

Abstract
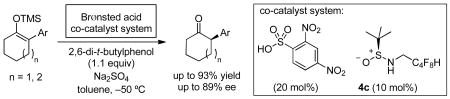
The application of chiral sulfinamides and achiral sulfonic acids as a co-catalyst system for enantioselective protonation reactions is described. Structurally simple, easily accessible sulfinamides were found to induce moderate-to-high ee's in the formation of 2-aryl-substituted cycloalkanones from the corresponding trimethylsilyl enol ethers.
Weak-to-moderately strong chiral Brønsted acids, ranging from diols to phosphoric acids, have been applied in a variety of catalytic enantioselective transformations. Particular success has been achieved in catalysis of addition reactions to relatively basic electrophiles such as imines.1 More recently, some effort has been directed towards accessing and utilizing stronger Brønsted acids, enabling expansion of the scope to the activation of carbonyl groups and certain olefins.2
We became interested in exploring the potential of the conjugate acids of chiral sulfinamides as a novel class of strong, chiral Brønsted acid catalysts. While sulfinamides find extensive use as chiral auxiliaries and ligands in asymmetric synthesis,3 applications of these privileged chiral structures as organocatalysts are less common.4,5 Our design was inspired by recent studies with sulfinamide–urea catalyst 1a, which revealed that the highly enantioselective addition of electron-rich alkenes to protioiminium ions can be achieved through a network of non-covalent interactions between the electrophile and the chiral urea-bound counteranion.5 In particular, spectroscopic and computational evidence was obtained for a hydrogen-bond interaction between the sulfinamide group of the catalyst and the N–H proton of the iminiumion intermediate (Figure 1A). We hypothesized that in the absence of the Lewis-basic imine, the combination of a sulfonic acid and sulfinamide urea catalyst 1a could produce a chiral acidic species capable of effecting enantioselective protonation reactions (Figure 1B). The proximity of the stereogenic sulfur to the proton would potentially enable high levels of stereochemical communication. Here we describe the development of this new approach for catalysis and its application to the enantioselective catalytic protonation of prochiral enol silanes as a method for the preparation of chiral, α-branched ketones.6,7
Figure 1.

(A) Schematic representation of the geometry and energy-minimized lowest energy transition structure for sulfinamide–urea/TfOH co-catalyzed Povarov reaction (from ref 5). (B) Schematic representation of the energy-minimized lowest energy ground state structure for the sulfinamide–urea 1a/TfOH ‘chiral acid’ complex. Structures calculated at the B3LYP/6-31G(d) level of density functional theory. Ar = 3,5-bis(CF3)C6H3.
Silyl enol ether 5a, derived from 2-phenylcyclohexanone, was selected as the model substrate (Scheme 1). A suitable achiral stoichiometric proton source was sought that would effect protonation of the sulfinamide catalyst scaffold without promoting a background racemic protonation pathway. It was found that 2,4-dinitrobenzene sulfonic acid (2,4-diNBSA) was well suited, as it is completely insoluble in toluene at −40 °C and, consequently, unreactive toward 5a under these conditions. However, in the presence of catalytic levels of sulfinamide–urea 1a, substrate protonation occurred to generate the corresponding ketone 6a in 67% ee. No reactivity toward 5a was displayed by 1a alone under these conditions. Ketone 6a was found to be configurationally stable under the catalytic conditions.
Scheme 1.
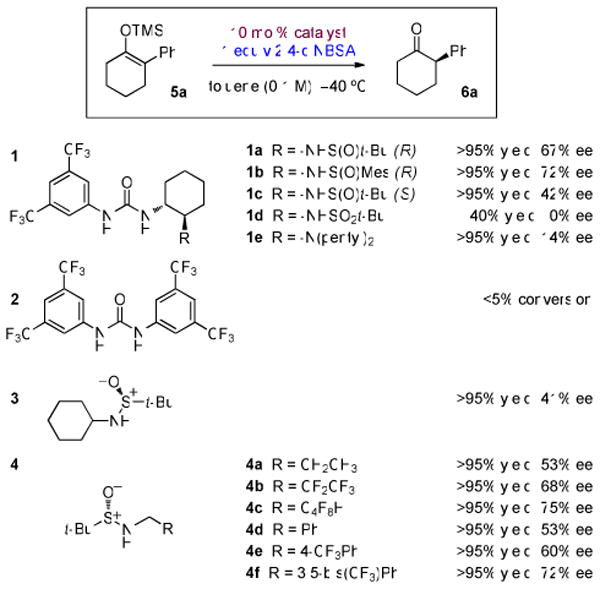
Evaluation of catalyst structuresa
a Yield determined by 1H NMR on a 0.05 mmol scale. Enantioselectivity determined by chiral HPLC.
Systematic variation of the catalyst structure revealed that replacement of the sulfinamide group with other basic functional groups such as sulfonamides (1d) or tertiary amines (1e) led to much less effective catalysts for the protonation of 5a with 2,4-diNBSA, and that urea derivatives such as 2, lacking a basic ancillary group, were completely unreactive (Scheme 1).
Examination of simple sulfinamide 3, which lacks a urea moiety, revealed that it was also catalytically active in the protonation of 5a, affording ketone 6a in >95% yield and 41% ee. The enantioselectivity observed with 3, while moderate, revealed that enantioselective catalysis could be achieved with compounds bearing only the sulfinamide moiety. The synthetic accessibility of these simple structures allowed for the rapid preparation and screening of a large array of substituted sulfinamide derivatives.8 Testing analogues of 3 demonstrated that branching at the carbon center adjacent to the sulfinamide nitrogen was deleterious to both reactivity and enantioselectivity, so efforts were focused on simple primary sulfinamide derivatives (Scheme 1, 4a–f). Interestingly, both simple alkyl- and benzyl-substituted catalysts performed comparably (4a vs. 4d). For both, however, a significant increase in enantioselectivity was observed with analogues bearing additional electron-withdrawing groups. This effect was especially pronounced with fluorinated analogues (4a vs. 4b and 4c; 4d vs. 4e and 4f).
The enantioselectivity was also found to be responsive to the identity of the sulfonic acid, even though none of the sulfonic acid derivatives examined displayed any background reactivity in the absence of catalyst 4c (Table 1, entries 2–5). Reactions with 2,4-dinitrobenzene sulfonic acid as the strong acid source afforded highest ee's. Further, it was observed that it is possible to use a catalytic quantity of the sulfonic acid as long a stoichiometric proton source such as water or a phenol is introduced (entries 6–8). In particular, reactions with hindered phenols such as 2,6-di-tert-butylphenol afforded product 6a in highest enantioselectivity (entry 8). Addition of a dessicant such as sodium sulfate to remove residual water associated with the hydroscopic sulfonic acids had a beneficial effect on both yield and ee (entries 1 vs. 2, and 8 vs. 9). Under optimal conditions, product 6a was obtained in 86% ee using 4c as the catalyst with 0.2 equiv 2,4-diNBSA, 1.1 equiv 2,6-di-tert-butylphenol and 2.0 equiv Na2SO4 in toluene at −50 °C (Table 2, entry 9).
Table 1. Effect of the proton source on enantioselectivitya.
 | |||||
|---|---|---|---|---|---|
| entry | R | equiv ArSO3H | H+ source | conversionc (%) | eed (%) |
| 1 | 2,4-(NO2)2 | 1.0 | - | >95 | 76 |
| 2b | 2,4-(NO2)2 | 1.2 | - | >95 | 85 |
| 3b | 2-NO2 | 1.2 | - | >95 | 69 |
| 4b | 3-NO2 | 1.2 | - | >95 | 55 |
| 5b | 2,4,5-Cl3 | 1.2 | - | >95 | 58 |
| 6 | 2,4-(NO2)2 | 0.1 | H2O | 43 | 69 |
| 7 | 2,4-(NO2)2 | 0.1 | PhOH | >95 | 76 |
| 8 | 2,4-(NO2)2 | 0.1 | 2,6-(t-Bu)C6H3OH | 70 | 83 |
| 9b | 2,4-(NO2)2 | 0.2 | 2,6-(t-Bu)C6H3OH | 78 | 86 |
Reactions were carried out on 0.05 mmol scale.
2 equiv Na2SO4 included.
Determined by 1H NMR.
Determined by HPLC using commercial chiral columns.
Table 2. Substrate scopea.
 | ||||||
|---|---|---|---|---|---|---|
| entry | enol silane | n | R | ketone | yieldb (%) | eec (%) |
| 1 | 5a | 1 | C6H5 | 6a | 91 | 86 |
| 2 | 5b | 1 | 4-MeC6H4 | 6b | 92 | 88 |
| 3 | 5c | 1 | 4-OMeC6H4 | 6c | 90 | 84 |
| 4 | 5d | 1 | 4-ClC6H4 | 6d | 89 | 89 |
| 5 | 5e | 1 | 4-FC6H4 | 6e | 88 | 81 |
| 6 | 5f | 1 | 4-CO2MeC6H4 | 6f | 84 | 78 |
| 7 | 5g | 1 | 3-MeC6H4 | 6g | 92 | 84 |
| 8 | 5h | 1 | 2-MeC6H4 | 6h | 91 | 85 |
| 9 | 5i | 1 | 2-Naphthyl | 6i | 93 | 82 |
| 10 | 5j | 2 | C6H5 | 6j | 88 | 73 |
Reactions were carried out on 0.15 mmol scale. Silyl enol ether and 4c were added as a solution in toluene to 2,4-diNBSA, 2,6-di-t-butyl phenol and Na2SO4 in toluene at −78 °C.
Isolated yield based on silyl enol ether.
Determined by HPLC using commercial chiral columns.
A variety of silyl enol ethers were examined under these conditions in the enantioselective protonation reaction (Table 2). Several 2-aryl-substituted cyclic ketones bearing electron-donating or withdrawing para substituents could be obtained in high yield and with ee's between 78 and 89% (entries 1–6). Substituents in the ortho and meta positions were also tolerated (entries 7–9). The cycloheptanone derivative 5j, however, underwent protonation with measurably lower enantioselectivity (entry 10).
While the sulfinamide appears to promote the protonation reaction by functioning as a solid-to-solution phase transfer catalyst for the insoluble 2,4-diNBSA,9 the basis for stereoinduction in these reactions is intriguing and not at all apparent. As outlined below, our preliminary mechanistic investigation suggests several interesting possibilities for how a catalyst as simple as 4c might participate in cooperative stabilizing interactions in the selectivity-determing transition structure.
A linear dependence of reaction enantioselectivity on the enantiopurity of 4c was observed, indicating the sulfinamide catalyst maintains a monomeric structure in the ground state and in the ee-determining transition state. Accordingly, our analyses considered only pathways involving one chiral catalyst molecule.
In principle, either proton transfer or silyl transfer may be rate- and enantiodetermining in the protonation of silyl enol ethers catalyzed by 4c.10 Both scenarios were evaluated computationally in the reaction of silyl enol ether 5a with protonated sulfinamide catalyst CF3CH2NHS(O)t-Bu (4g) (Figure 2). Given the structural and functional group simplicity of the chiral catalyst, we were especially interested in whether attractive noncovalent interactions might play a role in organizing the transition structures into energetically well-defined geometries.11
Figure 2.
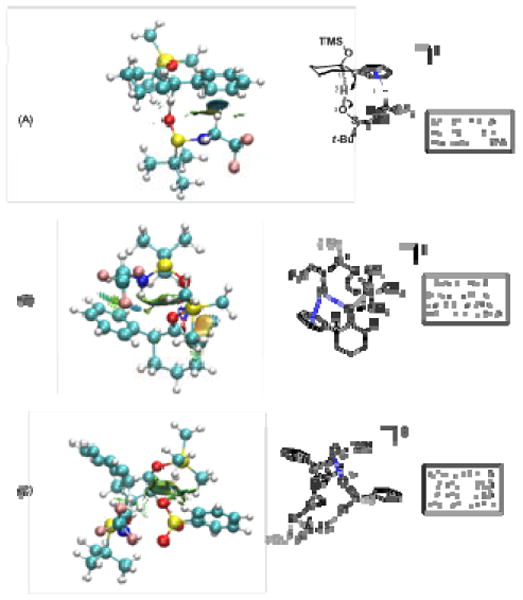
NCI (noncovalent interaction) analysis of calculated transition states. Gradient isosurfaces for noncovalent interactions are diplayed. The surfaces are colored according to the strength of the NCI which increase from green to blue with red signifying destabilizing nonbonded overlap. (A) proton transfer from sulfinamide to substrate; (B) silyl transfer from substrate to sulfinamide; (C) proton transfer with a sulfonate counterion included. Structures fully optimized at the M05-2X/6-31+g(d,p) level of theory.
Preliminary calculations indicated that proton transfer should occur from the oxygen atom of the sulfinamide, as NH-to-C proton transfer was significantly higher in energy. We examined a series of transition structures incorporating NH–π, CH–π or hydrogen bonding interactions, as well as others lacking secondary non-covalent interactions. Of these, the lowest energy structures were those that included CH–π interactions from the electron-deficient CH2 side chain of the catalyst to the substrate aryl ring (one representative structure is shown in Figure 2A). In this structure the distance from the closest hydrogen of catalyst to the centroid of the arene is 2.47 Å. Since the existence of a weak noncovalent interaction cannot be inferred from atomic distance only, analysis of the electron density and its derivatives was carried out using the NCIPLOT program recently developed by Yang and co-workers.12 This approach allows for the generation of gradient isosurfaces that indicate the location and strength of noncovalent interactions of all types.
Transition structures for silyl transfer from a C-protonated silyl enol ether intermediate to the sulfinamide oxygen were also modeled. In one such structure, hydrogen bonding from the NH of 4g to the incipient carbonyl and also to the arene is observed to provide a rigidifying framework (Figure 2B).
The identity of the sulfonate counterion has a measurable influence on enantioselectivity (Table 1). Modeling the proton transfer step with a benzene sulfonate counterion included (Figure 2C) also revealed a network of potential attractive interactions. In the most energetically accesible structures, the sulfonate appears to be held in place by hydrogen bonding to the sulfinamide NH. However, NCI analysis points to electrostatic attraction with the CO bond developing positive charge as the dominant force that positions the sulfonate.
At this stage, development of a rigorous stereochemical model is beyond the scope of this analysis, and would likely require a dynamic approach that considers an ensemble of structures. Nevertheless, intriguing possibilities have been identified for how the structurally simple sulfinamide catalysts might engage in nocovalent, attractive interactions that can play a critical role in transition state organization.
In summary, simple chiral sulfinamide derivatives used in conjunction with a strong achiral sulfonic acid are effective catalysts for enantioselective protonation of prochiral silyl enol ethers. The use of these sulfinamide catalysts as acid shuttles introduces a new role for these readily accessible compounds, and we anticipate extension of this reactivity principle to other types of synthetically interesting enantioselective protonation reactions.
Supplementary Material
Acknowledgments
This work was supported by the NIGMS (PO1 GM-69721 and RO1 GM-43214) and by a Mary Fieser Postdoctoral Fellowship to E.M.B. from Harvard University and an NIH postdoctoral fellowship to A.M.H. We thank Amanda Turek for catalyst synthesis and Stephan Zeund for providing the calculated structure in Figure 1B.
Footnotes
Supporting Information Available Full experimental procedures, syntheses of substrates and catalysts, characterization data for all new compounds, NMR spectra and HPLC traces for protonation products, additional optimization with details of additional sulfinamide scaffolds, methods for computational anaylsis, cartessian coordinates for calculated structures, and NCIPLOT structures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For general reviews on chiral Brønsted acids as organocatalysts see: Kampen D, Reisinger CM, List B. Top Curr Chem. 2010;291:395. doi: 10.1007/978-3-642-02815-1_1.Terada M. Synthesis. 2010;12:1929.Doyle AG, Jacobsen EN. Chem Rev. 2007;107:5713. doi: 10.1021/cr068373r.
- 2.(a) Nakashima D, Yamamoto H. J Am Chem Soc. 2006;128:9626. doi: 10.1021/ja062508t. [DOI] [PubMed] [Google Scholar]; (b) Hatano M, Maki T, Moriyama K, Arinobe M, Ishihara K. J Am Chem Soc. 2008;130:16858. doi: 10.1021/ja806875c. [DOI] [PubMed] [Google Scholar]; (c) García-García P, Lay F, García- García P, Rabalakos C, List B. Angew Chem Int Ed. 2009;48:4363. doi: 10.1002/anie.200901768. [DOI] [PubMed] [Google Scholar]; (d) Uragguachi D, Nakashima D, Ooi T. J Am Chem Soc. 2009;131:7242. doi: 10.1021/ja903271t. [DOI] [PubMed] [Google Scholar]; (e) Shapiro ND, Rauniyar V, Hamilton GL, Wu J, Toste FD. Nature. 2011;470:245. doi: 10.1038/nature09723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For a recent, comprehensive review of the synthesis and applications of tert-butanesulfinamide, see: Robak MT, Herbage MA, Ellman JA. Chem Rev. 2010;110:3600. doi: 10.1021/cr900382t. and for p-tolylsulfinamide see: Zhou P, Chen BC, Davis FA. Tetrahedron. 2004;60:8003.
- 4.For examples of sulfinamides as organocatalysts, see Pei D, Wang Z, Wei S, Zhang Y, Sun J. Org Lett. 2006;8:5913. doi: 10.1021/ol062633+.Tan KL, Jacobsen EN. Angew Chem Int Ed. 2007;46:1315. doi: 10.1002/anie.200603354.Wang C, Wu X, Zhou L, Sun J. Chem Eur J. 2008;14:8789. doi: 10.1002/chem.200801479.Pei D, Zhang Y, Wei S, Wang M, Sun J. Adv Synth Catal. 2008;350:619.Robak MT, Trincado M, Ellman JA. J Am Chem Soc. 2007;129:15110. doi: 10.1021/ja075653v.Kimmel KL, Robak MT, Ellman JA. J Am Chem Soc. 2009;131:8754. doi: 10.1021/ja903351a.
- 5.Xu H, Zuend SJ, Woll MG, Tao Y, Jacobsen EN. Science. 2010;327:986. doi: 10.1126/science.1182826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For previous examples of enantioselective protonation as a route to α-aryl cyclohexanones, see: Ishihara K, Nakamura S, Kaneeda M, Yamamoto H. J Am Chem Soc. 1996;118:12854.Nakamura S, Kaneeda M, Ishihara K, Yamamoto H. J Am Chem Soc. 2000;122:8120.Ishihara K, Nakashima D, Hiraiwa Y, Yamamoto H. J Am Chem Soc. 2003;125:24. doi: 10.1021/ja021000x.Yanagisawa A, Touge T, Arai T. Angew Chem Int Ed. 2005;44:1546. doi: 10.1002/anie.200462325.Cheon CH, Yamamoto H. J Am Chem Soc. 2008;130:9246. doi: 10.1021/ja8041542.
- 7.For enantioselective protonation of other silyl enol ether substrate classes, see: Poisson T, Dalla V, Marsais F, Dupas G, Oudeyer S, Levacher V. Angew Chem Int Ed. 2007;46:7090. doi: 10.1002/anie.200701683.Poisson T, Oudeyer S, Dalla V, Marsais F, Levacher V. Synlett. 2008:2447.Sugiura M, Nakai T. Angew Chem Int Ed. 1997;36:2366.Morita M, Drouin L, Motoki R, Kimura Y, Fujimori I, Kanai M, Shibasaki M. J Am Chem Soc. 2009;131:3858. doi: 10.1021/ja9005018.Uraguchi D, Kinoshita N, Ooi T. J Am Chem Soc. 2010;132:12240. doi: 10.1021/ja105945z.
- 8.For additional details of other sulfinamide structures and associated selectivities in the enantioselective protonation reaction, see supporting information.
- 9.The complexation behavior of the sulfinamide catalysts with 2,4-diNBSA was investigated by1H NMR. Peak integration of mixtures in C6D6 revealed that bifunctional catalysts 1a–e form a 1:1 complex with 2,4-diNBSA. In contrast, no dissolution of 2,4-diNBSA was observed with the simple urea 2 Simple sulfinamides 4c and 4f, lacking a urea component, also induced solubilization of the sulfonic acid. These data point to a role of the sulfinamides as solid-to-solution phase transfer catalysts.
- 10.For an example in which rate-limiting desilylation is proposed, see ref. 6e.
- 11.Accurately reproducing noncovalent interactions is a challenge for many density functional theory methods. We utilized Truhlar's M05-2X functional, which has been shown to be suitable in this respect: Zhou Y, Truhlar DG. Acc Chem Res. 2008;41:157. doi: 10.1021/ar700111a.
- 12.(a) Johnson ER, Keinan S, Mori-Sánchez P, Contreras-García J, Cohen AJ, Yang W. J Am Chem Soc. 2010;132:6498. doi: 10.1021/ja100936w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Contreras-García J, Johnson ER, Keinan S, Chaudret R, Piquemal JP, Beratan DN, Yang W. J Chem Theory Comput. 2011;7:625. doi: 10.1021/ct100641a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.