

Developmental processes like seed germination are characterized by massive transcriptome changes. This study compares seed transcriptome data sets of different Brassicaceae to identify stable expressed reference genes for cross-species quantitative RT-PCR normalization. A workflow is presented for improving RNA quality, quantitative RT-PCR performance, and normalization when analyzing expression changes across species.
Abstract
Comparative biology includes the comparison of transcriptome and quantitative real-time RT-PCR (qRT-PCR) data sets in a range of species to detect evolutionarily conserved and divergent processes. Transcript abundance analysis of target genes by qRT-PCR requires a highly accurate and robust workflow. This includes reference genes with high expression stability (i.e., low intersample transcript abundance variation) for correct target gene normalization. Cross-species qRT-PCR for proper comparative transcript quantification requires reference genes suitable for different species. We addressed this issue using tissue-specific transcriptome data sets of germinating Lepidium sativum seeds to identify new candidate reference genes. We investigated their expression stability in germinating seeds of L. sativum and Arabidopsis thaliana by qRT-PCR, combined with in silico analysis of Arabidopsis and Brassica napus microarray data sets. This revealed that reference gene expression stability is higher for a given developmental process between distinct species than for distinct developmental processes within a given single species. The identified superior cross-species reference genes may be used for family-wide comparative qRT-PCR analysis of Brassicaceae seed germination. Furthermore, using germinating seeds, we exemplify optimization of the qRT-PCR workflow for challenging tissues regarding RNA quality, transcript stability, and tissue abundance. Our work therefore can serve as a guideline for moving beyond Arabidopsis by establishing high-quality cross-species qRT-PCR.
INTRODUCTION
Quantitative real-time RT-PCR (qRT-PCR) has emerged as a gold standard technique in quantifying gene transcript abundances due to its high accuracy and resolution power. The principal ease of use of this technology, in terms of simply following a protocol to rapidly obtain quantitative values for steady state transcript abundance without the need for deeper understanding of the underlying mechanisms, made qRT-PCR the method of choice for a broad range of applications. Nevertheless, there are numerous pitfalls and potential difficulties that arise when using this powerful technique, of which several will be addressed in this introduction. Due to its high sensitivity, certain requirements must be met for each step to yield reliable and reproducible results. This is reflected in a flourish of publications dealing with the general workflow, quality assessment, performance, and standardization of various stages of the qRT-PCR procedure (Huggett et al., 2005; Nolan et al., 2006; Bustin et al., 2009, 2010; Rieu and Powers, 2009; Derveaux et al., 2010). Recently, this awareness has also caused debates in the plant research community and likewise led here to recommendations for refining qRT-PCR standards (Martin, 2008; Udvardi et al., 2008). These provide guidance for studying a defined set of RNA samples in a single species for which sufficient sequence information is available.
One of the most important issues in this debate is the requirement of a robust normalization strategy based on validated so-called reference or housekeeping genes, which are needed to normalize transcript expression data (Czechowski et al., 2004; Gutierrez et al., 2008b; Guénin et al., 2009). However, the choice of a reference gene becomes especially difficult when the transcriptomes differ strongly, as the case may be for different samples, developmental processes, or species. For example, the transcriptomes of pollen and seeds are known to be very different from most other plant tissues (Czechowski et al., 2004, 2005; Cadman et al., 2006; Wang et al., 2008; Linkies et al., 2009; Wei et al., 2010). Comparative biology in which cross-species approaches are used to investigate conservation and diversity in a phylogenetic context also include the comparison of genome-wide transcript expression patterns (Bergmann et al., 2004; Rensink et al., 2005; Schranz et al., 2007; Tirosh et al., 2007; Andersen et al., 2008; Vandenbroucke et al., 2008; Schreiber et al., 2009; Parikh et al., 2010). Multispecies global gene expression analysis (phylotranscriptomics) aims to use the evolutionary distance between organisms to its advantage (Vandepoele and Van de Peer, 2005; Kohonen et al., 2007; Fierro et al., 2008; Hashimshony and Yanai, 2010). Cross-species phylotranscriptomics of distinct developmental processes such as pollen or seed development, maturation, and germination require validated cross-species reference genes for normalization of comparative qRT-PCR experiments. Here, we provide a guideline for moving qRT-PCR work recommendations (Czechowski et al., 2005; Gutierrez et al., 2008a; Udvardi et al., 2008) beyond Arabidopsis thaliana to the cross-species level illustrated in a spatio-temporal and hormonal case study with seeds for which the experimental challenges start already with the extraction of high-quality total RNA.
General concerns in qRT-PCR analyses are the input RNA quality and the RT reaction performance, both of which have major impacts on reproducibility and quality of the detection of target transcripts as well as on the stability of reference gene expression and thereby on the output results (Pérez-Novo et al., 2005; Fleige and Pfaffl, 2006). RNA extraction from seed tissues is a highly demanding task. Seeds often contain large amounts of polysaccharides that in many cases cause clogging of matrices of commercial column-based RNA extraction kits. Furthermore, seeds possess high phenolic content that, if not removed by excessive purification steps, can negatively influence qRT-PCR performance at multiple levels (e.g., by RT inhibition and PCR efficiency decrease). In terms of quality, not only the degree of RNA degradation matters, but also contamination with residual genomic DNA strongly influences qRT-PCR results and must be accounted for (Vandesompele et al., 2002a). Not only the RNA itself but also the conditions of the RT reaction leading to the cDNA template have a major impact on qRT-PCR results (Ståhlberg et al., 2004; Stangegaard et al., 2006; Ross et al., 2008). In our experimental strategy, we covered what we consider to be the most influential factors for robust qRT-PCR analysis and show how RT reactions can be improved even for demanding plant tissues.
One of the most critical issues of the qRT-PCR run is the PCR efficiency (Pfaffl, 2001; Bustin, 2004; Guénin et al., 2009). To account for this, postrun data handling is of utmost importance and has a major influence on obtaining meaningful and reproducible results. In an ideal 100% efficient PCR run, the amount of DNA amplicon is doubled in each PCR cycle. This is often assumed to be true for any gene and sample in a qRT-PCR analysis. Therefore, non-normalized transcript abundance is often calculated as 2(−CT), where CT is the cycle threshold (i.e., the fractional PCR cycle number at which the fluorescence of a particular sample passes a certain threshold within the early exponential phase of the amplification curve). The assumption of a 100% efficient PCR in all investigated samples is highly questionable regarding the vast amount of possible factors negatively influencing this efficiency, which include poor RNA or cDNA quality, inhibitory contaminants, such as salts, phenolic compounds, and certain proteins, as well as primer design and concentration, amplicon size, and structure (Meijerink et al., 2001; Kontanis and Reed, 2006; Karlen et al., 2007). Small efficiency differences between a target and a reference gene of only 5% can lead to a profound under- or overestimation of the real expression ratio, as has been exemplified in detail by Pfaffl (2004). Therefore, it is necessary to determine PCR efficiency in individual qRT-PCR reactions to be able to correct for it in postrun calculations. There are several methods available to determine PCR efficiency. Traditionally, this can be done by means of a standard curve or by using recent algorithms, such as LinRegPCR (Ramakers et al., 2003) or PCR Miner (Zhao and Fernald, 2005). The latter ones are able to determine efficiency on a fluorescence per well basis for the individual qRT-PCR reactions. In this work, we address the effects of PCR efficiency correction and show how efficiency can be optimized.
To analyze gene expression in different RNA samples and to adjust for sample-to-sample variation, relative transcript abundance quantification is the most widely used method. Appropriate and robust normalization for this is required to obtain corrected quantitative values. One approach is to normalize the amount of the detected mRNA of interest against the total mRNA amount present in a certain sample. Since the amount of all mRNAs in a sample is usually unknown, reference transcripts thought to be representative for the total mRNA pool are used (Huggett et al., 2005). These transcripts are ideally constitutively present (i.e., no differential expression under any of the tested experimental conditions [tissues, treatments, etc.]). This is an often overlooked consideration, despite the vast number of publications pointing out that traditionally used reference genes are often not stably expressed under all possible circumstances, thereby highlighting the importance of validating reference genes (e.g., Volkov et al., 2003; Radonić et al., 2004; Nicot et al., 2005; Gutierrez et al., 2008b; Remans et al., 2008). Especially in the plant research field such validation is most often ignored, as noted by Gutierrez et al. (2008a), who showed that out of 188 different qRT-PCR analyses recently published in leading plant biology journals only 3.2% used validated reference genes. It has been shown in a number of studies that different environmental conditions, developmental stages, tissues, or treatments strongly affect the usability of potential reference genes by causing differential expression (Thellin et al., 1999; Suzuki et al., 2000; Lee et al., 2002; Czechowski et al., 2005). Therefore, conclusions drawn from nonvalidated data might reflect changes in reference rather than target gene expression and thus could be misleading. Another concern is that reference genes found to perform well in one species do not necessarily perform equally well in another species (Gutierrez et al., 2008b). The rational assumption that a reference gene of a model species like Arabidopsis or rice (Oryza sativa) can be used in another species seems to decrease with increasing phylogenetic distance between the species. This is especially important for cross-species approaches and/or if working with rather weakly established species in terms of molecular knowledge, where the applicability of cross-species reference genes becomes a major point of concern.
Czechowski et al. (2005) performed a global transcriptional comparison of different developmental stages of Arabidopsis to determine new stable reference genes performing consistently in a variety of different experimental conditions, tissues, and developmental stages. In this analysis, they concluded that the transcriptomes of seeds and pollen are highly distinct compared with other plant tissues. This conclusion is in agreement with recent findings that many reference genes known from vegetative tissues are not stably expressed in seeds and pollen (Paolacci et al., 2009; Chen et al., 2010; Wei et al., 2010). Gutierrez et al. (2008a, 2008b) demonstrated that up to 100-fold variation could be found for expression of a target gene in distinct plant tissues and species depending on the reference gene used for normalization (i.e., there is a huge potential scope for misinterpretation of the results). They concluded that a universal reference gene does not exist but that there is an urgent need for systematic validation of reference genes based on the actual experimental conditions under investigation.
Here, we provide a guideline for obtaining and validating superior cross-species reference genes exemplified by a seed germination study in the Brassicaceae family. The seed is a remarkable stage in a plant life cycle allowing for long distance and temporal displacement through diverse and harsh environments that need special adaptations. Dry seeds are known to store RNA (Dure and Waters, 1965; Comai et al., 1989; Ishibashi et al., 1990). Upon imbibition, dramatic changes in the transcriptional profiles take place from early germination on (Nakabayashi et al., 2005; Preston et al., 2009; Okamoto et al., 2010). Most traditional reference genes have been selected in the pregenomic era and were assumed to be ubiquitously and constitutively expressed based on their functions in vegetative tissues. Thus, there is a need for reference gene identification for other plant stages, like seed development and germination. For seeds, the situation gets even more complicated when specific seed tissues or organs are compared. Endosperm and embryo tissues show different transcriptional profiles due to their distinct functions during seed dormancy and germination (reviewed in Finch-Savage and Leubner-Metzger, 2006; Holdsworth et al., 2008; Linkies et al., 2010a). Analyses of specific tissues are therefore important, but this further complicates the identification of stable reference genes that can be used both for the specific seed tissues and for entire seeds. Another general issue of tissue-specific analysis is that the amount of RNA available for qRT-PCR analysis is often a limiting factor as it requires time-consuming dissection during sampling. Therefore, multiple challenges must be met when performing qRT-PCR analysis with seed samples. Here, we show how optimization at different experimental levels is possible to obtain high qRT-PCR signal intensity with a low RNA input, and we present a strategy for identification of superior cross-species reference genes for seeds. We thereby provide a guideline for how to establish cross-species state-of-the-art qRT-PCR analysis using seed germination as a demanding case study.
A stable cross-species reference gene must fulfill three main criteria for all developmental (tissues, organs, and life cycle processes) and physiological (treatments, times, environmental cues, and stresses) states: (1) comparable overall transcript abundance to the target genes; (2) low variation in transcript abundance (stable and constitutive) across all the samples; and (3) cross-species stability across a phylogenetic clade for the individual developmental and physiological states. As a starting point for identifying superior cross-species reference genes, we use our previous transcriptome analysis of garden cress (Lepidium sativum; Brassicaceae) seed germination (Linkies et al., 2009). The larger seed size compared with Arabidopsis was used to carry out a heterologous transcriptome analysis of distinct seed tissues at different times during germination and upon hormonal treatment. Using these L. sativum transcriptome data sets, we identified candidate reference genes with putative constitutive and sufficiently high expression during the germination process and validated their stable expression by qRT-PCR. We further used public microarray data sets to identify conserved expression patterns and compare the stability of reference gene expression across different Brassicaceae species. Our case study with seeds thereby provides a guideline for moving qRT-PCR work recommendations (Czechowski et al., 2005; Gutierrez et al., 2008a; Udvardi et al., 2008) beyond Arabidopsis to the cross-species level and for applying it in a spatio-temporal and hormonal manner to germinating seeds as a demanding example. The presented cross-species in silico analysis together with qRT-PCR validation in germinating seeds of L. sativum and Arabidopsis provided superior stable reference genes for use within the Brassicaceae family. This results in a robust normalization procedure for seed qRT-PCR of different Brassicaceae species, which has been lacking so far. Using qRT-PCR off the beaten path of mainstream applications and model species with a sequenced genome will become increasingly important for multispecies gene expression analysis by phylotranscriptomics that will be promoted by the rise of next-generation sequencing as discussed later.
RESULTS AND DISCUSSION
Establishment of a State-of-the-Art qRT-PCR Workflow: Quality and qRT-PCR Performance of cDNA Depends Strongly on the RNA Extraction Method and Improved Efficiency for the RT Conditions
The basis of our optimized qRT-PCR workflow with seeds are high-quality total RNA samples combined with improved efficiency of the RT reactions, both being prerequisites for accurate qRT-PCR analyses also outlined by Bustin and Nolan (2004) and Udvardi et al. (2008). High RNA purity requires rigid RNA quality control (see Supplemental Figure 1 online). Besides that, a low RNA yield can limit downstream reactions. The amount of total RNA available can be restricted depending on the source of extraction and the hands-on time needed to obtain and process the samples. This is especially true for our case study with specific seed tissues in which tissue dissection and RNA extraction are laborious. The extraction of 5 μg total RNA from L. sativum micropylar endosperm seed tissues requires a hands-on time of 4 h to collect tissue sufficient for one biological replicate. Extraction of total RNA from seeds, including those of L. sativum and Arabidopsis, and other problematic plant tissues, is highly demanding (e.g., Zeng and Yang, 2002; Birtíc and Kranner, 2006; Wang et al., 2008) due to large amounts of polysaccharides (mucilage and storage substances), phenolic (tannins, and testa pigments), and other secondary compounds, which can negatively affect RNA quality and reduce RNA yield. L. sativum has extensive mucilage, which makes the use of commercial kits for fast RNA extraction almost impossible (e.g., columns are clogged). Therefore, a cetyltrimethylammoniumbromide (CTAB)/polyvinylpyrrolidone-based RNA extraction protocol according to Chang et al. (1993) was modified and used with subsequent cleanup steps to obtain high-quality total RNA from seeds (see Methods and Supplemental Figure 1 online). The CTAB method combined with polyvinylpyrrolidone addition has also been named the pine tree method and can be successfully adapted to a variety of demanding phenol-rich plant tissues (Porebski et al., 1997; Zeng and Yang, 2002; Gasic et al., 2004). Since the required extensive extraction and cleanup steps also reduce the yield, total RNA amount became the most limiting factor for qRT-PCR analyses with specific L. sativum seed tissues. Thus, we sought to obtain a reasonable qRT-PCR signal (i.e., a low CT value) with the lowest possible cDNA input.
To increase the qRT-PCR detection limit without increasing the total RNA starting amount, we analyzed the effects of different priming methods for the RT reactions and of different primer concentrations for the actual qRT-PCR reactions. Total RNA from L. sativum seeds was reverse transcribed using different priming methods (Table 1). To compare the RT efficiencies of the different methods, qRT-PCR analysis was performed using gene-specific primers designed by state-of-the-art criteria (Udvardi et al., 2008) for elongation factor 1-α (EF1-α) and actin 7 (ACT7), two transcripts that are highly abundant in L. sativum seed tissues. Table 1 summarizes these results and shows that the RT efficiencies of the different priming methods differ considerably. For both genes, the 0.3-nmol pentadecamer RT reactions (R15d) yielded the best results (i.e., the highest signal strength based on the same RNA input). These findings with plant RNA pools are in agreement with work demonstrating that pentadecamers produced higher cDNA yields and better coverage of human RNA pools (Stangegaard et al., 2006; Ross et al., 2008). Especially when the amount of total RNA is limiting and/or low abundant transcripts are investigated, the highest possible efficiency of the RT reactions is desirable to provide sufficient input cDNAs for the qRT-PCR reactions. For low-copy-number transcripts, Superscript III has been shown to be one of the two best reverse transcriptases with respect to repeatability, reproducibility, and sensitivity of the RT reaction (Okello et al., 2010). We show for EF1-α and ACT7 that in reactions using Superscript III the increased RT efficiency with R15d was 18- and 35-fold, respectively, compared with the least efficient RT priming method using random hexamers (R6) (Table 1). Although in both cases R15d yielded the highest RT efficiency, the fold increase values were gene specific. Clearly, the RT priming method and the structure of the amplicon are important parameters for which optimization leads to substantial increases in qRT-PCR sensitivity important to obtain robust and reproducible results with a minimum amount of high-quality input RNA.
Table 1.
Impact of Different RT Priming Methods on the RT Efficiencies of EF1-α and ACT7 Measured as Output Apparent Transcript Abundance of the qRT-PCR Reactions
| Gene | RT Priming Methoda | Apparent Transcript Abundanceb | Fold Increase Compared to R6 |
| EF1-α | R6 | 4.3 × 10−6 ± 0.7 × 10−6 | 1.0 |
| R15 | 4.1 × 10−5 ± 1.2 × 10−5 | 9.4 | |
| R6+dT | 8.7 × 10−6 ± 2.4 × 10−6 | 2.0 | |
| R15+dT | 3.9 × 10−5 ± 1.3 × 10−5 | 8.9 | |
| R15d | 7.7 × 10−5 ± 1.9 × 10−5 | 17.5 | |
| ACT7 | R6 | 2.6 × 10−7 ± 0.8 × 10−7 | 1.0 |
| R15 | 2.8 × 10−6 ± 1.1 × 10−6 | 10.9 | |
| R6+dT | 8.9 × 10−7 ± 3.5 × 10−7 | 3.4 | |
| R15+dT | 2.7 × 10−6 ± 1.0 × 10−6 | 10.4 | |
| R15d | 9.1 × 10−6 ± 2.7 × 10−6 | 34.9 |
Different primer combinations and concentrations were used in RT reactions with 5 μg total RNA from dry L. sativum seeds: R6 = 0.14 nmol random hexamers, R15 = 0.14 nmol random pentadecamers, R6+dT = 0.14 nmol random hexamers + 0.05 nmol oligo(dT), R15+dT = 0.14 nmol random pentadecamers + 0.05 nmol oligo(dT), and R15d = 0.3 nmol pentadecamers (amount per reaction).
The output apparent transcript abundances were determined as (1 + EAverageofReplicates)(−CT) (see Methods) and are presented as mean values ± sd from four biological replicates. Equal RNA input amounts were used for the RT reactions, but different RT priming efficiencies resulted in different cDNA amounts. Of these, equal input volumes were used in the actual qRT-PCR reactions and generated based on differences in cDNA amounts different output apparent transcript abundances.
An alternative to circumvent RNA amount restrictions is to amplify RNA prior to qRT-PCR, but this amplification is error prone as it is not necessarily achieved in a linear manner for every transcript and, therefore, of doubtful use in qRT-PCR analyses (Derveaux et al., 2010). In contrast with well-directed diagnostic assays for which the linearity of RNA amplification may be verified for the few genes in question, exploratory qRT-PCR assays in plant research often rely on comparing relative transcript abundances of RNA samples for many genes. For our seed research, we therefore work with nonamplified RNA samples. Furthermore, to minimize variation and errors, the same RT master mix should be used to generate one cDNA batch of all RNA samples in one experiment. Certainly there are other influential factors, such as different RT enzymes, RT temperatures, extraction protocols, and other experimental details (described in Ståhlberg et al., 2004; Nolan et al., 2006; Udvardi et al., 2008), but we limited our analysis to what we consider as the most important factors, especially when working with seed tissues.
The Importance of PCR Efficiency Correction for Successful qRT-PCR Analysis and the Simplicity of Using Algorithms for Postrun PCR Efficiency Determination with Web-Based Tools Like PCR Miner
After optimization of the RNA extraction and the RT conditions for high qRT-PCR signal strength, we investigated the effect of the primer concentrations on PCR efficiency of the qRT-PCR reactions. PCR efficiencies measured for different gene-specific primer concentrations are compiled in Table 2. A decrease in primer concentration resulted in a decrease in PCR efficiency, with 140 nM yielding the best results for both transcripts tested. However, as for the RT efficiency, the extent of the decrease in PCR efficiency was gene specific. It caused a 1.2- and 1.4-fold increase in PCR efficiency for EF1-α and ACT7, respectively, when the lowest and highest primer concentrations are compared (Table 2). This gene-specific effect is in agreement with results by Karlen et al. (2007) who showed that the amplicon structure is the main cause for PCR efficiency variation.
Table 2.
Impact of Gene-Specific Primer Concentrations on the PCR Efficiencies of qRT-PCR Reactions for EF1-α and ACT7
| Gene | Primer Concentration (nM)a | PCR Efficiencyb | Fold Increase Compared to 35 nM |
| EF1-α | 35 | 0.74 ± 0.02 | 1.0 |
| 70 | 0.82 ± 0.04 | 1.1 | |
| 140 | 0.88 ± 0.03 | 1.2 | |
| ACT7 | 35 | 0.65 ± 0.02 | 1.0 |
| 70 | 0.83 ± 0.01 | 1.3 | |
| 140 | 0.91 ± 0.03 | 1.4 |
cDNA was obtained from RT reactions with 0.3 nmol pentadecamers (R15d in Table 1) and total RNA of combined CAP&RAD tissues dissected from 8-h imbibed L. sativum seeds (Figure 2). qRT-PCR reactions were performed with three different concentrations of gene-specific primers using the same amounts of input cDNA.
PCR efficiencies were determined for each reaction using the PCR Miner algorithm (Zhao and Fernald, 2005). A value of 1.0 corresponds to 100% PCR efficiency, which corresponds to an exact doubling of amplicon numbers in each PCR cycle. Mean PCR efficiency values ± sd are presented for four biological replicates.
Traditionally, PCR efficiency is determined via a dilution series of input cDNA. The CT values of the template dilution series are plotted against the input cDNA amounts, and the PCR efficiency (E) is calculated from the obtained standard curve as E = 10(−1/slope) (Pfaffl, 2001; Rutledge and Côté, 2003). This approach has the drawback that a lot of cDNA is wasted for the standard curve, which is especially important when working with small amounts of RNA. A faster and easier way for determining E is to estimate it directly from the fluorescence signal of each individual reaction. A vast number of algorithms to calculate PCR efficiencies from fluorescence data on a per-well basis is available. One of the first approaches was implemented in software called LinRegPCR (Ramakers et al., 2003), which handles absolute fluorescence data of the exponential amplification phase of each individual reaction to determine E. Karlen et al. (2007) showed that LinRegPCR and standard curves both provide good estimators for E. Czechowski et al. (2004) found that these two methods provide very comparable E values, and Čikoš et al. (2007) showed that the standard curve method and a variety of single-well fluorescence data-based methods (LinRegPCR and others) perform comparably well for quantifying transcript abundances when the average PCR efficiency per gene is determined. These findings show that single-well fluorescence-based PCR efficiency estimation is possible and can replace template-, time- and money-consuming standard curve analyses. Beyond LinRegPCR, today there are different algorithms available to determine PCR efficiency, and it is a matter of debate which of those is the best algorithm and might evolve to a gold standard in qRT-PCR analysis (see Ramakers et al., 2003; Tichopad et al., 2003; Wong and Medrano, 2005; Guescini et al., 2008; Rutledge and Stewart, 2008; Logan et al., 2009; Ruijter et al., 2009 and references therein).
We used the effect of different primer concentrations to demonstrate the impact of qRT-PCR efficiency correction in postrun analysis by mimicking any parameter that could alter PCR efficiency. For our qRT-PCR workflow, we used the Real-time PCR Miner algorithm (Zhao and Fernald, 2005), a state-of-the-art software tool available for postrun qRT-PCR efficiency determination, which calculates single-well PCR efficiencies and CT values. Table 2 shows PCR efficiencies determined by PCR Miner that we used to calculate the efficiency corrected apparent transcript abundances for EF1-α and ACT7 obtained for the different concentrations of gene-specific primers. We compared these corrected values to apparent transcript abundances without PCR efficiency correction (Figure 1). The effect of a lowered PCR efficiency is immediately evident from the amplification plots (insets in the top right corners) as a reduced steepness of the exponential phase of the amplification curve. The PCR Miner algorithm uses this information to calculate individual efficiencies on a per-well fluorescence curve basis without the need of a standard dilution curve. For each of these qRT-PCR reactions differing in primer concentrations, the same amount of input cDNA was used, but different apparent transcript abundances were obtained with and without PCR efficiency correction (Figure 1). When no efficiency correction was performed (i.e., when a 100% PCR efficiency is assumed), the determined apparent transcript levels can be up to a 100-fold underestimated (compare the 35 nM with the 140 nM results in Figure 1). By contrast, the PCR Miner algorithm was able to precisely determine the different PCR efficiencies, and based on this postrun PCR efficiency correction provided highly similar apparent transcript levels for the reactions that had the same input cDNA amounts but different primer concentrations (Figure 1).
Figure 1.

Effect of Different PCR Efficiencies on the Measured Transcript Abundance of EF1-α and ACT7 with and without Efficiency Correction.
Different PCR efficiencies were obtained by varying the concentrations of the gene-specific primers as described in Table 2. Lowered primer concentration results in reduction of PCR efficiency recognizable by decreased steepness of the exponential phases in the qRT-PCR fluorescence curves (insets in the top right corners; shown are individual amplification plots for three different primer concentrations, four biological replicates each). PCR efficiencies were determined by PCR Miner software. Efficiency-corrected transcript abundance was calculated as described in Methods using the average efficiency of all samples for each gene and each primer concentration. Nonefficiency-corrected transcript abundance was calculated from the CT values determined by PCR Miner assuming 100% PCR efficiency. Mean values ± sd of four biological replicates are presented.
The PCR Miner algorithm uses nonbaseline subtracted raw fluorescence data as input to determine the fluorescence baseline and the exponential phase via a complex multistep fitting approach (Zhao and Fernald, 2005). It has recently been shown that such a correct baseline determination is a prerequisite to exact PCR efficiency calculations (Ruijter et al., 2009). In contrast with some other algorithms, PCR Miner determines PCR efficiency largely independently of the platform used to obtain the raw fluorescence qRT-PCR data (Zhao and Fernald, 2005; Arikawa and Yang, 2007). We found CT values to be very similar when determined via PCR Miner and with Applied Biosystems’ SDS software (v 1.4) for real-time PCR platforms. Furthermore, Real-time PCR Miner is user friendly and freely available as a Web-based tool (www.miner.ewindup.info). In Figure 1, we showed how correction of transcript abundance with PCR efficiencies determined by PCR Miner is able to compensate for differences in CT values caused by differences in PCR efficiency rather than by different cDNA amounts. The use of PCR Miner and subsequent efficiency correction to calculate transcript abundance can therefore fully compensate for large differences in PCR efficiency and makes cDNA-requiring standard curves dispensable in qRT-PCR analyses.
This is especially useful when many different genes are analyzed in the same RNA samples. We investigated the dependence of efficiencies derived from our complete qRT-PCR data set on the amplified gene and on other factors potentially influencing PCR efficiency, such as different tissues or treatments. We calculated F-values and approximate Z-values of six different factors to assess their importance on PCR efficiency (Table 3). This analysis shows the highest approximate Z-value (i.e., the most important factor in explaining PCR efficiency variation) for the amplified gene. This finding is in full agreement with Karlen et al. (2007), who also showed that the best input DNA quantification model in terms of precision, robustness, and reliability relies on averaging efficiencies per amplicon. We therefore calculated non-normalized transcript abundances using averaged PCR efficiencies of all RNA samples for each gene (EAveragePerAmplicon) separately as (1 + EAveragePerAmplicon)− CTIndividualSample rather than using individual per-sample efficiencies or averaged efficiencies for a RNA sample class. Taken together, optimal design and concentration of the gene-specific primer pairs together with PCR efficiency correction using algorithms like PCR Miner are crucial for successful qRT-PCR analysis.
Table 3.
Factors Influencing PCR Efficiency as Determined by Analysis of Variance F-Tests on Efficiencies Obtained from 687 qRT-PCR Reactions of L. sativum
| Factora | Degrees of Freedom | F-Valueb | Approximate Z-Valuec | R2d | Adjusted R2e |
| Time | 4 | 13.03 | 5.98 | 0.071 | 0.066 |
| Tissue | 2 | 67.92 | 9.57 | 0.166 | 0.163 |
| Treatment | 1 | 20.48 | 4.15 | 0.029 | 0.028 |
| Sample | 59 | 2.48 | 5.83 | 0.190 | 0.113 |
| Replicate | 14 | 10.00 | 9.29 | 0.172 | 0.155 |
| Gene | 11 | 90.75 | 24.72 | 0.597 | 0.590 |
Dependence of PCR efficiency on different multilevel factors was tested. Corresponding to Figure 3, these factors are time in hours (0, 8, 18, 30, and 96), tissue (CAP, RAD, and CAP&RAD), treatment (CON and ABA), individual sample, biological replicates, and the amplified gene.
F-values were obtained by testing the linear model for PCR efficiency with each factor included individually against the null model, which includes the constant term only.
The approximate Z-values were obtained by applying the Wilson-Hilferty cube root normalizing transformation to the F-values and then standardizing, noting that because the denominator degrees of freedom is large in each case, the theoretical F distribution is effectively a scaled χ2 distribution. The approximate Z-values of different factors can be more directly compared than F-values. Larger Z-values indicate higher importance of a factor in explaining PCR efficiency variation.
R2 is the square of the correlation between the observed PCR efficiency values and the fitted values under each model. A value close to 1 corresponds to a high level of agreement between the fitted model and the observed values.
Adjusted R2 is defined in similar fashion to R2 but tends to be smaller than R2 when the model has a large number of parameters, as is the case with the factor sample. The table shows that the amplified gene is by some way the most important factor in explaining PCR efficiency variation in our data set. We have not reported P values as all were very small (<10−6 in all cases and <10−16 in some cases) and therefore are not useful for comparing the importance of the different factors.
Seed Transcriptomes Differ from Other Tissues, and Mining of Cross-Species Microarrays of L. sativum Seed Tissues Provided Suitable Candidate Reference Genes for L. sativum and Arabidopsis Germination
Dry seeds store mRNA, which was first shown for cotton (Gossypium hirsutum; Dure and Waters, 1965) and later found to be a general phenomenon of desiccated orthodox seeds (Comai et al., 1989; Ishibashi et al., 1990; Nakabayashi et al., 2005). Many of these transcripts may be important for late embryogenesis as well as for early seed germination. Over 10,000 stored different mRNAs were identified by global transcriptome analysis in dry Arabidopsis seeds (Nakabayashi et al., 2005; Kimura and Nambara, 2010). So far, the published dry seed transcriptomes are from whole seeds, but it is known that the different seed compartments (e.g., endosperm and embryo) accumulate different transcripts during seed development (Le et al., 2010). The mature seeds of most species, including the Brassicaceae L. sativum and Arabidopsis, have retained a single layer of endosperm between the embryo and the testa (Linkies et al., 2010a). In these cases, weakening of the micropylar endosperm covering the radicle/hypocotyl is an important process during seed germination that involves tissue interactions (e.g., Bewley, 1997; Finch-Savage and Leubner-Metzger, 2006; Holdsworth et al., 2008). During seed germination, massive transcriptome changes take place as shown, for example, in Arabidopsis (Nakabayashi et al., 2005; Preston et al., 2009), Brassica napus (Li et al., 2005), and barley (Hordeum vulgare; Sreenivasulu et al., 2008). Transcriptomes can differ considerably between specific seed tissues, which has been shown for L. sativum (Linkies et al., 2009) and barley (Barrero et al., 2009). Seed transcriptomes therefore not only exhibit massive temporal changes upon imbibition but are also highly distinct between seed tissues.
Czechowski et al. (2005) used Arabidopsis microarray data sets that differed in developmental processes, tissues, stress, or hormone treatments and identified new reference genes with better performance. They conclude from their analysis that pollen and seeds have transcriptomes that are very different from other tissues. This is indeed the case for germinating pollen (Grennan, 2007; Wang et al., 2008; Wei et al., 2010) and seeds (Nakabayashi et al., 2005; Cadman et al., 2006; Linkies et al., 2009). Therefore, reference genes that originate from work with vegetative tissues are unlikely to be optimal candidates for qRT-PCR normalization of seed germination as demonstrated in cereal grains (Paolacci et al., 2009) and, as we show below, in Brassicaceae seeds.
We used the extensive L. sativum seed transcriptome data sets described by Linkies et al. (2009) to identify new candidate reference genes for robust normalization of L. sativum and Arabidopsis cross-species qRT-PCR analyses during seed germination. These data sets consisted of specific seed tissues, treatments, and times prior to endosperm rupture (i.e., the completion of germination). The seeds of these two Brassicaceae species are similar in germination physiology and structure (Figure 2) but differ significantly in size. Linkies et al. (2009) used the bigger seed size of L. sativum to dissect specific seed tissues, as described in Figure 2B. The radicle/hypocotyl (RAD) and the micropylar endosperm (CAP) play different roles during the germination process: the radicle/hypocotyl elongates during germination, while the endosperm regulates germination by functioning as a restraint to radicle protrusion, which ruptures the endosperm when germination is completed (Ni and Bradford, 1993; Toorop et al., 2000; Leubner-Metzger, 2003; Müller et al., 2006). Abscisic acid (ABA) strongly delays endosperm rupture of L. sativum, Arabidopsis (Figure 2C), and other species (Kucera et al., 2005), at least in part by inhibiting the onset and rate of endosperm cap weakening (Figure 2C). Specific seed tissues were used to conduct time-course transcriptome analyses of the germination process with (ABA) and without (control; labeled CON) the addition of ABA to the medium (Figure 2C). Candidates were selected from the 22,025 transcripts present in the CON arrays that had stable and high expression across times and tissues, as described in Methods. From these stable and highly expressed genes, an overlap of 1604 genes was determined for the CON arrays (Figure 2A). The same procedure was applied to the 19,704 transcripts present in the ABA arrays, and an overlap of 266 genes was determined (Figure 2A). Both overlaps were compared and 15 transcripts were identified as present in both selections (Figure 2A; see Supplemental Data Set 1 online). Therefore, these represent genes with both a high and stable transcript expression level in the specific seed tissues during germination and whose expression stability and level was not appreciably affected by ABA.
Figure 2.

Selection of Constitutive (Stable) Expressed Transcripts as Reference Gene Candidates for qRT-PCR Analysis of L. sativum Seed Germination from Transcriptome Data Sets.
(A) Microarray data sets from Linkies et al. (2009) were used to select genes with constitutive (blue) and high (red) transcript expression in different seed tissues and at different times during seed germination without (CON arrays) and with ABA added to the medium (ABA arrays). Microarray expression values of the 15 reference gene candidates are listed in Supplemental Data Set 1 online.
(B) Seed structure and seed size comparison of L. sativum and Arabidopsis. CAP (micropylar endosperm) and RAD (radicle plus lower hypocotyl) tissues were dissected from L. sativum seeds. NME, nonmicropylar endosperm.
(C) Germination time courses of L. sativum and Arabidopsis. For L. sativum, the times for RNA extraction from specific seed tissues for the CON and ABA arrays and qRT-PCR analyses are indicated on top of graph. Only seeds prior to the completion of germination (i.e., with unruptured micropylar endosperm) were used for these analyses. Both Arabidopsis and L. sativum have a two-step germination process with testa rupture preceding endosperm rupture (Liu et al., 2005; Müller et al., 2006). ABA treatment of after-ripened seeds inhibits endosperm weakening and rupture but does not affect the kinetics of testa rupture.
We succeeded in cloning partial cDNAs for five of these L. sativum genes for further analysis by qRT-PCR. These candidate reference genes are the putative orthologs of the Arabidopsis genes At1G17210 (ILP1; zinc ion binding), At2G04660 (APC2; ubiquitin protein ligase), At2G19980 (allergen V5/Tpx-1-related family protein), At2G20000 (HBT; anaphase-promoting complex subunit), and At4G04320 (malonyl-CoA decarboxylase family protein). The cDNA sequences were submitted to GenBank and for reasons of comparability were given names that correspond in numbers to the orthologous Arabidopsis gene identifiers: LesaG17210, LesaG04660, LesaG19980, LesaG20000, and LesaG04320. The nucleotide similarities were between 77 and 92% (details and GenBank accession numbers are listed in Supplemental Table 1 online). The high sequence similarity values are in agreement with what we obtained before for other L. sativum and Arabidopsis sequence comparisons (Graeber et al., 2010; Linkies et al., 2010b). We therefore assume that the five L. sativum reference gene candidates are the putative orthologs of the Arabidopsis genes. For highest accuracy and comparability, qRT-PCR primers for both L. sativum and Arabidopsis genes were designed at identical or near-identical positions within the cDNAs (see Supplemental Table 2 online) for testing the cross-species usability of these five reference gene candidates in seed germination experiments with both species.
Rigid Expression Stability Validation Delivers Superior Reference Genes with Stable Transcript Abundances for Cross-Species qRT-PCR Analysis of L. sativum and Arabidopsis Seed Germination
We used qRT-PCR to determine the expression stability of the five reference gene candidates in specific seed tissues, RAD and CAP, of L. sativum and in whole seeds of Arabidopsis at different times during germination without (CON) and with ABA treatment (Figure 3). For the L. sativum seed tissues, we used the same time points during germination for sampling as in the original transcriptome analysis of Linkies et al. (2009). In addition, to consider the dry seed (0 h) state for which the two tissues cannot be separated, we also included combined CAP&RAD 0 h samples. For the Arabidopsis whole-seed samples, we investigated dry seeds (0 h) as well as different time points for the CON and ABA series (Figure 3). With these sampling times, we cover the entire germination time course for both species (Figure 2C). This resulted in 57 L. sativum and 22 Arabidopsis RNA samples for which we performed qRT-PCR (Figure 3). We compared the five new reference gene candidates selected from the L. sativum seed transcriptome analysis (see above) with seven traditionally used reference genes: cyclophilin1 (CYP1), 5.8S rRNA, ACT7, ACT8, EF1-α, cyclophilin5 (CYP5), and ubiquitin11 (UBQ11), which were chosen as they are frequently used for qRT-PCR normalization in seed research. Fluorescence raw data were analyzed with PCR Miner to determine well-specific CT values and efficiencies that were used to calculate (see Methods) the efficiency-corrected transcript abundances (Figure 3). In addition, no-RT controls, no-template controls, and inter-run controls were included in the analysis as described in Methods. In this way, we performed a precise comprehensive analysis as demanded by qRT-PCR state-of-the-art quality standards (Udvardi et al., 2008; Bustin et al., 2009, 2010; Guénin et al., 2009).
Figure 3.

Efficiency-Corrected Transcript Abundance of New and Traditionally Used Reference Genes for Brassicaceae Seed Germination as Determined by qRT-PCR.
(A) L. sativum seed tissue RNA samples, CAP (micropylar endosperm) and RAD (radicle plus lower hypocotyl), were dissected from seeds imbibed without (CON) and with ABA added, at the times indicated. In addition, to also consider the dry seed (0 h) state for which the two tissues cannot be separated, we also included combined CAP&RAD 0 h samples (and combined 8 h samples for the comparison with the separated 8 h tissues; all combined samples included also testa).
(B) Arabidopsis whole-seed RNA samples from the CON and ABA series. For both species, only seeds prior to the completion of germination (i.e., with unruptured micropylar endosperm) were used for these analyses (as described in Figure 2). PCR efficiency correction was performed as described in Methods. Single-sample results are presented, but for comparisons and downstream calculations, the three to four biological replicates (as indicated in the graph) can be combined. For new reference genes, the L. sativum sequence names and the Arabidopsis AGI codes are indicated. Traditionally used reference genes are indicated by “trivial name (AGI code).”
These results (Figure 3) indicate that transcript stability differs between genes, treatments, times, tissues, species, and even between biological replicates. The GeNORM software tool (Vandesompele et al., 2002b) represents one of the most commonly used algorithms to analyze these differences and to thereby validate reference genes by their average gene transcript expression stability across samples (Gutierrez et al., 2008a). It carries out a pairwise comparison of a given set of genes and expression values, resulting in determination of the average expression stability measure M. This value therefore allows ranking from the least (highest M value) to the most (lowest M value) stable gene. Using the results presented in Figure 3, we performed a GeNORM analysis for the reference gene candidates of L. sativum and Arabidopsis (Figure 4). These stability rankings clearly show that four (L. sativum) or three (Arabidopsis) of the five new putative reference genes perform better compared with all tested traditionally used genes across the analyzed samples for each of the two species. Furthermore, three of the new reference gene candidates (LesaG20000/At2G20000, LesaG17210/At1G17210, and LesaG04660/At2G04660) appear to be superior cross-species reference genes in both species as they are most stable during both L. sativum and Arabidopsis seed germination (Figure 4).
Figure 4.

Ranking of New and Traditional L. sativum and Arabidopsis Reference Genes for Seed Germination Based on Their Average Expression Stability Measure M.
M was determined by analyzing the efficiency-corrected transcript abundances across all samples (as determined by qRT-PCR; Figure 3) via GeNORM for L. sativum (A) and Arabidopsis (B) separately. Black bars indicate traditional and white bars new reference genes that were selected from L. sativum microarray analysis (Figure 2). Note that stability measure M decreases more steadily within all the L. sativum reference genes, whereas for Arabidopsis, there is a rapid decline within the three least stable genes and more stable genes do differ less among each other. A possible reason for this is the fact that due to the specific seed tissue samples, the L. sativum data set is more diverse, whereas whole-seed samples were used for Arabidopsis. The average expression stability rankings of the two species are highly similar. The orthologous pairs LesaG20000/At2G20000, LesaG04660/At2G04660, and LesaG17210/At1G17210 provide the most stable cross-species reference genes for qRT-PCR analyses of seed germination.
It is of interest to quantify the difference between expression stability rankings such as those presented in Figure 4. To do this, we used a simple Euclidean metric, as explained in Methods. This enabled us to calculate a distance, which we name the ranking distance, between any two rankings of the same set of items. The ranking distance is normalized so that the maximum possible distance between two rankings is 1. The minimum distance of 0 occurs when two rankings are identical. The ranking distance for the comparison between the two species shown in Figure 4 is 0.37, and this value also numerically visualizes their considerable similarity.
For L. sativum seed germination, the complex set of 57 samples that differed in tissue, time, and treatment allowed us to analyze reference gene performance in the treatments and tissues separately. Analysis of the CON and the ABA series (Table 4) showed that the stability rankings of the various reference genes differed from the overall analysis (Figure 4A) with the CON series being very similar to the overall analysis (ranking distance 0.21), the ABA series being rather different from the overall series (ranking distance 0.53), and an even higher difference between the CON and the ABA series (ranking distance 0.60). However, with the exception of LesaG17210 in the ABA series (underlining the importance for validating each treatment), the three best CON and ABA series reference genes for L. sativum (LesaG04320, LesaG04660, and LesaG20000) are also among the most stable reference genes in the overall analysis (Table 4, Figure 4A). The stability rankings of the various reference genes also differed (Table 4) between the CAP, RAD, and the overall series with a high similarity between the overall series (Figure 4A) and the RAD series (ranking distance 0.15, Table 4) and somewhat more dissimilarities between the overall series and the CAP series (ranking distance 0.31), as well as between the RAD and the CAP series (ranking distance 0.36). However, the four best reference genes of the overall analysis were also the most stable ones (Table 4, Figure 4A).
Table 4.
The Effect of Treatments and Tissues on the Average Expression Stability (M) Ranking of Traditional and New Reference Genes for L. sativum Seed Germination
| Subseries Treatments, Combined Tissue (CAP and RAD) Results |
Subseries Tissues, Combined Treatment (CON and ABA) Results |
||
| CON | ABA | CAP | RAD |
| LesaG04320/LesaG17210 (0.23) | LesaG04660/LesaG20000 (0.46) | LesaG17210/LesaG20000 (0.26) | LesaG17210/LesaG20000 (0.34) |
| LesaG20000 (0.39) | LesaG04320 (0.50) | LesaG04320 (0.31) | LesaG04320 (0.39) |
| CYP5 (0.54) | ACT7 (0.57) | LesaG04660 (0.41) | LesaG04660 (0.42) |
| EF-1α (0.64) | CYP5 (0.64) | UBQ11 (0.48) | CYP5 (0.46) |
| ACT7 (0.67) | ACT8 (0.73) | EF-1α (0.53) | ACT7 (0.49) |
| LesaG04660 (0.70) | CYP1 (0.80) | ACT7 (0.57) | EF-1α (0.50) |
| CYP1 (0.91) | 5.8s rRNA (0.91) | CYP5 (0.60) | CYP1 (0.57) |
| 5.8s rRNA (1.07) | UBQ11 (1.05) | 5.8s rRNA (0.71) | ACT8 (0.63) |
| ACT8 (1.19) | LesaG19980 (1.38) | ACT8 (0.82) | 5.8s rRNA (0.70) |
| UBQ11 (1.35) | LesaG17210 (2.07) | CYP1 (0.91) | LesaG19980 (0.78) |
| LesaG19980 (1.49) | EF-1α (2.65) | LesaG19980 (1.21) | UBQ11 (0.86) |
Distinct subseries of efficiency-corrected qRT-PCR results (Figure 3A) were analyzed with GeNORM to obtain average reference genes stability measures (M; numbers in parentheses). In the subseries treatments, the samples from different tissues (CAP and RAD) and times of the series without (CON) and with ABA addition are compared. In the subseries tissues, the samples from different treatments (CON and ABA) and times of the CAP and the RAD series are compared. For each treatment or tissue, the reference genes are ranked from most stable (top, low M values) to least stable (bottom, high M values). New reference genes derived from the L. sativum transcriptome analysis (Figures 3 and 4) are in bold. Note that GeNORM does not discriminate between the two most stable genes.
Taken together, transcriptome analyses can be successfully mined to provide candidate reference genes with high probability, but rigid validation for all tissues, times, and treatments is required to obtain reference genes with a proven performance. Most of the new reference genes that we obtained were superior to the traditionally used ones in seed germination: Two (LesaG20000 and LesaG04320) of the three best new reference genes from the overall analysis for L. sativum are also the two most stable ones upon treatments (CON versus ABA) and among seed tissues (CAP versus RAD) of L. sativum. Most interestingly, the putative orthologs LesaG20000/At2G20000, LesaG17210/At1G17210, and LesaG04660/At2G04660 appear to be superior cross-species reference genes for investigating L. sativum and Arabidopsis seed germination. In agreement with the distinct nature of seed transcriptomes from transcriptomes of other processes, none of them is present on any of the 10 Top 100 lists for superior reference genes of the above mentioned analysis of Czechowski et al. (2005).
From the ranking distance comparisons described above, we conclude that for seed germination the species and tissue differences are less critical for the reference gene stability rankings than the ABA treatment. That different treatments, including hormones and stresses, can have a strong effect on reference gene stability has also been shown by others (Volkov et al., 2003; Radonić et al., 2004; Nicot et al., 2005; Remans et al., 2008) and underlines the importance of reference gene validation with all the different RNA samples of an experiment. Despite these differences in the stability rankings, in all the different rankings (species, tissues, and treatments; Figure 4, Table 4), three to four of our new reference genes constitute the top group.
Comparison of Brassicaceae Seed Development and Germination Microarray Data Sets Revealed Phylogenetical Conservation of Reference Gene Stability for a Developmental Process across Species
We further broadened this seed-related cross-species approach and tested for its robustness using commonly available Arabidopsis seed microarray data. For this in silico analysis, we used the seed-specific eFP browser and the eNorthern tool (available at www.bar.utoronto.ca) to access diverse transcriptome data sets not only of Arabidopsis seed germination but also of seed development as a distinct developmental process (Toufighi et al., 2005; Winter et al., 2007; Bassel et al., 2008). Altogether, 101 different microarray experiments focusing on diverse treatments, mutants, tissues, and developmental processes of Arabidopsis seeds were analyzed (see Supplemental Data Set 2 online). Expression values for the new and traditional reference genes were obtained as described in Methods, ranked using GeNORM, and the result of this analysis is shown as the series Seed Total in Figure 5. A considerable similarity of the Arabidopsis reference gene expression stability rankings obtained from our qRT-PCR data (Figure 4B) and microarray data (Figure 5, Seed Total) is evident (ranking distance 0.35). For a more detailed analysis of the transcriptome data, we used GeNORM to rank expression data of the reference genes from experiments focusing only on the seed germination process (Figure 5, subseries Germination Total) and on the seed development process (Figure 5, subseries Development Total) individually. When comparing qRT-PCR data of Arabidopsis seed germination (Figure 4B) with microarray data of Arabidopsis seed germination (Figure 5, subseries Germination Total), we find extremely similar gene expression stability rankings (ranking distance 0.26), whereas when comparing this qRT-PCR data ranking to a ranking of microarray data on seed development (Figure 5, subseries Development Total), large differences become evident (ranking distance 0.68). This difference is also obvious when rankings based purely on microarray data of these two distinct developmental processes are compared, as indicated by the black arrows between subseries Germination Total and Development Total in Figure 5 (corresponding to a ranking distance of 0.69). In a cross-species comparison, this trend also is evident as indicated by a higher similarity of the qRT-PCR based gene ranking of L. sativum seed germination (Figure 4A) and the Arabidopsis Germination Total subseries (Figure 5, ranking distance 0.37) in contrast with a lower similarity of this L. sativum ranking to the Arabidopsis Development Total subseries (Figure 4, ranking distance 0.54).
Figure 5.

Comparative Gene Expression Stability Analysis of Seed-Related Brassicaceae Transcriptome Data Sets for Reference Genes Used in the Brassicaceae Cross-Species qRT-PCR Seed Germination Study.
For the traditional and new L. sativum and Arabidopsis reference genes (Figure 4), Arabidopsis microarray data of 101 diverse seed-related experiments (Seed Total data set) was partitioned into the Germination Total data set (containing only experiments focusing on the seed germination process) and the Development Total data set (containing only experiments focusing on embryogenesis/seed development). The latter was reduced into the Development Endosperm data set (containing only experiments focusing on endosperm development) indicated by arrows above columns and detailed in Methods and Supplemental Data Set 2 online. B. napus microarray data for endosperm development of putative orthologs of the Arabidopsis reference genes was analyzed (see Methods) and genes named accordingly. Both endosperm data sets consist of highly comparable developmental stages (see Methods). Each data set was analyzed by GeNORM to rank the genes by their average expression stability measure M (value in brackets; proportional grayscale color intensity). Black arrows between columns indicate differences in gene expression stability rankings between the two seed processes, germination, and development for Arabidopsis. Gray arrows between columns indicate differences between the two species Arabidopsis and B. napus for the endosperm development process. The putative B. napus orthologs of Arabidopsis genes At4G04320 and At2G04660 are not present on the Brassica microarray and are therefore left blank.
Taken together, this analysis showed that the three best new reference genes from our qRT-PCR analysis of Arabidopsis seed germination (Figure 4B; At2G20000, At2G04660, and At1G17210) were among the four most stable genes in the subseries Germination Total. At2G20000 and At2G04660 were also the most stable genes of the Seed Total series, and in general four of the five new reference genes performed better or at least as good as the traditional reference genes (Figure 5). However, while the reference gene stability rankings were highly similar across species and between qRT-PCR and microarray data sets of the same species when looking at the same developmental process, the reference gene stability rankings differed considerably within one species when looking at different developmental processes.
To further address the issue of the two distinct seed processes in a cross-species manner, we used a microarray data set of Brassica napus (Brassicaceae) seed development (Huang et al., 2009). The seed development microarray data sets for Arabidopsis (available via the BAR server; Le et al., 2010) and B. napus (available via the Gene Expression Omnibus [GEO], GSE14766; Huang et al., 2009) are highly similar with respect to the investigated developmental stages (see Methods). The B. napus data set was analyzed, putative orthologous sequences to the Arabidopsis reference genes were identified (as described in Methods), and their expression values were ranked with GeNORM. The B. napus data set contains only endosperm tissue samples. For reasons of comparability, we therefore excluded all embryo-containing samples from the Arabidopsis Development Total subseries, resulting in the Development Endosperm subseries (Figure 5). The reference gene stability rankings for these highly comparable data sets (in terms of developmental stages covered) of the two species are very similar, as indicated by the gray arrows in Figure 5 between the Development Endosperm subseries of Arabidopsis and B. napus.
Due to missing genes on the B. napus array, GeNORM rankings were repeated for Arabidopsis data sets with reduced gene numbers to calculate ranking distances (detailed in Supplemental Data Set 3 online). Analysis of these rankings showed more similarity in gene expression stability between B. napus and Arabidopsis endosperm development (ranking distance 0.57) than between B. napus Development Endosperm and Arabidopsis Germination Total (ranking distance 0.83) and between B. napus Development Endosperm and Arabidopsis Development Total (ranking distance 0.76), which also contains embryo tissue.
Taken together, our cross-species comparisons for seed germination (L. sativum versus Arabidopsis) and seed development (Arabidopsis versus B. napus) strongly suggest that there are common stable reference genes for the same developmental processes and tissues for different Brassicaceae species. By contrast, the within-species differences in the reference gene stability rankings are severe between the two distinct processes. We considered specific seed tissues, hormonal treatments, and times during seed germination to provide superior reference genes for cross-species qRT-PCR analysis of L. sativum and Arabidopsis. We propose that these candidate genes can also be used as reference genes to study seed germination of other Brassicaceae species, such as B. napus. We provide evidence that the evolutionary conservation of reference gene performance is higher for a given developmental process between distinct species than for distinct developmental processes within a given single species. Our cross-species approach therefore supports the hypothesis proposed by Czechowski et al. (2005) that putative gene orthologs of new reference genes derived from one species can serve the same purpose in other species. We are convinced that this approach can also be applied to determine stable reference genes for other developmental stages, which also show constitutive expression in a cross-species manner, and that this approach is also valid for other plant families besides the Brassicaceae.
Superior Cross-Species Reference Genes Allow qRT-PCR Studies of Expression Regulation Even with Minor Changes in Transcript Abundance
Kwon et al. (2009) used microarray data sets to identify and qRT-PCR to validate superior new reference genes for detecting even small differences in transcript abundance across a wide range of human RNA samples. As suggested by Vandesompele et al. (2002b), they used the geometric mean of several validated reference genes for successful qRT-PCR normalization. For Arabidopsis seed germination, we obtained three superior new reference genes (At2G2000, At2G04660, and At1G17210; Figure 4B) for which the efficiency-corrected non-normalized transcript abundances can be used for target gene normalization. To test if the transcript expression of weakly regulated genes (i.e., exhibiting minor but detectable changes in transcript abundance) can be analyzed with our new reference genes for Brassicaceae seed germination, we used the traditional reference gene UBQ11 as target gene. UBQ11 exhibited low average expression stability as already shown in Figure 4, and it therefore may provide a weakly regulated target gene rather than a reference gene. We calculated the normalized relative transcript abundances of UBQ11 from its efficiency-corrected non-normalized transcript abundances using the following formula:
The expression analysis of UBQ11 showed a decrease in normalized transcript abundance during early seed germination, and ABA did not affect the relative transcript abundance during the late seed germination phase of Arabidopsis (Figure 6A) and L. sativum. A completely independent verification of this regulatory pattern was possible by in silico analysis of Arabidopsis seed germination microarray data sets, which are based on a different normalization strategy (Figure 6B). Our superior reference genes were therefore suitable to investigate the temporal and ABA-related regulation of UBQ11 transcript expression.
Figure 6.

Comparison of Transcript Expression Studies of UBQ11 during Arabidopsis Seed Germination.
(A) Normalized qRT-PCR results. Efficiency-corrected transcript abundance of UBQ11 was normalized by the geometric mean of the three best-performing reference genes for Arabidopsis seed germination (At1G17210, At2G04660, and At2G20000; as indicated in Figure 4). Mean values ± sd; n = 3.
(B) Microarray-based results. Affymetrix GeneChip ATH1 data normalized using GCOS and the MAS5.0 algorithm with a target value (TGT) of 100 for UBQ11 during the Arabidopsis germination time course was obtained from the BAR server (Toufighi et al., 2005; Winter et al., 2007). Note: Microarray expression data compiled from different experiments (see Supplemental Data Set 2 online) using BAR server eNorthern raw expression values. Mean values ± sd; n = 2 to 4.
Polyubiquitination and proteasome-mediated hormone signaling are important for plant development, including seed germination (e.g., Ferreira et al., 1995; Sun and Callis, 1997; Graeber et al., 2010; Linkies et al., 2010b). Sun and Callis (1997) studied the transcript regulation of different polyubiquitin genes in different organs and in response to environmental changes and found that UBQ11 is a weakly regulated polyubiquitin gene. In our work on L. sativum, we found UBQ11 enriched in a subtractive cDNA library of the endosperm cap, which confirms that it is also regulated during seed germination of this species (Linkies et al., 2010b). We further showed in Linkies et al. (2010b) that an Arabidopsis SALK line for UBQ11 has a seed germination phenotype. Our finding that UBQ11 is weakly regulated in seeds is in agreement with a role of this gene in Brassicaceae seed germination and confirms that this traditional reference gene is not suitable during seed germination. By contrast, our newly identified cross-species Brassicaceae reference genes are superior as they can be used for normalizing qRT-PCR data during seed germination of weakly and strongly regulated genes.
We have shown that comparative approaches can deliver cross-species reference genes for a specific developmental process, which can be applied in the emerging field of phylotranscriptomics. We propose that our new reference genes for seed germination are not only superior in L. sativum and Arabidopsis but may be family-wide new cross-species reference genes for qRT-PCR studies of Brassicaceae seeds, although this needs to be experimentally verified.
Future Perspectives of Transcript Quantification Techniques in Comparative Biology and the Importance of Cross-Species qRT-PCR
The emerging next-generation sequencing technologies will make phylotranscriptomics more amenable and a rapidly evolving research field. Massive parallel sequencing of whole transcriptomes, so called RNA-seq approaches, provide a tempting alternative to microarray analyses especially in comparative biology. This is due to the fact that for RNA-seq no a priori sequence information is needed that allows studying nonmodel species transcriptomes with an unprecedented depth (Bräutigam and Gowik, 2010). By aligning and counting the output of a RNA-seq experiment (i.e., millions of short sequence reads), one obtains a quantitative measure of gene expression of the whole transcriptome (Wang et al., 2010).
It is tempting to speculate that quantitative transcript analyses on a transcriptome-wide level with high sensitivity using RNA-seq will replace global analysis techniques, such as microarrays, and focused single-gene quantification methods like RNA gel blots or qRT-PCR (Roy et al., 2011). Even though next-generation sequencing will undoubtedly revolutionize biological research, especially in the field of comparative biology, it is currently still a new technique under heavy development and the user faces multiple challenges (Wang et al., 2010). Sequencing costs are still too high for most labs to use RNA-seq as a standard method that replaces microarrays for quantifying global gene expression. Global transcriptome analysis of RNA samples for an affordable core set of biological comparisons by either microarrays or RNA-seq is usually followed by the comparative expression analysis of a few genes in a larger set of RNA samples. If the quantification of transcript expression of only a few genes are the focus of interest (e.g., in a large set of RNA samples from different treatments or tissues), qRT-PCR is clearly the more cost- and time-effective method.
Another important issue to consider before using RNA-seq is the extensive bioinformatics needed to analyze the data (Wang et al., 2009). In contrast with microarrays and qRT-PCR (this work and references cited), there is still a lack of established user-friendly pipelines for the bioinformatics of RNA-seq data. Furthermore, besides the yet to be assessed bias in transcript quantification introduced by RNA-seq due to library preparation and amplification protocols, the sensitivity of RNA-seq is a heavily debated topic in the research community (Marioni et al., 2008; Fu et al., 2009; Roy et al., 2011). In a comparison of different technologies, Liu et al. (2011) showed that when investigating differentially expressed genes between species, RNA-seq tends to miss low abundant transcripts that are detectable with qRT-PCR. Complex transcriptomes of higher eukaryotes require extremely high sequencing coverage to detect low abundant transcripts (Marguerat and Bähler, 2010). The sensitivity of transcript quantification by RNA-seq can be enhanced by higher sequencing coverage, but this results in increased sequencing costs (Wang et al., 2009).
Global transcriptome analyses by microarrays or RNA-seq are often used for hypothesis generation. These hypotheses require independent validation by other means, for example, by using techniques such as qRT-PCR (Liu et al., 2011) or proteomics (Fu et al., 2009). Thus, even though the new sequencing technologies will yield fundamental new insights on a large scale in comparative biology, the need for alternative and complementary approaches will persist and includes normalization strategies. Due to its flexibility, speed, reliability, cost effectiveness, and ease of use based on established workflows, qRT-PCR is still the gold standard for transcript quantification. It is the technique of choice in comparative biology that complements phylotranscriptomics, conducted by either microarrays or RNA-seq, but must be based on the use of validated cross-species reference genes.
METHODS
Plant Material and Germination Assays
After-ripened Lepidium sativum ('Keimsprossen') seeds (Juliwa) were incubated in Petri dishes on two layers of filter paper with 6 mL 1/10 Murashige-Skoog salts in continuous white light (~100 μmol s−1 m−2) at 24°C as described (Müller et al., 2006; Linkies et al., 2009). After-ripened Arabidopsis thaliana ecotype Columbia seeds were incubated without cold stratification in continuous light also on filter paper for RNA extraction or on the same medium solidified with 1% (w/v) agar-agar at 24°C for germination kinetics. Where indicated, cis-S(+)-ABA (Duchefa) was added. Testa rupture and endosperm rupture were scored using a binocular microscope. Puncture force measurements were performed as described by Linkies et al. (2009).
RNA Extraction from Seed Tissues
For each sample, 200 micropylar endosperm caps (CAP) or 200 radicles plus lower hypocotyl (RAD) from imbibed or 100 CAP&RAD tissues (including testa) from dry after-ripened seeds of L. sativum or 100 μL whole after-ripened seeds of Arabidopsis ecotype Columbia were collected at the times indicated, frozen in liquid nitrogen, and stored at −80°C. Total RNA extraction was performed as described by Chang et al. (1993) with the following modifications. Sample extraction was done in 2-mL tubes; after addition of CTAB buffer, the tissue was kept at 65°C for 15 min. All chloroform:isoamylalcohol steps were repeated three times. After these RNA extraction steps, the remaining genomic DNA was removed by DNase-I (Qiagen) digestion in solution, which was followed by additional cleanup step using columns (Qiagen RNeasy kit). For RNA quality control, RNA integrity was checked by gel electrophoresis (see Supplemental Figure 1 online) followed by quantity and purity determination with the Nanodrop spectrophotometer ND-1000 (Peqlab). Only high-quality samples with OD ratios of at least 2 (260/280 nm) and 1.8 (260/230 nm) were used for further analysis (see Supplemental Figure 1 online). At least three biological replicate RNA samples were used for downstream applications.
Cloning of L. sativum cDNA Sequences
Total RNA extraction from dry seeds of L. sativum was performed as described by Chang et al. (1993) with the modifications described in the section above. RNA was reverse transcribed as described in the section below. Primers for subsequent cloning of transcript fragments from the new L. sativum reference genes were designed based on the putative orthologous Arabidopsis sequences. Partial cDNA sequences were obtained, submitted to a BLAST search to confirm correct fragment amplification, and have been submitted to GenBank. qRT-PCR primers were designed on these sequences. For traditional reference genes, primers based on Arabidopsis sequences were tested directly in qRT-PCR reactions with L. sativum cDNA as a template, their amplicons were purified, sequenced, and submitted to a BLAST search to ensure amplification of the correct reference gene. Accession numbers, highest scoring BLAST hits, and percentage of similarity to Arabidopsis for every L. sativum sequence are listed in Supplemental Table 1 online.
Quantitative Real-Time RT-PCR Reaction
Transcript expression of selected genes was quantified by qRT-PCR, which was performed according to the requirements described by Udvardi et al. (2008). A minimum of three, in most cases four, biological replicates of total RNA from every time point and treatment were reverse transcribed. For this, 5 μg RNA was used in a 20-μL reaction with 0.3 nmol random pentadecamers (custom made; Operon) per reaction [if not stated differently in the text; random hexamers (Qiagen); oligo(dT)16 (custom made; Operon)] according to the Superscript III kit instructions (Invitrogen). In brief, 5 μg RNA, 1 μL deoxynucleotide triphosphate (10 mM), 0.3 nmol pentadecamers in 13 μL RNase-free water were heated 5 min at 65°C, and chilled on ice for 1 min. Four microliters of 5× first-strand buffer, 1 μL 0.1 M DTT, 1 μL RNaseOUT (40 units/μL), and 1 μL SuperScript III (200 units/μL) were added. The thermal treatment was 5 min at 25°C, 60 min at 50°C, and 15 min at 70°C. The cDNA was then digested with 1 μL RNase H (Ambion) and diluted to obtain a final amount of 100 ng reverse-transcribed RNA per microliter. cDNA Aliquots of 1 μL were used for each quantitative PCR reaction. For quantification with the ABI PRISM 7300 sequence detection system (Applied Biosystems), the ABsolute QPCR SYBR Green ROX Mix (Thermo Fisher) was used according to the manufacturer’s instructions. Gene-specific primers (140 nM; listed in Supplemental Table 2 online) were used per qRT-PCR (if not stated differently in the text). The thermal treatment was 15 min at 95°C, followed by 40 cycles of 15 s at 95°C, 30 s at 60°C, and 30 s at 72°C. Single product amplification was validated by melting curve analysis.
Postrun qRT-PCR Data Analysis
The qRT-PCR efficiency (E) and CT value for individual reactions were determined by analysis of raw fluorescence data (without baseline correction) using the freely available software PCR Miner (Zhao and Fernald, 2005; www.miner.ewindup.info). For all samples, average efficiency per amplicon (i.e., per primer pair) was calculated and used to determine the efficiency-corrected transcript abundance for each sample as (1 +EAveragePerAmplicon) −CTIndividualSample. For quality controls, three no-template controls for each primer pair were included per qRT-PCR plate, and sample data resulting from these primer pairs were only taken when no no-template control amplification was evident. Four inter-run controls were included per qRT-PCR plate. If necessary, a calibration factor resulting from the inter-run control was calculated to account for inter-plate variations. No-RT controls were performed as suggested by Vandesompele et al. (2002a) to control for absence of genomic DNA contamination. Determination of gene expression stability was performed using the GeNORM tool as described by Vandesompele et al. (2002b). Efficiency-corrected transcript abundance values of at least three biological replicates for all samples were used for GeNORM analysis.
Ranking Distance Analysis
To quantify the difference between expression stability rankings, we used a simple Euclidean metric to calculate a ranking distance value. Suppose we wish to quantify the difference between two rankings R1,R2 of the same set of n items. We can represent the two rankings by 1, 2, …, n and p1, p2, …, pn, where p1, p2, …, pn is a permutation of 1,2, …, n. We then define the ranking distance between the two rankings to be
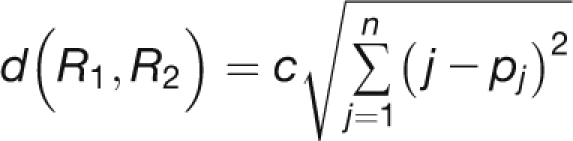 |
where 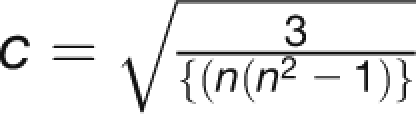 is chosen so that the maximum possible distance between R1 and R2 is 1. If ties are present, we specify each tied rank to be the average rank of the tied items. It can be shown that the ranking distance does not depend on which ranking we label 1, 2, …, n and which ranking we label p1,…, pn.
is chosen so that the maximum possible distance between R1 and R2 is 1. If ties are present, we specify each tied rank to be the average rank of the tied items. It can be shown that the ranking distance does not depend on which ranking we label 1, 2, …, n and which ranking we label p1,…, pn.
Microarray Data Analysis and Candidate Reference Gene Mining
L. sativum
Heterologous L. sativum microarray data sets from radicles plus lower hypocotyl (RAD), micropylar endosperm (CAP), and nonmicropylar endosperm (NME) tissues were taken from Linkies et al. (2009). Data were validated as described by Linkies et al. (2009) and can be found in ArrayExpress (www.ebi.ac.uk/microarray/) under accession number E-TABM-745 (CON arrays) and E-TABM-743 (ABA arrays). The individual lists contained 22,025 (CON) and 19,794 (ABA) genes. These gene lists were used for the identification of candidate reference genes. Expression stability was determined by comparing the log2-transformed transcript expression difference values (coefficients) between tissues and times. To select candidate reference genes from the 22,025 transcripts present in the CON arrays, all genes were determined that had stable (putatively constitutive) and high expression across times and tissues (RAD, CAP, and NME): Only genes for which transcript expression coefficients had adjusted P values between 0.8 and 1.0 in the F-tests across all samples and in all the t tests between two samples (see Supplemental Data Set 3 in Linkies et al., 2009) were selected. Expression strength was determined by comparing the normalized log2-transformed mean per-spot intensity values, which ranged from 6.4 to 16.0 (see Supplemental Data Set 1 in Linkies et al., 2009). Only genes with a minimum per-spot intensity of 7.5 were selected, and an overlap of 1604 genes was determined (Figure 2A). The same procedure was applied to the 19,704 transcripts present in the ABA arrays (RAD and CAP at four time points; see Supplemental Data Sets 2 and 4 in Linkies et al., 2009), and an overlap of 266 genes was determined (Figure 2A). Both overlaps were compared and 15 transcripts were present in both selections (Figure 2A) that did not differ by more than 0.7 in their transcript expression coefficients. These 15 reference gene candidates are listed in Supplemental Data Set 1 online.
Arabidopsis
Using the eNorthern with Expression Browser tool from the Bio-Array Resource database (http://bar.utoronto.ca/; Toufighi et al., 2005), Affymetrix GeneChip ATH1 expression data of all Arabidopsis genes used in our qRT-PCR analysis (Figure 4B) except 5.8S rRNA (which is not present on the ATH1 chip) was obtained. The Average of Replicate Treatments option was used to obtain expression data of averaged biological replicates for all microarray experiments in the AtGenExpress seed series, which resulted in data from 101 different microarray experiments compiled from different sources on different developmental stages, treatments, and mutants of Arabidopsis seeds. A list of all analyzed microarray experiments can be found in Supplemental Data Set 2 online. The complete microarray data set (Seed Total, 101 experiments) was partitioned in different subseries: Germination Total (67 experiments focusing on seed germination, based on different sources), Development Total (34 experiments focusing on seed development, based on the Goldberg-Harada data set; http://seedgenenetwork.net/; Le et al., 2010; Zuber et al., 2010) (also available via GEO, http://www.ncbi.nlm.nih.gov/geo, accession number GSE12404), and Development Endosperm (24 experiments, a subseries of the Development Total data set focusing only on endosperm-derived data). Based on embryo development, the Arabidopsis endosperm data set contains preglobular, globular, heart, linear cotyledon, and mature green stages. Experiments included in different subseries are detailed in Supplemental Data Set 2 online. We used these microarray expression data sets to perform individual gene expression stability analysis via GeNORM.
Brassica napus
Data from a B. napus microarray consisting of 10,642 amplicons of unisequences of seed-specific cDNA sequences of Brassica spp (Xiang et al., 2008) deposited as platform in GEO (accession number GPL8090) was used. Huang et al. (2009) hybridized RNA from B. napus endosperm at different developmental stages during seed development to this microarray, which resulted in 24 data sets (available via GEO; accession number GSE14766). Based on embryo development, the B. napus endosperm data set contains globular, heart, and cotyledon stages. These data sets were analyzed using Genedata Expressionist Analyst 2.1 software. The normalized expression data of the putative Arabidopsis reference gene orthologs were analyzed via GeNORM to determine the average expression stability measure, M. The selection procedure to identify B. napus microarray gene probes that most likely bind putative B. napus orthologs of Arabidopsis reference genes is described in detail in the Supplemental Methods online.
Accession Numbers
The L. sativum cDNAs isolated and described here have been deposited in GenBank/EMBL data libraries under the following accession numbers: LesaG17210 (HQ912755), LesaG04660 (HQ912754), LesaG19980 (HQ912756), LesaG20000 (HQ912757), LesaG04320 (HQ912753), LesaACT7 (HS981849), LesaACT8 (HS981850), LesaCYP1 (HS981851), LesaCYP5 (HS981852), LesaEF1-α (HS981853), LesaUBQ11 (HS981854), and Lesa5.8SrRNA (X78126.1). Arabidopsis Genome Initiative codes of Arabidopsis genes are as follows: CYP1 (At4G38740), ACT7 (At5G09810), ACT8 (At1G49240), EF1-α (At5G60390), CYP5 (At2G29960), UBQ11 (At4G05050), ILP1 (At1G17210), APC2 (At2G04660), allergen V5/Tpx-1-related family protein (At2G19980), HBT (At2G20000), and malonyl-CoA decarboxylase family protein (At4G04320). B. napus Genbank accession numbers are as follows: BrCYP1 (EE541625), BrG17210 (EE462267), BrUBQ11 (CN735823), BrEF1-α (EE439665), BrG20000 (CX270495), BrCYP5 (CN736026), BrG19980 (CN727945), BrACT7 (CN733524), and BrACT8 (CN727783). Microarray data used in this study can be found in ArrayExpress (www.ebi.ac.uk/microarray/) under accession numbers E-TABM-745 (CON arrays) and E-TABM-743 (ABA arrays).
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. Determination of RNA Integrity via Gel Electrophoresis.
Supplemental Table 1. Sequence Similarities to Arabidopsis and Accession Numbers of L. sativum Reference Gene cDNA Sequences.
Supplemental Table 2. Primer Sequences Used for qRT-PCR.
Supplemental Methods. Detailed Selection Procedure for Identification of Putative Brassica napus Orthologs of Arabidopsis Reference Genes on a Brassica Microarray for Comparative Gene Expression Stability Analysis as Shown in Figure 5.
Supplemental Data Set 1. Transcript Expression Values of the 15 Seed Germination Reference Gene Candidates Mined from the L. sativum Microarrays.
Supplemental Data Set 2. Arabidopsis Seed-Related Microarray Experiments Used for Expression Analysis and GeNORM Expression Stability Rankings of Reference Genes.
Supplemental Data Set 3. Expression Stability Ranking Comparisons and Ranking Distance Measures of Lepidium sativum, Arabidopsis, and Brassica napus Genes.
Supplementary Material
Acknowledgments
Our work is funded by grants from the Deutsche Forschungsgemeinschaft (Grants DFG LE720/6 and LE720/7) to G.L.-M. and from the ERA-NET Plant Genomics vSEED Project (BBG02488X1) to A.T.A.W. For more information: The Seed Biology Place, www.seedbiology.de.
References
- Andersen M.R., Vongsangnak W., Panagiotou G., Salazar M.P., Lehmann L., Nielsen J. (2008). A trispecies Aspergillus microarray: Comparative transcriptomics of three Aspergillus species. Proc. Natl. Acad. Sci. USA 105: 4387–4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arikawa E., Yang J. (2007). Comparing algorithms for calculating amplification efficiencies of real-time PCR. In Cambridge Healthtech Institute's Third Annual Quantitative PCR, Microarrays, and Biological Validation: Capturing the Complete Biological Story, October 15-October 17, 2007, Providence, RI (Frederick, MD: SuperArray Bioscience Corporation; ). [Google Scholar]
- Barrero J.M., Talbot M.J., White R.G., Jacobsen J.V., Gubler F. (2009). Anatomical and transcriptomic studies of the coleorhiza reveal the importance of this tissue in regulating dormancy in barley. Plant Physiol. 150: 1006–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassel G.W., Fung P., Chow T.-F., Foong J.A., Provart N.J., Cutler S.R. (2008). Elucidating the germination transcriptional program using small molecules. Plant Physiol. 147: 143–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann S., Ihmels J., Barkai N. (2004). Similarities and differences in genome-wide expression data of six organisms. PLoS Biol. 2: E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewley J.D. (1997). Breaking down the walls - A role for endo-β-mannanase in release from seed dormancy? Trends Plant Sci. 2: 464–469 [Google Scholar]
- Birtić S., Kranner I. (2006). Isolation of high-quality RNA from polyphenol-, polysaccharide- and lipid-rich seeds. Phytochem. Anal. 17: 144–148 [DOI] [PubMed] [Google Scholar]
- Bräutigam A., Gowik U. (2010). What can next generation sequencing do for you? Next generation sequencing as a valuable tool in plant research. Plant Biol. 12: 831–841 [DOI] [PubMed] [Google Scholar]
- Bustin S. (2004). A-Z of Quantitative PCR. (La Jolla, CA: International University Line; ). [Google Scholar]
- Bustin S.A., Beaulieu J.-F., Huggett J., Jaggi R., Kibenge F.S., Olsvik P.A., Penning L.C., Toegel S. (2010). MIQE précis: Practical implementation of minimum standard guidelines for fluorescence-based quantitative real-time PCR experiments. BMC Plant Biol. 11: 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin S.A., Benes V., Garson J.A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M.W., Shipley G.L., Vandesompele J., Wittwer C.T. (2009). The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55: 611–622 [DOI] [PubMed] [Google Scholar]
- Bustin S.A., Nolan T. (2004). Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J. Biomol. Tech. 15: 155–166 [PMC free article] [PubMed] [Google Scholar]
- Cadman C.S.C., Toorop P.E., Hilhorst H.W.M., Finch-Savage W.E. (2006). Gene expression profiles of Arabidopsis Cvi seeds during dormancy cycling indicate a common underlying dormancy control mechanism. Plant J. 46: 805–822 [DOI] [PubMed] [Google Scholar]
- Chang S., Puryear J., Cairney J. (1993). A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 11: 113–116 [Google Scholar]
- Chen X., Truksa M., Shah S., Weselake R.J. (2010). A survey of quantitative real-time polymerase chain reaction internal reference genes for expression studies in Brassica napus. Anal. Biochem. 405: 138–140 [DOI] [PubMed] [Google Scholar]
- Čikoš Š., Bukovská A., Koppel J. (2007). Relative quantification of mRNA: Comparison of methods currently used for real-time PCR data analysis. BMC Mol. Biol. 8: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comai L., Dietrich R.A., Maslyar D.J., Baden C.S., Harada J.J. (1989). Coordinate expression of transcriptionally regulated isocitrate lyase and malate synthase genes in Brassica napus L. Plant Cell 1: 293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czechowski T., Bari R.P., Stitt M., Scheible W.-R., Udvardi M.K. (2004). Real-time RT-PCR profiling of over 1400 Arabidopsis transcription factors: Unprecedented sensitivity reveals novel root- and shoot-specific genes. Plant J. 38: 366–379 [DOI] [PubMed] [Google Scholar]
- Czechowski T., Stitt M., Altmann T., Udvardi M.K., Scheible W.-R. (2005). Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 139: 5–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derveaux S., Vandesompele J., Hellemans J. (2010). How to do successful gene expression analysis using real-time PCR. Methods 50: 227–230 [DOI] [PubMed] [Google Scholar]
- Dure L., Waters L. (1965). Long-lived messenger RNA: Evidence from cotton seed germination. Science 147: 410–412 [DOI] [PubMed] [Google Scholar]
- Ferreira R.M.B., Ramos P.C.R., Franco E., Ricardo C.P.P., Teixeira A.R.N. (1995). Changes in ubiquitin and ubiquitin-protein conjugates during seed formation and germination. J. Exp. Bot. 46: 211–219 [Google Scholar]
- Fierro A.C., Vandenbussche F., Engelen K., Van de Peer Y., Marchal K. (2008). Meta analysis of gene expression data within and across species. Curr. Genomics 9: 525–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch-Savage W.E., Leubner-Metzger G. (2006). Seed dormancy and the control of germination. New Phytol. 171: 501–523 [DOI] [PubMed] [Google Scholar]
- Fleige S., Pfaffl M.W. (2006). RNA integrity and the effect on the real-time qRT-PCR performance. Mol. Aspects Med. 27: 126–139 [DOI] [PubMed] [Google Scholar]
- Fu X., Fu N., Guo S., Yan Z., Xu Y., Hu H., Menzel C., Chen W., Li Y., Zeng R., Khaitovich P. (2009). Estimating accuracy of RNA-Seq and microarrays with proteomics. BMC Genomics 10: 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasic K., Hernandez A., Korban S. (2004). RNA extraction from different apple tissues rich in polyphenols and polysaccharides for cDNA library construction. Plant Mol. Biol. Rep. 22: 437–438 [Google Scholar]
- Graeber K., Linkies A., Müller K., Wunchova A., Rott A., Leubner-Metzger G. (2010). Cross-species approaches to seed dormancy and germination: conservation and biodiversity of ABA-regulated mechanisms and the Brassicaceae DOG1 genes. Plant Mol. Biol. 73: 67–87 [DOI] [PubMed] [Google Scholar]
- Grennan A.K. (2007). An analysis of the Arabidopsis pollen transcriptome. Plant Physiol. 145: 3–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guénin S., Mauriat M., Pelloux J., Van Wuytswinkel O., Bellini C., Gutierrez L. (2009). Normalization of qRT-PCR data: The necessity of adopting a systematic, experimental conditions-specific, validation of references. J. Exp. Bot. 60: 487–493 [DOI] [PubMed] [Google Scholar]
- Guescini M., Sisti D., Rocchi M.B., Stocchi L., Stocchi V. (2008). A new real-time PCR method to overcome significant quantitative inaccuracy due to slight amplification inhibition. BMC Bioinformatics 9: 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez L., Mauriat M., Guénin S., Pelloux J., Lefebvre J.-F., Louvet R., Rusterucci C., Moritz T., Guerineau F., Bellini C., Van Wuytswinkel O. (2008b). The lack of a systematic validation of reference genes: A serious pitfall undervalued in reverse transcription-polymerase chain reaction (RT-PCR) analysis in plants. Plant Biotechnol. J. 6: 609–618 [DOI] [PubMed] [Google Scholar]
- Gutierrez L., Mauriat M., Pelloux J., Bellini C., Van Wuytswinkel O. (2008a). Towards a systematic validation of references in real-time RT-PCR. Plant Cell 20: 1734–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony T., Yanai I. (2010). Revealing developmental networks by comparative transcriptomics. Transcr. 1: 154–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdsworth M.J., Bentsink L., Soppe W.J.J. (2008). Molecular networks regulating Arabidopsis seed maturation, after-ripening, dormancy and germination. New Phytol. 179: 33–54 [DOI] [PubMed] [Google Scholar]
- Huang Y., Chen L., Wang L., Vijayan K., Phan S., Liu Z., Wan L., Ross A., Xiang D., Datla R., Pan Y., Zou J. (2009). Probing the endosperm gene expression landscape in Brassica napus. BMC Genomics 10: 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggett J., Dheda K., Bustin S., Zumla A. (2005). Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 6: 279–284 [DOI] [PubMed] [Google Scholar]
- Ishibashi N., Yamauchi D., Minamikawa T. (1990). Stored mRNA in cotyledons of Vigna unguiculata seeds: Nucleotide sequence of cloned cDNA for a stored mRNA and induction of its synthesis by precocious germination. Plant Mol. Biol. 15: 59–64 [DOI] [PubMed] [Google Scholar]
- Karlen Y., McNair A., Perseguers S., Mazza C., Mermod N. (2007). Statistical significance of quantitative PCR. BMC Bioinformatics 8: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M., Nambara E. (2010). Stored and neosynthesized mRNA in Arabidopsis seeds: Effects of cycloheximide and controlled deterioration treatment on the resumption of transcription during imbibition. Plant Mol. Biol. 73: 119–129 [DOI] [PubMed] [Google Scholar]
- Kohonen P., Nera K.P., Lassila O. (2007). Avian model for B-cell immunology—New genomes and phylotranscriptomics. Scand. J. Immunol. 66: 113–121 [DOI] [PubMed] [Google Scholar]
- Kontanis E.J., Reed F.A. (2006). Evaluation of real-time PCR amplification efficiencies to detect PCR inhibitors. J. Forensic Sci. 51: 795–804 [DOI] [PubMed] [Google Scholar]
- Kucera B., Cohn M.A., Leubner-Metzger G. (2005). Plant hormone interactions during seed dormancy release and germination. Seed Sci. Res. 15: 281–307 [Google Scholar]
- Kwon M.J., Oh E., Lee S., Roh M.R., Kim S.E., Lee Y., Choi Y.-L., In Y.-H., Park T., Koh S.S., Shin Y.K. (2009). Identification of novel reference genes using multiplatform expression data and their validation for quantitative gene expression analysis. PLoS ONE 4: e6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le B.H., et al. (2010). Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc. Natl. Acad. Sci. USA 107: 8063–8070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P.D., Sladek R., Greenwood C.M.T., Hudson T.J. (2002). Control genes and variability: Absence of ubiquitous reference transcripts in diverse mammalian expression studies. Genome Res. 12: 292–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leubner-Metzger G. (2003). Functions and regulation of β-1,3-glucanases during seed germination, dormancy release and after-ripening. Seed Sci. Res. 13: 17–34 [Google Scholar]
- Li F., Wu X., Tsang E., Cutler A.J. (2005). Transcriptional profiling of imbibed Brassica napus seed. Genomics 86: 718–730 [DOI] [PubMed] [Google Scholar]
- Linkies A., Graeber K., Knight C., Leubner-Metzger G. (2010a). The evolution of seeds. New Phytol. 186: 817–831 [DOI] [PubMed] [Google Scholar]
- Linkies A., Müller K., Morris K., Turecková V., Wenk M., Cadman C.S.C., Corbineau F., Strnad M., Lynn J.R., Finch-Savage W.E., Leubner-Metzger G. (2009). Ethylene interacts with abscisic acid to regulate endosperm rupture during germination: A comparative approach using Lepidium sativum and Arabidopsis thaliana. Plant Cell 21: 3803–3822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkies A., Schuster-Sherpa U., Tintelnot S., Leubner-Metzger G., Müller K. (2010b). Peroxidases identified in a subtractive cDNA library approach show tissue-specific transcript abundance and enzyme activity during seed germination of Lepidium sativum. J. Exp. Bot. 61: 491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.-P., Koizuka N., Homrichhausen T.M., Hewitt J.R., Martin R.C., Nonogaki H. (2005). Large-scale screening of Arabidopsis enhancer-trap lines for seed germination-associated genes. Plant J. 41: 936–944 [DOI] [PubMed] [Google Scholar]
- Liu S., Lin L., Jiang P., Wang D., Xing Y. (2011). A comparison of RNA-Seq and high-density exon array for detecting differential gene expression between closely related species. Nucleic Acids Res. 39: 578–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan J., Edwards K., Saunders N. (2009). Real-Time PCR: Current Technology and Applications. (London: Caister Academic Press; ). [Google Scholar]
- Marguerat S., Bähler J. (2010). RNA-seq: From technology to biology. Cell. Mol. Life Sci. 67: 569–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni J.C., Mason C.E., Mane S.M., Stephens M., Gilad Y. (2008). RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 18: 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C. (2008). Refining our standards. Plant Cell 20: 1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijerink J., Mandigers C., van de Locht L., Tönnissen E., Goodsaid F., Raemaekers J. (2001). A novel method to compensate for different amplification efficiencies between patient DNA samples in quantitative real-time PCR. J. Mol. Diagn. 3: 55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller K., Tintelnot S., Leubner-Metzger G. (2006). Endosperm-limited Brassicaceae seed germination: Abscisic acid inhibits embryo-induced endosperm weakening of Lepidium sativum (cress) and endosperm rupture of cress and Arabidopsis thaliana. Plant Cell Physiol. 47: 864–877 [DOI] [PubMed] [Google Scholar]
- Nakabayashi K., Okamoto M., Koshiba T., Kamiya Y., Nambara E. (2005). Genome-wide profiling of stored mRNA in Arabidopsis thaliana seed germination: Epigenetic and genetic regulation of transcription in seed. Plant J. 41: 697–709 [DOI] [PubMed] [Google Scholar]
- Ni B.R., Bradford K.J. (1993). Germination and dormancy of abscisic acid- and gibberellin-deficient mutant tomato (Lycopersicon esculentum) seeds (sensitivity of germination to abscisic acid, gibberellin, and water potential). Plant Physiol. 101: 607–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicot N., Hausman J.F., Hoffmann L., Evers D. (2005). Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. J. Exp. Bot. 56: 2907–2914 [DOI] [PubMed] [Google Scholar]
- Nolan T., Hands R.E., Bustin S.A. (2006). Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 1: 1559–1582 [DOI] [PubMed] [Google Scholar]
- Okamoto M., et al. (2010). Genome-wide analysis of endogenous abscisic acid-mediated transcription in dry and imbibed seeds of Arabidopsis using tiling arrays. Plant J. 62: 39–51 [DOI] [PubMed] [Google Scholar]
- Okello J.B., Rodriguez L., Poinar D., Bos K., Okwi A.L., Bimenya G.S., Sewankambo N.K., Henry K.R., Kuch M., Poinar H.N. (2010). Quantitative assessment of the sensitivity of various commercial reverse transcriptases based on armored HIV RNA. PLoS ONE 5: e13931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolacci A.R., Tanzarella O.A., Porceddu E., Ciaffi M. (2009). Identification and validation of reference genes for quantitative RT-PCR normalization in wheat. BMC Mol. Biol. 10: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh A., et al. (2010). Conserved developmental transcriptomes in evolutionarily divergent species. Genome Biol. 11: R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Novo C.A., Claeys C., Speleman F., Van Cauwenberge P., Bachert C., Vandesompele J. (2005). Impact of RNA quality on reference gene expression stability. Biotechniques 39: 52–56, 54, 56 [DOI] [PubMed] [Google Scholar]
- Pfaffl M.W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl M.W. (2004). Quantification Strategies in Real-Time PCR. (La Jolla, CA: International University Line; ). [Google Scholar]
- Porebski S., Bailey L., Baum B. (1997). Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 15: 8–15 [Google Scholar]
- Preston J., Tatematsu K., Kanno Y., Hobo T., Kimura M., Jikumaru Y., Yano R., Kamiya Y., Nambara E. (2009). Temporal expression patterns of hormone metabolism genes during imbibition of Arabidopsis thaliana seeds: A comparative study on dormant and non-dormant accessions. Plant Cell Physiol. 50: 1786–1800 [DOI] [PubMed] [Google Scholar]
- Radonić A., Thulke S., Mackay I.M., Landt O., Siegert W., Nitsche A. (2004). Guideline to reference gene selection for quantitative real-time PCR. Biochem. Biophys. Res. Commun. 313: 856–862 [DOI] [PubMed] [Google Scholar]
- Ramakers C., Ruijter J.M., Deprez R.H., Moorman A.F. (2003). Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 339: 62–66 [DOI] [PubMed] [Google Scholar]
- Remans T., Smeets K., Opdenakker K., Mathijsen D., Vangronsveld J., Cuypers A. (2008). Normalisation of real-time RT-PCR gene expression measurements in Arabidopsis thaliana exposed to increased metal concentrations. Planta 227: 1343–1349 [DOI] [PubMed] [Google Scholar]
- Rensink W.A., Lee Y., Liu J., Iobst S., Ouyang S., Buell C.R. (2005). Comparative analyses of six solanaceous transcriptomes reveal a high degree of sequence conservation and species-specific transcripts. BMC Genomics 6: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieu I., Powers S.J. (2009). Real-time quantitative RT-PCR: Design, calculations, and statistics. Plant Cell 21: 1031–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross D.M., Watkins D.B., Hughes T.P., Branford S. (2008). Reverse transcription with random pentadecamer primers improves the detection limit of a quantitative PCR assay for BCR-ABL transcripts in chronic myeloid leukemia: Implications for defining sensitivity in minimal residual disease. Clin. Chem. 54: 1568–1571 [DOI] [PubMed] [Google Scholar]
- Roy N.C., Altermann E., Park Z.A., McNabb W.C. (2011). A comparison of analog and Next-Generation transcriptomic tools for mammalian studies. Brief Funct. Genomics 10: 135–150 [DOI] [PubMed] [Google Scholar]
- Ruijter J.M., Ramakers C., Hoogaars W.M., Karlen Y., Bakker O., van den Hoff M.J., Moorman A.F. (2009). Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 37: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge R.G., Côté C. (2003). Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Res. 31: e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge R.G., Stewart D. (2008). Critical evaluation of methods used to determine amplification efficiency refutes the exponential character of real-time PCR. BMC Mol. Biol. 9: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schranz M.E., Song B.H., Windsor A.J., Mitchell-Olds T. (2007). Comparative genomics in the Brassicaceae: A family-wide perspective. Curr. Opin. Plant Biol. 10: 168–175 [DOI] [PubMed] [Google Scholar]
- Schreiber A.W., Sutton T., Caldo R.A., Kalashyan E., Lovell B., Mayo G., Muehlbauer G.J., Druka A., Waugh R., Wise R.P., Langridge P., Baumann U. (2009). Comparative transcriptomics in the Triticeae. BMC Genomics 10: 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreenivasulu N., Usadel B., Winter A., Radchuk V., Scholz U., Stein N., Weschke W., Strickert M., Close T.J., Stitt M., Graner A., Wobus U. (2008). Barley grain maturation and germination: metabolic pathway and regulatory network commonalities and differences highlighted by new MapMan/PageMan profiling tools. Plant Physiol. 146: 1738–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ståhlberg A., Håkansson J., Xian X., Semb H., Kubista M. (2004). Properties of the reverse transcription reaction in mRNA quantification. Clin. Chem. 50: 509–515 [DOI] [PubMed] [Google Scholar]
- Stangegaard M., Dufva I.H., Dufva M. (2006). Reverse transcription using random pentadecamer primers increases yield and quality of resulting cDNA. Biotechniques 40: 649–657 [DOI] [PubMed] [Google Scholar]
- Sun C.-W., Callis J. (1997). Independent modulation of Arabidopsis thaliana polyubiquitin mRNAs in different organs and in response to environmental changes. Plant J. 11: 1017–1027 [DOI] [PubMed] [Google Scholar]
- Suzuki T., Higgins P.J., Crawford D.R. (2000). Control selection for RNA quantitation. Biotechniques 29: 332–337 [DOI] [PubMed] [Google Scholar]
- Thellin O., Zorzi W., Lakaye B., De Borman B., Coumans B., Hennen G., Grisar T., Igout A., Heinen E. (1999). Housekeeping genes as internal standards: use and limits. J. Biotechnol. 75: 291–295 [DOI] [PubMed] [Google Scholar]
- Tichopad A., Dilger M., Schwarz G., Pfaffl M.W. (2003). Standardized determination of real-time PCR efficiency from a single reaction set-up. Nucleic Acids Res. 31: e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I., Bilu Y., Barkai N. (2007). Comparative biology: Beyond sequence analysis. Curr. Opin. Biotechnol. 18: 371–377 [DOI] [PubMed] [Google Scholar]
- Toorop P.E., van Aelst A.C., Hilhorst H.W.M. (2000). The second step of the biphasic endosperm cap weakening that mediates tomato (Lycopersicon esculentum) seed germination is under control of ABA. J. Exp. Bot. 51: 1371–1379 [PubMed] [Google Scholar]
- Toufighi K., Brady S.M., Austin R., Ly E., Provart N.J. (2005). The botany array resource: e-Northerns, expression angling, and promoter analyses. Plant J. 43: 153–163 [DOI] [PubMed] [Google Scholar]
- Udvardi M.K., Czechowski T., Scheible W.-R. (2008). Eleven golden rules of quantitative RT-PCR. Plant Cell 20: 1736–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbroucke K., Robbens S., Vandepoele K., Inzé D., Van de Peer Y., Van Breusegem F. (2008). Hydrogen peroxide-induced gene expression across kingdoms: A comparative analysis. Mol. Biol. Evol. 25: 507–516 [DOI] [PubMed] [Google Scholar]
- Vandepoele K., Van de Peer Y. (2005). Exploring the plant transcriptome through phylogenetic profiling. Plant Physiol. 137: 31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J., De Paepe A., Speleman F. (2002a). Elimination of primer-dimer artifacts and genomic coamplification using a two-step SYBR green I real-time RT-PCR. Anal. Biochem. 303: 95–98 [DOI] [PubMed] [Google Scholar]
- Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. (2002b). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3: RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov R.A., Panchuk I.I., Schöffl F. (2003). Heat-stress-dependency and developmental modulation of gene expression: The potential of house-keeping genes as internal standards in mRNA expression profiling using real-time RT-PCR. J. Exp. Bot. 54: 2343–2349 [DOI] [PubMed] [Google Scholar]
- Wang L., Li P., Brutnell T.P. (2010). Exploring plant transcriptomes using ultra high-throughput sequencing. Brief Funct. Genomics 9: 118–128 [DOI] [PubMed] [Google Scholar]
- Wang Y., Zhang W.Z., Song L.F., Zou J.J., Su Z., Wu W.H. (2008). Transcriptome analyses show changes in gene expression to accompany pollen germination and tube growth in Arabidopsis. Plant Physiol. 148: 1201–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Gerstein M., Snyder M. (2009). RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 10: 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L.Q., Xu W.Y., Deng Z.Y., Su Z., Xue Y., Wang T. (2010). Genome-scale analysis and comparison of gene expression profiles in developing and germinated pollen in Oryza sativa. BMC Genomics 11: 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter D., Vinegar B., Nahal H., Ammar R., Wilson G.V., Provart N.J. (2007). An “Electronic Fluorescent Pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS ONE 2: e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M.L., Medrano J.F. (2005). Real-time PCR for mRNA quantitation. Biotechniques 39: 75–85 [DOI] [PubMed] [Google Scholar]
- Xiang D., et al. (2008). Development of a Brassica seed cDNA microarray. Genome 51: 236–242 [DOI] [PubMed] [Google Scholar]
- Zeng Y., Yang T. (2002). RNA isolation from highly viscous samples rich in polyphenols and polysaccharides. Plant Mol. Biol. Rep. 20: 417a–417e [Google Scholar]
- Zhao S., Fernald R.D. (2005). Comprehensive algorithm for quantitative real-time polymerase chain reaction. J. Comput. Biol. 12: 1047–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber H., Davidian J.-C., Aubert G., Aimé D., Belghazi M., Lugan R., Heintz D., Wirtz M., Hell R., Thompson R., Gallardo K. (2010). The seed composition of Arabidopsis mutants for the group 3 sulfate transporters indicates a role in sulfate translocation within developing seeds. Plant Physiol. 154: 913–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.