants. Vision Res. 2009;49:479–489. doi: 10.1016/j.visres.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hudak LM, Lunt S, Chang CH, Winkler E, Flammer H, Lindsey M, Perkins B. The Intraflagellar Transport Protein Ift80 is essential for Photoreceptor Survival in a Zebrafish Model of Jeune Asphyxiating Thoracic Dystrophy. Invest Ophthalmol Vis Sci. 2010 doi: 10.1167/iovs.09-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Omori Y, Zhao C, Saras A, Mukhopadhyay S, Kim W, Furukawa T, Sengupta P, Veraksa A, Malicki J. Elipsa is an early determinant of ciliogenesis that links the IFT particle to membrane-associated small GTPase Rab8. Nat Cell Biol. 2008;10:437–444. doi: 10.1038/ncb1706. [DOI] [PubMed] [Google Scholar]
- 129.Doerre G, Malicki J. Genetic analysis of photoreceptor cell development in the zebrafish retina. Mech Dev. 2002;110:125–138. doi: 10.1016/s0925-4773(01)00571-8. [DOI] [PubMed] [Google Scholar]
- 130.Krock BL, Mills-Henry I, Perkins BD. Retrograde intraflagellar transport by cytoplasmic dynein-2 is required for outer segment extension in vertebrate photoreceptors but not arrestin translocation. Invest Ophthalmol Vis Sci. 2009;50:5463–5471. doi: 10.1167/iovs.09-3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Blacque OE, Leroux MR. Bardet-Biedl syndrome: an emerging pathomechanism of intracellular transport. Cell Mol Life Sci. 2006;63:2145–2161. doi: 10.1007/s00018-006-6180-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Muto A, Orger MB, Wehman AM, Smear MC, Kay JN, Page-McCaw PS, Gahtan E, Xiao T, Nevin LM, Gosse NJ, Staub W, Finger-Baier K, Baier H. Forward genetic analysis of visual behavior in zebrafish. PLoS Genet. 2005;1:e66. doi: 10.1371/journal.pgen.0010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Brockerhoff SE, Hurley JB, Janssen-Bienhold U, Neuhauss SC, Driever W, Dowling JE. A behavioral screen for isolating zebrafish mutants with visual system defects. Proc Natl Acad Sci U S A. 1995;92:10545–10549. doi: 10.1073/pnas.92.23.10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li L. Zebrafish mutants: behavioral genetic studies of visual system defects. Dev Dyn. 2001;221:365–372. doi: 10.1002/dvdy.1159. [DOI] [PubMed] [Google Scholar]
- 135.Neuhauss SC, Biehlmaier O, Seeliger MW, Das T, Kohler K, Harris WA, Baier H. Genetic disorders of vision revealed by a behavioral screen of 400 essential loci in zebrafish. J Neurosci. 1999;19:8603–8615. doi: 10.1523/JNEUROSCI.19-19-08603.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Taylor MR, Hurley JB, Van Epps HA, Brockerhoff SE. A zebrafish model for pyruvate dehydrogenase deficiency: rescue of neurological dysfunction and embryonic lethality using a ketogenic diet. Proc Natl Acad Sci U S A. 2004;101:4584–4589. doi: 10.1073/pnas.0307074101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Maaswinkel H, Riesbeck LE, Riley ME, Carr AL, Mullin JP, Nakamoto AT, Li L. Behavioral screening for nightblindness mutants in zebrafish reveals three new loci that cause dominant photoreceptor cell degeneration. Mech Ageing Dev. 2005;126:1079–1089. doi: 10.1016/j.mad.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 138.Li L, Dowling JE. A dominant form of inherited retinal degeneration caused by a non-photoreceptor cell-specific mutation. Proc Natl Acad Sci U S A. 1997;94:11645–11650. doi: 10.1073/pnas.94.21.11645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Maaswinkel H, Ren JQ, Li L. Slow-progressing photoreceptor cell degeneration in night blindness c mutant zebrafish. J Neurocytol. 2003;32:1107–1116. doi: 10.1023/B:NEUR.0000021905.33091.f1. [DOI] [PubMed] [Google Scholar]
- 140.Maaswinkel H, Mason B, Li L. ENU-induced late-onset night blindness associated with rod photoreceptor cell degeneration in zebrafish. Mech Ageing Dev. 2003;124:1065–1071. doi: 10.1016/j.mad.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 141.Li L, Dowling JE. Disruption of the olfactoretinal centrifugal pathway may relate to the visual system defect in night blindness b mutant zebrafish. J Neurosci. 2000;20:1883–1892. doi: 10.1523/JNEUROSCI.20-05-01883.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Baehr W, Wu SM, Bird AC, Palczewski K. The retinoid cycle and retina disease. Vision Res. 2003;43:2957–2958. doi: 10.1016/j.visres.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 143.Kevany BM, Palczewski K. Phagocytosis of retinal rod and cone photoreceptors. Physiology (Bethesda) 2010;25:8–15. doi: 10.1152/physiol.00038.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85:845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- 145.Krock BL, Bilotta J, Perkins BD. Noncell-autonomous photoreceptor degeneration in a zebrafish model of choroideremia. Proc Natl Acad Sci U S A. 2007;104:4600–4605. doi: 10.1073/pnas.0605818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Moosajee M, Tulloch M, Baron RA, Gregory-Evans CY, Pereira-Leal JB, Seabra MC. Single choroideremia gene in nonmammalian vertebrates explains early embryonic lethality of the zebrafish model of choroideremia. Invest Ophthalmol Vis Sci. 2009;50:3009–3016. doi: 10.1167/iovs.08-2755. [DOI] [PubMed] [Google Scholar]
- 147.Raposo G, Marks MS. The dark side of lysosome-related organelles: specialization of the endocytic pathway for melanosome biogenesis. Traffic. 2002;3:237–248. doi: 10.1034/j.1600-0854.2002.030401.x. [DOI] [PubMed] [Google Scholar]
- 148.Schonthaler HB, Fleisch VC, Biehlmaier O, Makhankov Y, Rinner O, Bahadori R, Geisler R, Schwarz H, Neuhauss SC, Dahm R. The zebrafish mutant lbk/vam6 resembles human multisystemic disorders caused by aberrant trafficking of endosomal vesicles. Development. 2008;135:387–399. doi: 10.1242/dev.006098. [DOI] [PubMed] [Google Scholar]
- 149.Maldonado E, Hernandez F, Lozano C, Castro ME, Navarro RE. The zebrafish mutant vps18 as a model for vesicle-traffic related hypopigmentation diseases. Pigment Cell Res. 2006;19:315–326. doi: 10.1111/j.1600-0749.2006.00320.x. [DOI] [PubMed] [Google Scholar]
- 150.Marmorstein AD, Finnemann SC, Bonilha VL, Rodriguez-Boulan E. Morphogenesis of the retinal pigment epithelium: toward understanding retinal degenerative diseases. Ann N Y Acad Sci. 1998;857:1–12. doi: 10.1111/j.1749-6632.1998.tb10102.x. [DOI] [PubMed] [Google Scholar]
- 151.Finnemann SC, Leung LW, Rodriguez-Boulan E. The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2002;99:3842–3847. doi: 10.1073/pnas.052025899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bergmann M, Schutt F, Holz FG, Kopitz J. Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB J. 2004;18:562–564. doi: 10.1096/fj.03-0289fje. [DOI] [PubMed] [Google Scholar]
- 153.Nuckels RJ, Ng A, Darland T, Gross JM. The vacuolar-ATPase complex regulates retinoblast proliferation and survival, photoreceptor morphogenesis, and pigmentation in the zebrafish eye. Invest Ophthalmol Vis Sci. 2009;50:893–905. doi: 10.1167/iovs.08-2743. [DOI] [PubMed] [Google Scholar]
- 154.Biehlmaier O, Neuhauss SC, Kohler K. Double cone dystrophy and RPE degeneration in the retina of the zebrafish gnn mutant. Invest Ophthalmol Vis Sci. 2003;44:1287–1298. doi: 10.1167/iovs.02-0363. [DOI] [PubMed] [Google Scholar]
- 155.Neuhauss SC, Seeliger MW, Schepp CP, Biehlmaier O. Retinal defects in the zebrafish bleached mutant. Doc Ophthalmol. 2003;107:71–78. doi: 10.1023/a:1024492029629. [DOI] [PubMed] [Google Scholar]
- 156.Bahadori R, Rinner O, Schonthaler HB, Biehlmaier O, Makhankov YV, Rao P, Jagadeeswaran P, Neuhauss SC. The Zebrafish fade out mutant: a novel genetic model for Hermansky-Pudlak syndrome. Invest Ophthalmol Vis Sci. 2006;47:4523–4531. doi: 10.1167/iovs.05-1596. [DOI] [PubMed] [Google Scholar]
- 157.Kelsh RN, Brand M, Jiang YJ, Heisenberg CP, Lin S, Haffter P, Odenthal J, Mullins MC, van Eeden FJ, Furutani-Seiki M, Granato M, Hammerschmidt M, Kane DA, Warga RM, Beuchle D, Vogelsang L, Nusslein-Volhard C. Zebrafish pigmentation mutations and the processes of neural crest development. Development. 1996;123:369–389. doi: 10.1242/dev.123.1.369. [DOI] [PubMed] [Google Scholar]
- 158.Schonthaler HB, Lampert JM, von Lintig J, Schwarz H, Geisler R, Neuhauss SC. A mutation in the silver gene leads to defects in melanosome biogenesis and alterations in the visual system in the zebrafish mutant fading vision. Dev Biol. 2005;284:421–436. doi: 10.1016/j.ydbio.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 159.Spanakis E, Lamina P, Bennett DC. Effects of the developmental colour mutations silver and recessive spotting on proliferation of diploid and immortal mouse melanocytes in culture. Development. 1992;114:675–680. doi: 10.1242/dev.114.3.675. [DOI] [PubMed] [Google Scholar]
- 160.Theos AC, Truschel ST, Raposo G, Marks MS. The Silver locus product Pmel17/gp100/Silv/ME20: controversial in name and in function. Pigment Cell Res. 2005;18:322–336. doi: 10.1111/j.1600-0749.2005.00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Johnson SL, Weston JA. Temperature-sensitive mutations that cause stage-specific defects in Zebrafish fin regeneration. Genetics. 1995;141:1583–1595. doi: 10.1093/genetics/141.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298:2188–2190. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- 163.Vihtelic TS, Hyde DR. Light-induced rod and cone cell death and regeneration in the adult albino zebrafish (Danio rerio) retina. J Neurobiol. 2000;44:289–307. doi: 10.1002/1097-4695(20000905)44:3<289::aid-neu1>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 164.Wehman AM, Staub W, Meyers JR, Raymond PA, Baier H. Genetic dissection of the zebrafish retinal stem-cell compartment. Dev Biol. 2005;281:53–65. doi: 10.1016/j.ydbio.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 165.Bibliowicz J, Gross JM. Expanded progenitor populations, vitreo-retinal abnormalities, and Muller glial reactivity in the zebrafish leprechaun/patched2 retina. BMC Dev Biol. 2009;9:52. doi: 10.1186/1471-213X-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gonzalez-Nunez V, Nocco V, Budd A. Characterization of DrCol15a1b, a Novel Component of the Stem-Cell Niche in the Zebrafish Retina. Stem Cells. 2010 doi: 10.1002/stem.461. [DOI] [PubMed] [Google Scholar]
- 167.Cerveny KL, Cavodeassi F, Turner KJ, de Jong-Curtain TA, Heath JK, Wilson SW. The zebrafish flotte lotte mutant reveals that the local retinal environment promotes the differentiation of proliferating precursors emerging from their stem cell niche. Development. 2010;137:2107–2115. doi: 10.1242/dev.047753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Stephens WZ, Senecal M, Nguyen M, Piotrowski T. Loss of adenomatous polyposis coli (apc) results in an expanded ciliary marginal zone in the zebrafish eye. Dev Dyn. 2010;239:2066–2077. doi: 10.1002/dvdy.22325. [DOI] [PubMed] [Google Scholar]
- 169.Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A. Muller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25:397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 170.Fischer AJ, Reh TA. Muller glia are a potential source of neural regeneration in the postnatal chicken retina. Nat Neurosci. 2001;4:247–252. doi: 10.1038/85090. [DOI] [PubMed] [Google Scholar]
- 171.Cameron DA. Cellular proliferation and neurogenesis in the injured retina of adult zebrafish. Vis Neurosci. 2000;17:789–797. doi: 10.1017/s0952523800175121. [DOI] [PubMed] [Google Scholar]
- 172.Black GC, Mazerolle CJ, Wang Y, Campsall KD, Petrin D, Leonard BC, Damji KF, Evans DG, McLeod D, Wallace VA. Abnormalities of the vitreoretinal interface caused by dysregulated Hedgehog signaling during retinal development. Hum Mol Genet. 2003;12:3269–3276. doi: 10.1093/hmg/ddg356. [DOI] [PubMed] [Google Scholar]
- 173.Raymond PA, Barthel LK, Bernardos RL, Perkowski JJ. Molecular characterization of retinal stem cells and their niches in adult zebrafish. BMC Dev Biol. 2006;6:36. doi: 10.1186/1471-213X-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Raymond PA, Rivlin PK. Germinal cells in the goldfish retina that produce rod photoreceptors. Dev Biol. 1987;122:120–138. doi: 10.1016/0012-1606(87)90338-1. [DOI] [PubMed] [Google Scholar]
- 175.Julian D, Ennis K, Korenbrot JI. Birth and fate of proliferative cells in the inner nuclear layer of the mature fish retina. J Comp Neurol. 1998;394:271–282. [PubMed] [Google Scholar]
- 176.Bernardos RL, Barthel LK, Meyers JR, Raymond PA. Late-stage neuronal progenitors in the retina are radial Muller glia that function as retinal stem cells. J Neurosci. 2007;27:7028–7040. doi: 10.1523/JNEUROSCI.1624-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Fausett BV, Goldman D. A role for alpha1 tubulin-expressing Muller glia in regeneration of the injured zebrafish retina. J Neurosci. 2006;26:6303–6313. doi: 10.1523/JNEUROSCI.0332-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kassen SC, Ramanan V, Montgomery JE, C TB, Liu CG, Vihtelic TS, Hyde DR. Time course analysis of gene expression during light-induced photoreceptor cell death and regeneration in albino zebrafish. Dev Neurobiol. 2007;67:1009–1031. doi: 10.1002/dneu.20362. [DOI] [PubMed] [Google Scholar]
- 179.Qin Z, Barthel LK, Raymond PA. Genetic evidence for shared mechanisms of epimorphic regeneration in zebrafish. Proc Natl Acad Sci U S A. 2009;106:9310–9315. doi: 10.1073/pnas.0811186106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wangsa-Wirawan ND, Linsenmeier RA. Retinal oxygen: fundamental and clinical aspects. Arch Ophthalmol. 2003;121:547–557. doi: 10.1001/archopht.121.4.547. [DOI] [PubMed] [Google Scholar]
- 181.Saint-Geniez M, D'Amore PA. Development and pathology of the hyaloid, choroidal and retinal vasculature. Int J Dev Biol. 2004;48:1045–1058. doi: 10.1387/ijdb.041895ms. [DOI] [PubMed] [Google Scholar]
- 182.Alvarez Y, Chen K, Reynolds AL, Waghorne N, O'Connor JJ, Kennedy BN. Predominant cone photoreceptor dysfunction in a hyperglycaemic model of non-proliferative diabetic retinopathy. Dis Model Mech. 2010;3:236–245. doi: 10.1242/dmm.003772. [DOI] [PubMed] [Google Scholar]
- 183.Gleeson M, Connaughton V, Arneson LS. Induction of hyperglycaemia in zebrafish (Danio rerio) leads to morphological changes in the retina. Acta Diabetol. 2007;44:157–163. doi: 10.1007/s00592-007-0257-3. [DOI] [PubMed] [Google Scholar]
- 184.Zhu M, Provis JM, Penfold PL. The human hyaloid system: cellular phenotypes and inter-relationships. Exp Eye Res. 1999;68:553–563. doi: 10.1006/exer.1998.0632. [DOI] [PubMed] [Google Scholar]
- 185.Alvarez Y, Cederlund ML, Cottell DC, Bill BR, Ekker SC, Torres-Vazquez J, Weinstein BM, Hyde DR, Vihtelic TS, Kennedy BN. Genetic determinants of hyaloid and retinal vasculature in zebrafish. BMC Dev Biol. 2007;7:114. doi: 10.1186/1471-213X-7-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Skarie JM, Link BA. FoxC1 is essential for vascular basement membrane integrity and hyaloid vessel morphogenesis. Invest Ophthalmol Vis Sci. 2009;50:5026–5034. doi: 10.1167/iovs.09-3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Jonasova K, Kozmik Z. Eye evolution: lens and cornea as an upgrade of animal visual system. Semin Cell Dev Biol. 2008;19:71–81. doi: 10.1016/j.semcdb.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 188.Congdon N, West SK, Buhrmann RR, Kouzis A, Munoz B, Mkocha H. Prevalence of the different types of age-related cataract in an African population. Invest Ophthalmol Vis Sci. 2001;42:2478–2482. [PubMed] [Google Scholar]
- 189.Hejtmancik JF. Congenital cataracts and their molecular genetics. Semin Cell Dev Biol. 2008;19:134–149. doi: 10.1016/j.semcdb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Graw J. Congenital hereditary cataracts. Int J Dev Biol. 2004;48:1031–1044. doi: 10.1387/ijdb.041854jg. [DOI] [PubMed] [Google Scholar]
- 191.Kuszak JR, Costello J. In: The Structure of the Vertebrate Lens. FJ L, ML R, editors. Cambridge University Press: Development of the Ocular Lens; 2004. pp. 71–118. [Google Scholar]
- 192.O'Rahilly R. The prenatal development of the human eye. Exp Eye Res. 1975;21:93–112. doi: 10.1016/0014-4835(75)90075-5. [DOI] [PubMed] [Google Scholar]
- 193.Rafferty NS, Rafferty KA., Jr Cell population kinetics of the mouse lens epithelium. J Cell Physiol. 1981;107:309–315. doi: 10.1002/jcp.1041070302. [DOI] [PubMed] [Google Scholar]
- 194.Bassnett S, Beebe D. In: Lens Fiber Differentiation. Lovicu FJ, Robinson ML, editors. Cambridge University Press: Development of the Ocular Lens; 2004. pp. 214–244. [Google Scholar]
- 195.Beebe DC, Compart PJ, Johnson MC, Feagans DE, Feinberg RN. The mechanism of cell elongation during lens fiber cell differentiation. Dev Biol. 1982;92:54–59. doi: 10.1016/0012-1606(82)90149-x. [DOI] [PubMed] [Google Scholar]
- 196.Land MF, Fernald RD. The evolution of eyes. Annu Rev Neurosci. 1992;15:1–29. doi: 10.1146/annurev.ne.15.030192.000245. [DOI] [PubMed] [Google Scholar]
- 197.Leonard DW, Meek KM. Refractive indices of the collagen fibrils and extrafibrillar material of the corneal stroma. Biophys J. 1997;72:1382–1387. doi: 10.1016/S0006-3495(97)78784-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Semina EV, Brownell I, Mintz-Hittner HA, Murray JC, Jamrich M. Mutations in the human forkhead transcription factor FOXE3 associated with anterior segment ocular dysgenesis and cataracts. Hum Mol Genet. 2001;10:231–236. doi: 10.1093/hmg/10.3.231. [DOI] [PubMed] [Google Scholar]
- 199.Semina EV, Ferrell RE, Mintz-Hittner HA, Bitoun P, Alward WL, Reiter RS, Funkhauser C, Daack-Hirsch S, Murray JC. A novel homeobox gene PITX3 is mutated in families with autosomal-dominant cataracts and ASMD. Nat Genet. 1998;19:167–170. doi: 10.1038/527. [DOI] [PubMed] [Google Scholar]
- 200.Dutta S, Dietrich JE, Aspock G, Burdine RD, Schier A, Westerfield M, Varga ZM. pitx3 defines an equivalence domain for lens and anterior pituitary placode. Development. 2005;132:1579–1590. doi: 10.1242/dev.01723. [DOI] [PubMed] [Google Scholar]
- 201.Shi X, Bosenko DV, Zinkevich NS, Foley S, Hyde DR, Semina EV, Vihtelic TS. Zebrafish pitx3 is necessary for normal lens and retinal development. Mech Dev. 2005;122:513–527. doi: 10.1016/j.mod.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 202.Shi X, Luo Y, Howley S, Dzialo A, Foley S, Hyde DR, Vihtelic TS. Zebrafish foxe3: roles in ocular lens morphogenesis through interaction with pitx3. Mech Dev. 2006;123:761–782. doi: 10.1016/j.mod.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 203.Swindell EC, Zilinski CA, Hashimoto R, Shah R, Lane ME, Jamrich M. Regulation and function of foxe3 during early zebrafish development. Genesis. 2008;46:177–183. doi: 10.1002/dvg.20380. [DOI] [PubMed] [Google Scholar]
- 204.Graw J. Genetics of crystallins: cataract and beyond. Exp Eye Res. 2009;88:173–189. doi: 10.1016/j.exer.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 205.Goishi K, Shimizu A, Najarro G, Watanabe S, Rogers R, Zon LI, Klagsbrun M. AlphaA-crystallin expression prevents gamma-crystallin insolubility and cataract formation in the zebrafish cloche mutant lens. Development. 2006;133:2585–2593. doi: 10.1242/dev.02424. [DOI] [PubMed] [Google Scholar]
- 206.Stainier DY, Weinstein BM, Detrich HW, 3rd, Zon LI, Fishman MC. Cloche, an early acting zebrafish gene, is required by both the endothelial and hematopoietic lineages. Development. 1995;121:3141–3150. doi: 10.1242/dev.121.10.3141. [DOI] [PubMed] [Google Scholar]
- 207.Danysh BP, Duncan MK. The lens capsule. Exp Eye Res. 2009;88:151–164. doi: 10.1016/j.exer.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Timpl R, Brown JC. The laminins. Matrix Biol. 1994;14:275–281. doi: 10.1016/0945-053x(94)90192-9. [DOI] [PubMed] [Google Scholar]
- 209.Semina EV, Bosenko DV, Zinkevich NC, Soules KA, Hyde DR, Vihtelic TS, Willer GB, Gregg RG, Link BA. Mutations in laminin alpha 1 result in complex, lens-independent ocular phenotypes in zebrafish. Dev Biol. 2006;299:63–77. doi: 10.1016/j.ydbio.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 210.Zinkevich NS, Bosenko DV, Link BA, Semina EV. laminin alpha 1 gene is essential for normal lens development in zebrafish. BMC Dev Biol. 2006;6:13. doi: 10.1186/1471-213X-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Bystrom B, Virtanen I, Rousselle P, Gullberg D, Pedrosa-Domellof F. Distribution of laminins in the developing human eye. Invest Ophthalmol Vis Sci. 2006;47:777–785. doi: 10.1167/iovs.05-0367. [DOI] [PubMed] [Google Scholar]
- 212.Kohno T, Sorgente N, Ishibashi T, Goodnight R, Ryan SJ. Immunofluorescent studies of fibronectin and laminin in the human eye. Invest Ophthalmol Vis Sci. 1987;28:506–514. [PubMed] [Google Scholar]
- 213.Zenker M, Tralau T, Lennert T, Pitz S, Mark K, Madlon H, Dotsch J, Reis A, Muntefering H, Neumann LM. Congenital nephrosis, mesangial sclerosis, and distinct eye abnormalities with microcoria: an autosomal recessive syndrome. Am J Med Genet A. 2004;130A:138–145. doi: 10.1002/ajmg.a.30310. [DOI] [PubMed] [Google Scholar]
- 214.Zenker M, Aigner T, Wendler O, Tralau T, Muntefering H, Fenski R, Pitz S, Schumacher V, Royer-Pokora B, Wuhl E, Cochat P, Bouvier R, Kraus C, Mark K, Madlon H, Dotsch J, Rascher W, Maruniak-Chudek I, Lennert T, Neumann LM, Reis A. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet. 2004;13:2625–2632. doi: 10.1093/hmg/ddh284. [DOI] [PubMed] [Google Scholar]
- 215.Vihtelic TS, Yamamoto Y, Sweeney MT, Jeffery WR, Hyde DR. Arrested differentiation and epithelial cell degeneration in zebrafish lens mutants. Dev Dyn. 2001;222:625–636. doi: 10.1002/dvdy.1217. [DOI] [PubMed] [Google Scholar]
- 216.Stemple DL, Solnica-Krezel L, Zwartkruis F, Neuhauss SC, Schier AF, Malicki J, Stainier DY, Abdelilah S, Rangini Z, Mountcastle-Shah E, Driever W. Mutations affecting development of the notochord in zebrafish. Development. 1996;123:117–128. doi: 10.1242/dev.123.1.117. [DOI] [PubMed] [Google Scholar]
- 217.Moosajee M, Gregory-Evans K, Ellis CD, Seabra MC, Gregory-Evans CY. Translational bypass of nonsense mutations in zebrafish rep1, pax2.1 and lamb1 highlights a viable therapeutic option for untreatable genetic eye disease. Hum Mol Genet. 2008;17:3987–4000. doi: 10.1093/hmg/ddn302. [DOI] [PubMed] [Google Scholar]
- 218.Paulus JD, Halloran MC. Zebrafish bashful/laminin-alpha 1 mutants exhibit multiple axon guidance defects. Dev Dyn. 2006;235:213–224. doi: 10.1002/dvdy.20604. [DOI] [PubMed] [Google Scholar]
- 219.Biehlmaier O, Makhankov Y, Neuhauss SC. Impaired retinal differentiation and maintenance in zebrafish laminin mutants. Invest Ophthalmol Vis Sci. 2007;48:2887–2894. doi: 10.1167/iovs.06-1212. [DOI] [PubMed] [Google Scholar]
- 220.Klintworth GK. Corneal dystrophies. Orphanet J Rare Dis. 2009;4:7. doi: 10.1186/1750-1172-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Klyce S, Beuerman R. Structure and Function of the Cornea. The Cornea. 1998:3–50. [Google Scholar]
- 222.Piatigorsky J. Enigma of the abundant water-soluble cytoplasmic proteins of the cornea: the "refracton" hypothesis. Cornea. 2001;20:853–858. doi: 10.1097/00003226-200111000-00015. [DOI] [PubMed] [Google Scholar]
- 223.Cuthbertson RA, Tomarev SI, Piatigorsky J. Taxon-specific recruitment of enzymes as major soluble proteins in the corneal epithelium of three mammals, chicken, and squid. Proc Natl Acad Sci U S A. 1992;89:4004–4008. doi: 10.1073/pnas.89.9.4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Xu YS, Kantorow M, Davis J, Piatigorsky J. Evidence for gelsolin as a corneal crystallin in zebrafish. J Biol Chem. 2000;275:24645–24652. doi: 10.1074/jbc.M001159200. [DOI] [PubMed] [Google Scholar]
- 225.Li S, Tiab L, Jiao X, Munier FL, Zografos L, Frueh BE, Sergeev Y, Smith J, Rubin B, Meallet MA, Forster RK, Hejtmancik JF, Schorderet DF. Mutations in PIP5K3 are associated with Francois-Neetens mouchetee fleck corneal dystrophy. Am J Hum Genet. 2005;77:54–63. doi: 10.1086/431346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Boisset G, Polok BK, Schorderet DF. Characterization of pip5k3 fleck corneal dystrophy-linked gene in zebrafish. Gene Expr Patterns. 2008;8:404–410. doi: 10.1016/j.gep.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 227.Yeh LK, Liu CY, Chien CL, Converse RL, Kao WW, Chen MS, Hu FR, Hsieh FJ, Wang IJ. Molecular analysis and characterization of zebrafish keratocan (zKera) gene. J Biol Chem. 2008;283:506–517. doi: 10.1074/jbc.M707656200. [DOI] [PubMed] [Google Scholar]
- 228.Pellegata NS, Dieguez-Lucena JL, Joensuu T, Lau S, Montgomery KT, Krahe R, Kivela T, Kucherlapati R, Forsius H, de la Chapelle A. Mutations in KERA, encoding keratocan, cause cornea plana. Nat Genet. 2000;25:91–95. doi: 10.1038/75664. [DOI] [PubMed] [Google Scholar]
- 229.Ritch R, Shields M, Krupin T. 1996 The Glaucomas. [Google Scholar]
- 230.Gould DB, John SW. Anterior segment dysgenesis and the developmental glaucomas are complex traits. Hum Mol Genet. 2002;11:1185–1193. doi: 10.1093/hmg/11.10.1185. [DOI] [PubMed] [Google Scholar]
- 231.Gordon MO, Beiser JA, Brandt JD, Heuer DK, Higginbotham EJ, Johnson CA, Keltner JL, Miller JP, Parrish RK, 2nd, Wilson MR, Kass MA. The Ocular Hypertension Treatment Study: baseline factors that predict the onset of primary open-angle glaucoma. Arch Ophthalmol. 2002;120:714–720. doi: 10.1001/archopht.120.6.714. discussion 829–730. [DOI] [PubMed] [Google Scholar]
- 232.Le A, Mukesh BN, McCarty CA, Taylor HR. Risk factors associated with the incidence of open-angle glaucoma: the visual impairment project. Invest Ophthalmol Vis Sci. 2003;44:3783–3789. doi: 10.1167/iovs.03-0077. [DOI] [PubMed] [Google Scholar]
- 233.Civan MM, Macknight AD. The ins and outs of aqueous humour secretion. Exp Eye Res. 2004;78:625–631. doi: 10.1016/j.exer.2003.09.021. [DOI] [PubMed] [Google Scholar]
- 234.Gabelt BT, Kaufman PL. Pharmacologic Enhancement of Aqueous Humor Outflow. In: Buskirk M, Shields M, editors. 100 years of progress in glaucoma. Philadelphia: Lippincott-Raven; 1997. [Google Scholar]
- 235.Tamm ER. The trabecular meshwork outflow pathways: structural and functional aspects. Exp Eye Res. 2009;88:648–655. doi: 10.1016/j.exer.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 236.McMahon C, Semina EV, Link BA. Using zebrafish to study the complex genetics of glaucoma. Comp Biochem Physiol C Toxicol Pharmacol. 2004;138:343–350. doi: 10.1016/j.cca.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 237.Mears AJ, Jordan T, Mirzayans F, Dubois S, Kume T, Parlee M, Ritch R, Koop B, Kuo WL, Collins C, Marshall J, Gould DB, Pearce W, Carlsson P, Enerback S, Morissette J, Bhattacharya S, Hogan B, Raymond V, Walter MA. Mutations of the forkhead/winged-helix gene, FKHL7, in patients with Axenfeld-Rieger anomaly. Am J Hum Genet. 1998;63:1316–1328. doi: 10.1086/302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Mirzayans F, Gould DB, Heon E, Billingsley GD, Cheung JC, Mears AJ, Walter MA. Axenfeld-Rieger syndrome resulting from mutation of the FKHL7 gene on chromosome 6p25. Eur J Hum Genet. 2000;8:71–74. doi: 10.1038/sj.ejhg.5200354. [DOI] [PubMed] [Google Scholar]
- 239.Nishimura DY, Swiderski RE, Alward WL, Searby CC, Patil SR, Bennet SR, Kanis AB, Gastier JM, Stone EM, Sheffield VC. The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat Genet. 1998;19:140–147. doi: 10.1038/493. [DOI] [PubMed] [Google Scholar]
- 240.Tamimi Y, Skarie JM, Footz T, Berry FB, Link BA, Walter MA. FGF19 is a target for FOXC1 regulation in ciliary body-derived cells. Hum Mol Genet. 2006;15:3229–3240. doi: 10.1093/hmg/ddl400. [DOI] [PubMed] [Google Scholar]
- 241.Berry FB, Skarie JM, Mirzayans F, Fortin Y, Hudson TJ, Raymond V, Link BA, Walter MA. FOXC1 is required for cell viability and resistance to oxidative stress in the eye through the transcriptional regulation of FOXO1A. Hum Mol Genet. 2008;17:490–505. doi: 10.1093/hmg/ddm326. [DOI] [PubMed] [Google Scholar]
- 242.Vollrath D, Jaramillo-Babb VL, Clough MV, McIntosh I, Scott KM, Lichter PR, Richards JE. Loss-of-function mutations in the LIM-homeodomain gene, LMX1B, in nail-patella syndrome. Hum Mol Genet. 1998;7:1091–1098. doi: 10.1093/hmg/7.7.1091. [DOI] [PubMed] [Google Scholar]
- 243.Dunston JA, Reimschisel T, Ding YQ, Sweeney E, Johnson RL, Chen ZF, McIntosh I. A neurological phenotype in nail patella syndrome (NPS) patients illuminated by studies of murine Lmx1b expression. Eur J Hum Genet. 2005;13:330–335. doi: 10.1038/sj.ejhg.5201332. [DOI] [PubMed] [Google Scholar]
- 244.Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, Siegel-Bartelt J, Bierke-Nelson D, Bitoun P, Zabel BU, Carey JC, Murray JC. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996;14:392–399. doi: 10.1038/ng1296-392. [DOI] [PubMed] [Google Scholar]
- 245.Hjalt TA, Semina EV. Current molecular understanding of Axenfeld-Rieger syndrome. Expert Rev Mol Med. 2005;7:1–17. doi: 10.1017/S1462399405010082. [DOI] [PubMed] [Google Scholar]
- 246.Ng D, Thakker N, Corcoran CM, Donnai D, Perveen R, Schneider A, Hadley DW, Tifft C, Zhang L, Wilkie AO, van der Smagt JJ, Gorlin RJ, Burgess SM, Bardwell VJ, Black GC, Biesecker LG. Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat Genet. 2004;36:411–416. doi: 10.1038/ng1321. [DOI] [PubMed] [Google Scholar]
- 247.Masai I, Lele Z, Yamaguchi M, Komori A, Nakata A, Nishiwaki Y, Wada H, Tanaka H, Nojima Y, Hammerschmidt M, Wilson SW, Okamoto H. N-cadherin mediates retinal lamination, maintenance of forebrain compartments and patterning of retinal neurites. Development. 2003;130:2479–2494. doi: 10.1242/dev.00465. [DOI] [PubMed] [Google Scholar]
- 248.Chao R, Nevin L, Agarwal P, Riemer J, Bai X, Delaney A, Akana M, JimenezLopez N, Bardakjian T, Schneider A, Chassaing N, Schorderet DF, FitzPatrick D, Kwok PY, Ellgaard L, Gould DB, Zhang Y, Malicki J, Baier H, Slavotinek A. A male with unilateral microphthalmia reveals a role for TMX3 in eye development. PLoS One. 5:e10565. doi: 10.1371/journal.pone.0010565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Gestri G, Osborne RJ, Wyatt AW, Gerrelli D, Gribble S, Stewart H, Fryer A, Bunyan DJ, Prescott K, Collin JR, Fitzgerald T, Robinson D, Carter NP, Wilson SW, Ragge NK. Reduced TFAP2A function causes variable optic fissure closure and retinal defects and sensitizes eye development to mutations in other morphogenetic regulators. Hum Genet. 2009 doi: 10.1007/s00439-009-0730-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Brockerhoff SE, Rieke F, Matthews HR, Taylor MR, Kennedy B, Ankoudinova I, Niemi GA, Tucker CL, Xiao M, Cilluffo MC, Fain GL, Hurley JB. Light stimulates a transducin-independent increase of cytoplasmic Ca2+ and suppression of current in cones from the zebrafish mutant nof. J Neurosci. 2003;23:470–480. doi: 10.1523/JNEUROSCI.23-02-00470.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Del Bene F, Wehman AM, Link BA, Baier H. Regulation of neurogenesis by interkinetic nuclear migration through an apical-basal notch gradient. Cell. 2008;134:1055–1065. doi: 10.1016/j.cell.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Pathak N, Obara T, Mangos S, Liu Y, Drummond IA. The zebrafish fleer gene encodes an essential regulator of cilia tubulin polyglutamylation. Mol Biol Cell. 2007;18:4353–4364. doi: 10.1091/mbc.E07-06-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Tsujikawa M, Malicki J. Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron. 2004;42:703–716. doi: 10.1016/s0896-6273(04)00268-5. [DOI] [PubMed] [Google Scholar]
- 254.Wei X, Malicki J. nagie oko, encoding a MAGUK-family protein, is essential for cellular patterning of the retina. Nat Genet. 2002;31:150–157. doi: 10.1038/ng883. [DOI] [PubMed] [Google Scholar]
- 255.Zou J, Lathrop KL, Sun M, Wei X. Intact retinal pigment epithelium maintained by Nok is essential for retinal epithelial polarity and cellular patterning in zebrafish. J Neurosci. 2008;28:13684–13695. doi: 10.1523/JNEUROSCI.4333-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Brockerhoff SE, Hurley JB, Niemi GA, Dowling JE. A new form of inherited red-blindness identified in zebrafish. J Neurosci. 1997;17:4236–4242. doi: 10.1523/JNEUROSCI.17-11-04236.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Taylor MR, Kikkawa S, Diez-Juan A, Ramamurthy V, Kawakami K, Carmeliet P, Brockerhoff SE. The zebrafish pob gene encodes a novel protein required for survival of red cone photoreceptor cells. Genetics. 2005;170:263–273. doi: 10.1534/genetics.104.036434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Nishiwaki Y, Komori A, Sagara H, Suzuki E, Manabe T, Hosoya T, Nojima Y, Wada H, Tanaka H, Okamoto H, Masai I. Mutation of cGMP phosphodiesterase 6alpha'-subunit gene causes progressive degeneration of cone photoreceptors in zebrafish. Mech Dev. 2008;125:932–946. doi: 10.1016/j.mod.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 259.Cui S, Otten C, Rohr S, Abdelilah-Seyfried S, Link BA. Analysis of aPKClambda and aPKCzeta reveals multiple and redundant functions during vertebrate retinogenesis. Mol Cell Neurosci. 2007;34:431–444. doi: 10.1016/j.mcn.2006.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Seiler C, Finger-Baier KC, Rinner O, Makhankov YV, Schwarz H, Neuhauss SC, Nicolson T. Duplicated genes with split functions: independent roles of protocadherin15 orthologues in zebrafish hearing and vision. Development. 2005;132:615–623. doi: 10.1242/dev.01591. [DOI] [PubMed] [Google Scholar]
- 261.Otteson DC, Tsujikawa M, Gunatilaka T, Malicki J, Zack DJ. Genomic organization of zebrafish cone-rod homeobox gene and exclusion as a candidate gene for retinal degeneration in niezerka and mikre oko. Mol Vis. 2005;11:986–995. [PMC free article] [PubMed] [Google Scholar]
- 262.Faucherre A, Taylor GS, Overvoorde J, Dixon JE, Hertog J. Zebrafish pten genes have overlapping and non-redundant functions in tumorigenesis and embryonic development. Oncogene. 2008;27:1079–1086. doi: 10.1038/sj.onc.1210730. [DOI] [PubMed] [Google Scholar]
- 263.Babb SG, Kotradi SM, Shah B, Chiappini-Williamson C, Bell LN, Schmeiser G, Chen E, Liu Q, Marrs JA. Zebrafish R-cadherin (Cdh4) controls visual system development and differentiation. Dev Dyn. 2005;233:930–945. doi: 10.1002/dvdy.20431. [DOI] [PubMed] [Google Scholar]
- 264.Vihtelic TS, Hyde DR. Zebrafish mutagenesis yields eye morphological mutants with retinal and lens defects. Vision Res. 2002;42:535–540. doi: 10.1016/s0042-6989(01)00261-9. [DOI] [PubMed] [Google Scholar]
- 265.Vihtelic TS, Murphy TR, Watson CT, Willer GB, Gregg RG, Hyde DR. Zebrafish lens opaque (lop) mutation mapping and gene identification. Invest Ophthalmol Vis Sci. 2007;48 [Google Scholar]
- 266.Vihtelic TS, Yamamoto Y, Springer SS, Jeffery WR, Hyde DR. Lens opacity and photoreceptor degeneration in the zebrafish lens opaque mutant. Dev Dyn. 2005;233:52–65. doi: 10.1002/dvdy.20294. [DOI] [PubMed] [Google Scholar]
- 267.Cheng S, Shakespeare T, Mui R, White TW, Valdimarsson G. Connexin 48.5 is required for normal cardiovascular function and lens development in zebrafish embryos. J Biol Chem. 2004;279:36993–37003. doi: 10.1074/jbc.M401355200. [DOI] [PubMed] [Google Scholar]
- 268.Ye M, Berry-Wynne KM, Asai-Coakwell M, Sundaresan P, Footz T, French CR, Abitbol M, Fleisch VC, Corbett N, Allison WT, Drummond G, Walter MA, Underhill TM, Waskiewicz AJ, Lehmann OJ. Mutation of the bone morphogenetic protein GDF3 causes ocular and skeletal anomalies. Hum Mol Genet. 2010;19:287–298. doi: 10.1093/hmg/ddp496. [DOI] [PubMed] [Google Scholar]
- 269.Nakayama Y, Miyake A, Nakagawa Y, Mido T, Yoshikawa M, Konishi M, Itoh N. Fgf19 is required for zebrafish lens and retina development. Dev Biol. 2008;313:752–766. doi: 10.1016/j.ydbio.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 270.Asai-Coakwell M, French CR, Berry KM, Ye M, Koss R, Somerville M, Mueller R, van Heyningen V, Waskiewicz AJ, Lehmann OJ. GDF6, a novel locus for a spectrum of ocular developmental anomalies. Am J Hum Genet. 2007;80:306–315. doi: 10.1086/511280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.French CR, Erickson T, French DV, Pilgrim DB, Waskiewicz AJ. Gdf6a is required for the initiation of dorsal-ventral retinal patterning and lens development. Dev Biol. 2009;333:37–47. doi: 10.1016/j.ydbio.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 272.Evans TG, Yamamoto Y, Jeffery WR, Krone PH. Zebrafish Hsp70 is required for embryonic lens formation. Cell Stress Chaperones. 2005;10:66–78. doi: 10.1379/CSC-79R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Evans TG, Belak Z, Ovsenek N, Krone PH. Heat shock factor 1 is required for constitutive Hsp70 expression and normal lens development in embryonic zebrafish. Comp Biochem Physiol A Mol Integr Physiol. 2007;146:131–140. doi: 10.1016/j.cbpa.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 274.Harding RL, Howley S, Baker LJ, Murphy TR, Archer WE, Wistow G, Hyde DR, Vihtelic TS. Lengsin expression and function during zebrafish lens formation. Exp Eye Res. 2008;86:807–818. doi: 10.1016/j.exer.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Kennedy BN, Stearns GW, Smyth VA, Ramamurthy V, van Eeden F, Ankoudinova I, Raible D, Hurley JB, Brockerhoff SE. Zebrafish rx3 and mab21l2 are required during eye morphogenesis. Dev Biol. 2004;270:336–349. doi: 10.1016/j.ydbio.2004.02.026. [DOI] [PubMed] [Google Scholar]
- 276.Kleinjan DA, Bancewicz RM, Gautier P, Dahm R, Schonthaler HB, Damante G, Seawright A, Hever AM, Yeyati PL, van Heyningen V, Coutinho P. Subfunctionalization of duplicated zebrafish pax6 genes by cis-regulatory divergence. PLoS Genet. 2008;4:e29. doi: 10.1371/journal.pgen.0040029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Yeyati PL, Bancewicz RM, Maule J, van Heyningen V. Hsp90 selectively modulates phenotype in vertebrate development. PLoS Genet. 2007;3:e43. doi: 10.1371/journal.pgen.0030043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Heisenberg CP, Brand M, Jiang YJ, Warga RM, Beuchle D, van Eeden FJ, Furutani-Seiki M, Granato M, Haffter P, Hammerschmidt M, Kane DA, Kelsh RN, Mullins MC, Odenthal J, Nusslein-Volhard C. Genes involved in forebrain development in the zebrafish, Danio rerio. Development. 1996;123:191–203. doi: 10.1242/dev.123.1.191. [DOI] [PubMed] [Google Scholar]
- 279.Stigloher C, Ninkovic J, Laplante M, Geling A, Tannhauser B, Topp S, Kikuta H, Becker TS, Houart C, Bally-Cuif L. Segregation of telencephalic and eye-field identities inside the zebrafish forebrain territory is controlled by Rx3. Development. 2006;133:2925–2935. doi: 10.1242/dev.02450. [DOI] [PubMed] [Google Scholar]
- 280.Skarie JM, Link BA. The primary open-angle glaucoma gene WDR36 functions in ribosomal RNA processing and interacts with the p53 stress-response pathway. Hum Mol Genet. 2008;17:2474–2485. doi: 10.1093/hmg/ddn147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Schonthaler HB, Franz-Odendaal TA, Hodel C, Gehring I, Geisler R, Schwarz H, Neuhauss SC, Dahm R. The zebrafish mutant bumper shows a hyperproliferation of lens epithelial cells and fibre cell degeneration leading to functional blindness. Mech Dev. 127:203–219. doi: 10.1016/j.mod.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 282.Avanesov A, Dahm R, Sewell WF, Malicki JJ. Mutations that affect the survival of selected amacrine cell subpopulations define a new class of genetic defects in the vertebrate retina. Dev Biol. 2005;285:138–155. doi: 10.1016/j.ydbio.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 283.Catalano AE, Raymond PA, Goldman D, Wei X. Zebrafish dou yan mutation causes patterning defects and extensive cell death in the retina. Dev Dyn. 2007;236:1295–1306. doi: 10.1002/dvdy.21148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Mullins MC, Hammerschmidt M, Haffter P, Nusslein-Volhard C. Large-scale mutagenesis in the zebrafish: in search of genes controlling development in a vertebrate. Curr Biol. 1994;4:189–202. doi: 10.1016/s0960-9822(00)00048-8. [DOI] [PubMed] [Google Scholar]
- 285.Patton EE, Zon LI. The art and design of genetic screens: zebrafish. Nat Rev Genet. 2001;2:956–966. doi: 10.1038/35103567. [DOI] [PubMed] [Google Scholar]
- 286.Li Z, Joseph NM, Easter SS., Jr The morphogenesis of the zebrafish eye, including a fate map of the optic vesicle. Dev Dyn. 2000;218:175–188. doi: 10.1002/(SICI)1097-0177(200005)218:1<175::AID-DVDY15>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 287.Insinna C, Besharse JC. Intraflagellar transport and the sensory outer segment of vertebrate photoreceptors. Dev Dyn. 2008;237:1982–1992. doi: 10.1002/dvdy.21554. [DOI] [PMC free article] [PubMed] [Google Scholar]