

Abstract
Baicalein (Ba) was found to be subject to serious first-pass metabolism after oral administration. We previously revealed the important role of intestine in the low oral bioavailability of Ba. The present study aims to evaluate the hepatic metabolism and disposition of Ba. Ba was given to Sprague–Dawley rats through bolus or infusion via intravenous or intra-portal route of administrations. Both plasma and bile samples at different time intervals were obtained. Concentrations of Ba and potential metabolites in the collected samples were analyzed with HPLC/UV and identified by LC/MS/MS, respectively. Plasma concentration versus time profiles of Ba obtained from intravenous and intra-portal administrations were compared to estimate the extent of hepatic metabolism. In addition, transport studies of baicalein-7-glucuronide (BG), one of the major metabolites of Ba, were carried out using transfected cell systems overexpressing various human organic anion-transporting polypeptide (OATP) isoforms to estimate the specific transporters involved in the hepatic disposition of Ba metabolites. The results showed that liver, in addition to intestine, also conferred extensive metabolism to Ba. Several mono- and di-conjugates of Ba, which were mainly glucuronides, sulfates, and methylates, were found in bile. The transport study demonstrated that besides MRPs and BCRP, human OATP2B1 and OATP1B3 in liver might also mediate the secretion of BG to bile. In summary, liver plays an important role in the metabolism of Ba and transport of its conjugated metabolites.
KEY WORDS: baicalein, conjugation, hepatic metabolism, transporters
INTRODUCTION
Baicalein (Ba; Fig. 1) is a bioactive flavonoid present in Scutellariae radix, a traditional Chinese medicinal herb derived from the root of Scutellaria baicalensis Georgi (1). Ba has been demonstrated to have diverse pharmacological activities including anti-inflammatory (2,3), anti-allergic (4), antioxidative activities (5), antiviral (6,7), etc. In recent years, the anticancer activities of Baicalein have received great attention (8–10).
Fig. 1.

Chemical structures of Ba, BG, and their proposed metabolites in bile. BGG baicalein-O-di-glucuronide, BGS baicalein-O-glucuronide-O-sulfate, BGGlu baicalein-O-glucose-O-glucuronoide, BG baicalein-7-O-glucuronide, MeBG methyl-O-baicalein-O-glucuronide, BG′ baicalein-6-O-glucuronide, BS baicalein-O-sulfate, MeBS methyl-O-baicalein-O-sulfate
Despite the variety of beneficial effects of Ba, investigations on its metabolism and disposition are not complete. Early studies have demonstrated that there is an extensive first-pass metabolism of Ba and that conjugated metabolites were predominant in systemic circulation after oral administration of Ba in rats (11,12). Since intestinal and hepatic metabolisms are generally responsible for the major first-pass effect for most of the ingested drugs or other xenobiotics, we first conducted studies on the metabolism and disposition of Ba in the intestine. By using a single-pass rat intestinal perfusion model, it was found that intensive glucuronidation of Ba took place in the small intestinal wall and that over 90% of Ba was converted to baicalein-7-glucuronide (BG), which was actively transported to the mesenteric blood (13). In addition, in vitro metabolic study revealed that glucuronidation and sulfation in hepatic microsomes were much more rapid than that in the intestinal microsome (14). The available evidence suggests that hepatic metabolism may also play a critical role in the extensive first-pass effect of Ba. However, detailed mechanistic investigations on the liver metabolism of Ba are lacking. In addition to the intensive first-pass metabolism, previous studies also demonstrated that a number of transporters mediated the dispositions of the conjugated metabolites of Ba. It was found that several efflux transporters in the intestine such as MRP1, 2, 3, and BCRP are essential for the transport of the glucuronide of Ba to both gut lumen and mesenteric circulation (15). Studies by other investigators on the absorption of different flavonoids also suggested the importance of efflux transporters in the intestinal disposition of flavonoids (16–18). Since similar transporters were found to be expressed in the liver, it is warranted to further investigate the contribution of these transporters to the disposition of the conjugated metabolites of Ba in the liver. Therefore, the present study aims to perform a mechanistic investigation on the hepatic metabolism of Ba and transport of its conjugated metabolites.
METHOD
Chemicals
Baicalein and baicalin (BG) were purchased from Aldrich Chem. Co. (Milwaukee, WI, USA). Probenecid and glucuronidase/sulfatase (type H-1, Helix pomatia) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). 6-Hydroxyflavanone, the internal standard for the high-performance liquid chromatography (HPLC) assay, was obtained from Indofine Chemical Co. (Hillsborough, NJ, USA). MK 571 was kindly supplied by Merck Frosst Canada & Co. Waters Oasis hydrophilic lipophilic-balanced (HLB, 1 ml) copolymer extraction cartridges were purchased from Waters (Milford, MA, USA). Acetonitrile (HPLC analytic grade) and methanol (HPLC analytic grade) were obtained from Labscan (Labscan Asia, Bangkok, Thailand). All other compounds were of analytical grade. Distilled and deionized water was used for the preparation of all aqueous solutions.
In Vivo Animal Experiments
The animal experiments were approved by both the Department of Health of the Government of the Hong Kong SAR and the Animal Experimentation Ethics Committee of the Chinese University of Hong Kong. Male Sprague–Dawley rats (body weight, 260–280 g) were supplied by the Laboratory Animal Services Center at the Chinese University of Hong Kong. The rats were housed in an air-conditioned room (24°C) under a 12/12-h light/dark cycle. Rats were fasted overnight with free access to water and were anesthetized with an intramuscular injection of a cocktail containing 60 mg/kg ketamine and 6 mg/kg xylazine before surgery. Right jugular vein or pyloric vein were cannulated with a polyethylene tubing (0.5-mm ID, 1-mm OD or 0.4-mm ID, 0.8-mm OD, respectively, Portex Ltd., Hythe, Kent, England) for intravenous (i.v.) or intra-portal (i.p.v.) dosing, respectively. The bile duct and left femoral artery were cannulated with a polyethylene tubing (0.4-mm ID, 0.8-mm OD) for bile and blood samplings, respectively. During the surgical process, the body temperature was maintained at 37°C by heating lamp. Ba and BG were dissolved in the mixture of PEG 400 and saline (with 10% Pluronic F-127; 1:1, v/v). The dosing regimens include:
An i.v. or i.p.v. bolus injection of Ba at 5.5 mg/kg (21.1 μmol/kg) or 2.2 mg/kg (8.46 μmol/kg). Blood samples were taken at 0.25, 0.5, 1, 3, 10, 30, and 60 min post-dosing. The bile samples were continuously collected at 0–15, 15–30, 30–45, 45–60, and 60–75 min.
An i.v. or i.p.v. infusion of Ba at rates of 0.148 mg min−1 kg−1 (low) and 0.296 mg min−1 kg−1 (high) with a syringe pump (Cole Parmer 74900 series, New Hope, PA, USA). Moreover, a loading dose via i.v. or i.p.v. bolus injection at 2.5 or 5 mg/kg was given prior to infusions at low and high doses, respectively, to accelerate the occurrence of steady state. The blood and bile samples were collected every 15 min for 90 min.
An i.v. bolus of BG at the dose of 1.1 mg/kg (2.47 μmol/kg) was performed with or without co-administration of MRP inhibitors (100 mg/kg of probenecid or 50 mg/kg MK 571) for the investigation of the transporters’ contribution to the disposition of BG. Blood samples were taken at 3, 5, 10, 20, 30, 45, 60, and 75 min, and bile samples were continuously collected at every 15-min interval for 75 min.
Sample Preparation
The plasma samples were obtained by centrifuging the collected blood samples at 13,000 rpm for 10 min. To determine the plasma concentrations of Ba or BG, the plasma samples were purified with the Oasis® HLB cartridge as described previously (19). Briefly, an aliquot of 50 μl of internal standard (10 μg/ml) was added into 50 μl of plasma sample. The sample was then diluted with 1 ml of 35% methanol in 25 mM sodium dihydrogen phosphate buffer (pH 2.5) containing 1% ascorbic acid. After vortex mixing and centrifugation at 16,000×g for 10 min, the supernatant was loaded on the preconditioned HLB cartridge. The cartridge was flushed subsequently and the analytes eluted from the cartridge by 1 ml methanol. The eluent was dried and the residue was reconstituted with 100 μl of 35% methanol in 25 mM sodium dihydrogen phosphate buffer (pH 2.5) containing 1% ascorbic acid. An aliquot (50 μl) of the resultant solution was injected into the HPLC system for analysis.
To determine the plasma concentration of total Ba, an aliquot of 50 μl of plasma was hydrolyzed by 50 μl gluronidase/sulfatase (4,000 U/ml, dissolved in 0.46 M sodium acetate buffer, pH 5.0) containing 2.5 μl of 20% ascorbic acid at 37°C for 2 h. After hydrolysis, the sample was extracted in the same manner as described above.
The collected bile samples were diluted with 35% methanol in 25 mM sodium dihydrogen phosphate buffer (pH 2.5) containing 1% ascorbic acid, and the resultant solution (50 μl) was directly subjected to HPLC. Similarly, the bile concentrations of total Ba were measured after enzymatic hydrolysis with the same protocol as described for plasma samples. The amount of glucuronide/sulfate was estimated by the difference between the amount of Ba in the sample before and after gluronidase/sulfatase hydrolysis
Quantitative Analysis of Ba and its Metabolites by HPLC/UV
The HPLC/UV method developed from our previous study was used for the quantitative analysis of Ba and its identified metabolites, with a minor modification (19). The HPLC system is composed of a Waters 2690 separation module and Waters 996 photodiode array detector. The chromatographic separation of Ba, its metabolites, and internal standard was achieved using a reversed-phase HPLC column (BDS reversed-phase column, 25-cm × 4.6-mm I.D., 5-mm particle size, Thermo Hypersil) connected with a protective guard column (Delta-Pak C18 Guard-Pak, Waters). For the plasma sample analysis, the mobile phase, consisting of a mixture of 20 mM sodium dihydrogen phosphate buffer (pH 4.6) (A), methanol (B), and acetonitrile (C), was run using a linear gradient elution program. The gradient began with 80% A, 5% B, and 15% C, changed linearly to 57% A, 8% B, and 35% C in the first 10 min; then 40% A, 0% B, and 60% C in the next 2 min; and then back to the initial composition in 6 min followed by 7 min for re-equilibration. The flow rate was set at 1 ml/min. As for the bile sample analysis, the mobile phase consisted of 1 mM of aqueous ammonia formate (A) and acetonitrile (B). The gradient elution began with 90% A and was changed to 70% A in 10 min, then 40% A in 2 min and kept for 6 min, and then back to 90% A followed by 6 min of equilibration before next injection. All analytes were detected at 320 nm.
Identification of Ba Metabolites by LC/MS/MS
To determine the metabolites of Ba formed, API Q-Trap (Applied Biosystems, Toronto, ON, Canada) equipped with two Perkin-Elmer PE-200 series micro-pumps and an autosampler was used to perform the analysis. The LC condition is identical to that for the bile samples described above. Twenty percent of eluent was introduced into the mass spectrometer and the other 80% was split off. Negative ion mode of the mass spectrometer was set for the analysis. The other working mass spectrometer parameters were: ion spray voltage, −4500 V; declustering potential, −80 V; entrance potential, −10 V; collision cell exit potential −4 V; collision energy, −43 V; curtain gas, 40 psi; nebulizer gases, 70 psi; auxiliary gas, 70 psi; and source temperature, 400°C.
In Vitro Methylation of Ba by Rat Liver Cytosol
Ba (0.3–55 μM) was pre-incubated with a pooled rat liver cytosolic fraction at a final protein concentration of 1 mg/ml at 37°C for 10 min in 50 mM Tris buffer (pH 7.4) containing 5 mM MgCl2, 8 mM dithiothreitol, and 0.0625% BSA. The reaction was initiated by the addition of 200 μM of S-adenosyl-l-methionine and lasted for 10 min. All the experiments were performed in triplicate.
Uptake Transport Study of BG in OATPs Transfected Cell Lines
The uptake transport study was performed as described previously (20,21). The inhibitory effects of BG on uptake transport of [3H]estrone-3-sulfate were estimated in the CHO-OATP1B1 and MDCKII-OATP2B1 transfected cell line, while the inhibitory effect on the uptake transport of Fluo3 was studied in CHO-OATP1B3 (Solvo Biotechnology, Szeged, HU) in the presence of 10 and 100 μM of BG. Values were presented on a relative scale with 100% defined as transport in the absence of BG (no inhibition). Each concentration was tested in duplicate.
Data Analyses
The pharmacokinetic parameters of Ba, BG, and total Ba were estimated using the WinNonlin program (Pharsight, Mountain View, CA) with a non-compartmental approach.
The systemic clearance (CL) of drug was calculated as: CL = Dose/AUC. The biliary clearance (CLbile) of drug was obtained as: CLbile = Amount of biliary secretion/AUC. The hepatic extraction ratio (ER) of drug was estimated as: ER = 1 − AUCipv/AUCiv. The steady-state systemic clearance (CLss) of drug was given by: CL = K0/Css, where K0 is the infusion rate and Css is the steady-state plasma concentration.
Reported values for in vivo and uptake transporter studies are presented as mean ± SD. Statistically significant difference between two groups or more than two groups was evaluated by Student’s t test and one-way ANOVA, respectively. A p < 0.05 was considered significant for all tests. Data for the in vitro enzymatic study are presented as the mean ± SE. The kinetic parameters of methylation were calculated with Prism (GraphPad Software, Inc.).
RESULTS
IV or IPV Administrations of Ba in Rats
Findings from Bolus Injection
Ba was found to rapidly convert to several conjugated metabolites in systemic circulation after either i.v. or i.p.v. administration (Fig. 2). By treating samples with beta-gluronidase and sulfatase, the glucuronides and sulfates of Ba were quantified. It was found that glucuronidation and sulfation seem to be the major pathway for the metabolism of Ba since Ba is the predominant product after hydrolysis with relative small fraction transformed through methylation (MeBa; Fig. 2). By comparing the AUCBa and AUCglu/sul of its glucuronide or sulfate conjugates, the metabolism of Ba was intensive in the body, and conjugates were the predominant forms of Ba in the body at most time points. In addition, Ba was disposed through bile in the form of various phase II conjugates without detectable Ba in the bile samples (Fig. 3). As indicated by LC/MS/MS, the formed conjugated metabolites in the bile were found to be mainly glucuronides, sulfates, and methylates of Ba.
Fig. 2.

HPLC chromatograms of plasma samples obtained 1 min after i.v. (top) and i.p.v. (middle) bolus administrations, as well as hydrolyzed plasma sample by beta-glucuronidase/sulfatase (bottom). Ba baicalein, BG baicalin, MeBa methyl-O-baicalein, BG′ baicalein-6-O-glucuronide, MeBG methyl-O-baicalein-O-glucuronide, IS internal standard
Fig. 3.
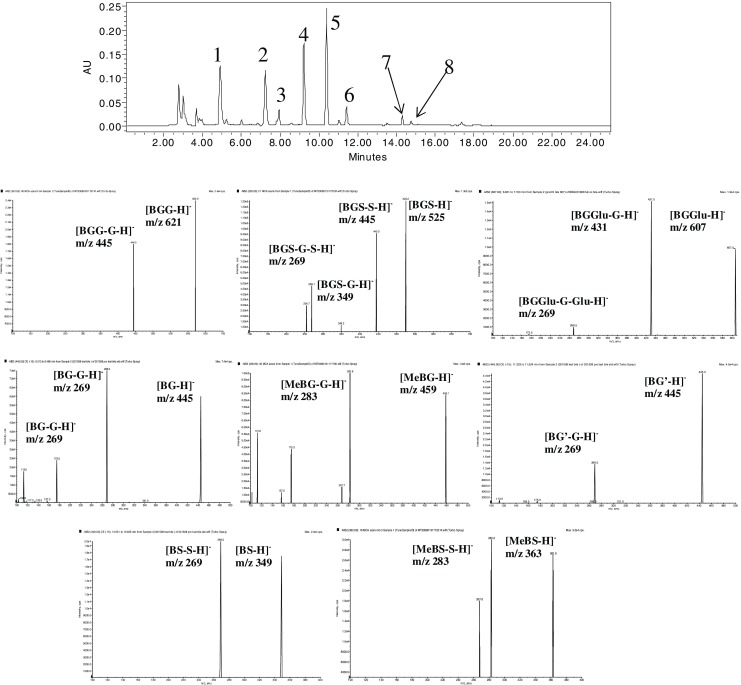
HPLC chromatograms and mass spectra of metabolites of Ba in the collected bile sample. 1 BGG (621/445): Baicalein-O-di-glucuronide. 2 BGS (525/445): Baicalein-O-glucuronide-O-sulfate. 3 BGGlu (607/431): Baicalein-O-glucose-O-glucuronoide. 4 BG (445/269): Baicalein-7-O-glucuronide. 5 MeBG (459/283): Methyl-O-baicalein-O-glucuronide. 6 BG′ (445/269): Baicalein-6-O-glucuronide. 7 BS (349/269): Baicalein-O-sulfate. 8 MeBS (349/269): Methyl-O-baicalein-O-sulfate
The pharmacokinetic profiles and parameters of Ba after i.v. or i.p.v. administration were summarized in Fig. 4 and Table I, respectively. After i.v. or i.p.v. administration, Ba existed in the systemic circulation for only a few minutes with a t1/2 of about or less than 1 min at both low and high doses of Ba (Table I). To estimate the hepatic ER of Ba, AUCi.p.v. was compared with AUCi.v. at both high and low doses. It was found that the hepatic ER of Ba was dose-dependent. There was over 90% of Ba (ER > 0.9) that was extracted in the liver at a low dose of Ba, whereas ER was significantly reduced to 0.15 at a higher dose of Ba (Table I).
Fig. 4.

Plasma concentrations of Ba (upper) and its metabolites versus time profiles (middle) as well as biliary secretion of conjugates (lower) after i.v. and i.p.v. administrations of Ba low (2.2 mg/kg, left column) and high doses (5.5 mg/kg, right column; n = 5)
Table I.
Pharmacokinetic Parameters of Ba and Conjugate Metabolites After i.v. or i.p.v. Administration (n = 5)
| t 1/2 (min) | AUC (nmol/ml min) | Vd or Vd/Fa (ml/kg) | CL or CL/Fa (ml/min/kg) | ERb | |||
|---|---|---|---|---|---|---|---|
| 2.2 mg/kg | i.v. | Parent | 0.778 ± 0.043** | 44.2 ± 4.5*** | 215 ± 12* | 193 ± 18* | 0.92 |
| Glu + Sul | 15.1 ± 4.2* | 328 ± 41 | |||||
| BG | 9.88 ± 3.09* | 158 ± 12 | |||||
| i.p.v. | Parent | 0.490 ± 0.128 | 3.37 ± 1.49 | (2.12 ± 1.05) × 103 | (2.88 ± 1.12) × 103 | ||
| Glu + Sul | 21.7 ± 2.4 | 292 ± 79 | |||||
| BG | 24.9 ± 8.3 | 95.7 ± 44.4 | |||||
| 5.5 mg/kg | i.v. | Parent | 1.07 ± 0.12 | 79.0 ± 15.8 | 425 ± 95 | 275 ± 54 | 0.15 |
| Glu + Sul | 19.0 ± 1.8 | 746 ± 176 | |||||
| BG | 14.3 ± 2.0 | 393 ± 109* | |||||
| i.p.v. | Parent | 0.964 ± 0.393 | 66.9 ± 13.2 | 445 ± 177 | 324 ± 61 | ||
| Glu + Sul | 24.9 ± 5.5 | 920 ± 260 | |||||
| BG | 15.6 ± 4.5 | 182 ± 68 |
Comparison between i.v. and i.p.v. groups: *p < 0.05; **p < 0.01; ***p < 0.0001
aFor i.p.v. administration
bER = 1 − AUCi.p.v./AUCi.v.
Elimination of glucuronide or sulfate conjugates was also fast, with t1/2 ranging from 15 to 25 min. Efficient biliary secretion of Ba conjugates may be responsible for their rapid excretion. Based on the hydrolyzed bile samples, we found that most of the biliary secretion occurred in the first 15 min. Around 55.5 ± 10.1% (1.27 ± 0.23 × 103 nmol, i.v.) and 53.6 ± 9.7% of dose (1.22 ± 0.22 × 103 nmol, ipv) of Ba given at a low dose and 60.1 ± 9.1% (3.42 ± 0.52 × 103 nmol, i.v.) and 64.4 ± 7.5% of dose (3.67 ± 0.43 × 103 nmol, ipv) of Ba given at a high dose were secreted via the bile as glucuronide/sulfate in 75 min. As demonstrated in Fig. 4, the cumulative biliary secretion profiles of glucuronide/sulfate were similar between the two (i.v. and i.p.v.) administration pathways.
Findings from Infusion
We also conducted i.v. or i.p.v. infusion of Ba to mimic the situation of Ba after oral administration. It was found that the plasma concentrations of Ba began to reach a steady state after 15 min of i.v. or i.p.v. infusion (Fig. 5). The plasma concentrations of Ba at steady state after i.v. infusion was 2.5-fold higher than those when performing an i.p.v. infusion at a low dose, and the difference reduced to 2.0-fold at a high-dose infusion (Table II). The hepatic extraction ratio was found to be 0.60 and 0.49 at low- and high-dose infusions, respectively. The steady-state systemic clearance of Ba (CLss) during i.p.v. infusion was 2.6-fold higher than that of i.v. infusion at a low dose. In general, the plasma concentrations and AUC of glucuronide/sulfate at steady state were similar between i.v. and i.p.v. infusions at both high and low doses.
Fig. 5.
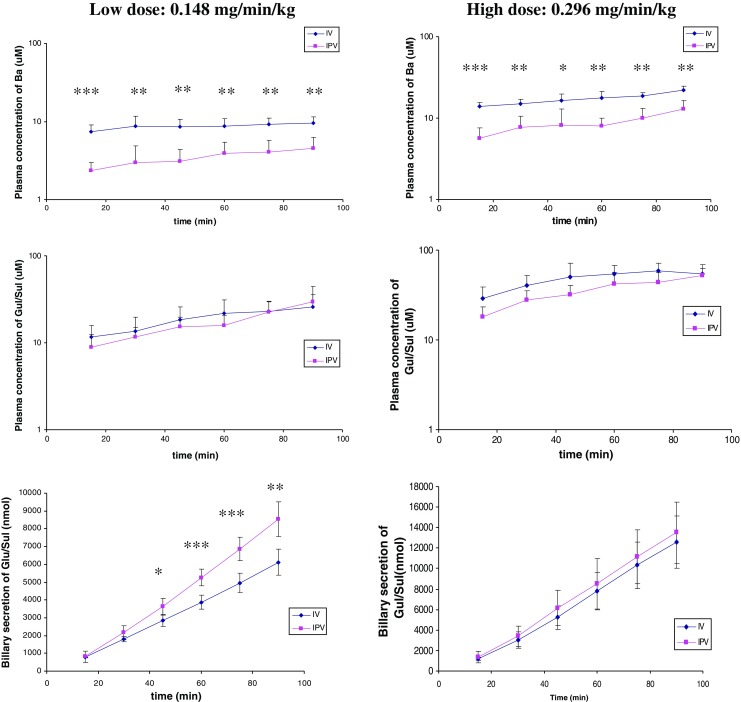
Plasma concentrations of Ba (upper), glucuronides (Glu), and sulfates (Sul) of Ba (middle) as well as accumulative biliary secretion of Glu/Sul (bottom) after i.v. and i.p.v. infusions of Ba at low (0.148 mg min−1 kg−1, left column) and high doses (0.296 mg min−1 kg−1, right column). *p < 0.05; **<p < 0.01; ***p < 0.001 (n = 5)
Table II.
Pharmacokinetic Parameters of Ba and Conjugate Metabolites after i.v. or i.p.v. Infusion (n = 5)
| 0.148 mg min−1 kg−1 | 0.296 mg min−1 kg−1 | |||
|---|---|---|---|---|
| i.v. | i.p.v. | i.v. | i.p.v. | |
| Css Ba (μM) | 8.70 ± 1.68*** | 3.49 ± 1.07 | 17.2 ± 2.1** | 8.77 ± 2.67 |
| CLss or CLss/Fa Ba (ml min−1 kg−1) | 67.5 ± 13.5** | 178 ± 59 | 66.8 ± 7.8 | 146 ± 68 |
| AUC Ba15–90 min (nmol/ml min) | 656 ± 129*** | 263 ± 86 | 1.28 ± 0.17 × 103** | 0.65 ± 0.20 × 103 |
| AUC glu/Sul15–90 min (nmol/ml min) | 1.43 ± 0.52 × 103 | 1.27 ± 0.33 × 103 | 3.67 ± 0.88 × 103 | 2.72 ± 0.46 × 103 |
| Billiary secretion (Glu + Sul, nmol) | 6.12 ± 0.71 × 103** | 8.54 ± 0.98 × 103 | 1.26 ± 0.26 × 104 | 1.35 ± 0.30 × 104 |
| Hepatic ER | 0.60 | 0.49 | ||
Comparison between i.v. and i.p.v. groups: *p < 0.05; **p < 0.01; ***p < 0.0001
aFor i.p.v. administration
Similar to i.v. and i.p.v. bolus, Ba given through i.v. or i.p.v. infusion were also disposed via the bile mainly in the form of glucuronides and sulfate conjugates. Based on the linear relationship between accumulative biliary secretion of glucuronide/sulfate and time course of infusion (Fig. 5), it was suggested that the biliary excretion rates of glucuronide and sulfate of Ba were basically constant during the whole process. Although at high dose there was no difference in the amounts of the conjugates secreted between the two infusion manners, faster biliary secretion rate was observed when dosing via i.p.v. at a low dose.
In Vitro Methylation Conjugation by Rat Liver Cytosol
To further explain why there were more glucuronides and sulfates than methylate in the body after venous administration, the in vitro methylation of Ba catalyzed by human liver cytosol were conducted and compared with the kinetics of glucuronidation and sulfation. Methylation of Ba followed the substrate inhibition profile (Ki = 11.23 μM; Fig. 6). Comparing with glucuronidation and sulfation (KmUGT = 77.1 μM, KmSULT = 4.40 μM) (14), Ba has a higher affinity but lower capacity to the catechol-O-methyltransferase (COMT; KmCOMT = 1.93 μM), which is responsible for the methylation of Ba. Based on the Clint value, which was usually used to estimate catalytical efficiency, it was suggested that glucuronidation (Clint = 436 μl min−1 mg−1) and sulfation (Clint = 1601 μl min−1 mg−1) are still more efficient pathways than methylation (Clint = 87 μl min−1 mg−1) for the biotransformation of Ba in the liver.
Fig. 6.

Formation rate of methylated Ba versus substrate concentration plots for human COMT (n = 3)
Role of Transporters in the Disposition of the Metabolites of Ba
Inhibition of Biliary Secretion of BG
Co-administration of MK 571 and probenecid substantially reduced the elimination and biliary secretion of BG (Fig. 7). In addition, the t1/2 of BG was prolonged in the presence of MK (65 min) and probenicid (38 min, p < 0.05). Both CL and CLbile of BG were reduced in the presence of both MK and probenecid (p < 0.05; Table III).
Fig. 7.

Inhibitory effect of MK 571 (MK) and probenecid (Pro) on the plasma clearance and biliary secretion of BG after i.v. administration of Ba at 1.1 mg/kg (n = 4)
Table III.
Inhibition of MK 571 and Probenecid on the Elimination of BG after Intravenous Administration of BG at 1.1 mg/kg (2.466 μmol/kg, n = 4)
| Treatment groups | AUC (nmol/ml min) | t 1/2 (min) | Billiary secretion of BG (nmol) | CL bile (ml min−1 kg−1) | CL (ml min−1 kg−1) |
|---|---|---|---|---|---|
| Control | 0.103 ± 0.020 × 103 | 7.56 ± 1.96 | 126 ± 22 | 4.64 ± 1.24 | 24.6 ± 4.7 |
| MK 571 | 1.38 ± 0.28 × 103*** | 64.7 ± 21.8*** | 64.0 ± 15.3*** | 0.178 ± 0.059*** | 1.84 ± 0.36*** |
| Probenecid | 0.701 ± 0.331 × 103* | 37.8 ± 10.8* | 80.8 ± 9.26** | 0.501 ± 0.229*** | 4.09 ± 1.69*** |
Comparison between group with inhibitor and control group: *p < 0.05; **p < 0.01; ***p < 0.0001
Uptake Transport Study of BG in OATP Transfected Cell Lines
As demonstrated in Table IV, BG exhibited the most potent inhibition on the uptake of probe substrates in organic anion-transporting polypeptide (OATP) 2B1, followed by that in OATP 1B3. The influence of BG on the uptake of probe substrate in OATP 1B1 is marginal.
Table IV.
Inhibition of BG to Probe Substrates in Various Human OATP Transfected Cells (n = 3)
| % of uptake inhibition | ||
|---|---|---|
| 10 μM BG | 100 μM BG | |
| OATP 1B1 (uptake of ES) | 6.1 ± 0.5 | 5.3 ± 0.1 |
| OATP 2B1 (uptake of ES) | 83.7 ± 15.4 | 89.5 ± 25.8 |
| OATP 1B3 (uptake of Fluo3) | 40.1 ± 3.7 | 37.8 ± 8.8 |
DISCUSSIONS
Intensive first-pass metabolism was found previously after ingestion of Ba (11). Anatomically, the intestine and liver should be most likely responsible for the remarkable metabolism of Ba. Our previous study showed that Ba was subjected to rapid glucuronidation in the intestine. To avoid the involvement of the intestinal first-pass metabolism, our present study mainly compared the metabolism and disposition of Ba after i.v. and i.p.v. administrations. It was found that over 90% of Ba was extracted by the liver at a lower dose of i.p.v. bolus administration, which allowed significant hepatic metabolism of Ba. In addition, as shown in Table I, AUCGlu/Sul (10-fold) were much greater than AUCBa after i.p.v. administration, and only Ba conjugates were found in the bile, which once again indicated the intensive hepatic metabolism of Ba. Although t1/2 of Ba after i.p.v. administration was shorter than that after i.v. administration, the half-lives of Ba of both routes of administration are so short, which serve as an additional evidence for the rapid hepatic and extrahepatic metabolism of Ba. The extrahepatic metabolism of Ba could be supported by our previous studies with the rat intestinal perfusion model and Caco-2 cell model, which consistently showed intensive metabolism of Ba in the intestine.
Similar to our previous findings in intestinal perfusion study, BG was still one of the major metabolites in bile and blood circulation after i.p.v. administration. However, there were abundant di-conjugations in the bile which was not observed during intestinal perfusion of Ba. In addition, it was found previously that liver cytosol, but not intestine cytosol, catalyzed the sulfation of Ba (14). Consistently, a remarkable amount of sulfate conjugates in mono- or di-conjugates in the bile and all of them were absent in intestinal perfusion study. In the present study, we demonstrated that glucuronides and sulfates of Ba were more predominant than its methylate conjugates when comparing the amount of Ba and MeBa in both plasma or hydrolyzed bile samples. Consistently, it was also demonstrated by the in vitro metabolism study that glucuronidation and sulfation were more efficient pathways than methylation for the elimination of Ba. It is likely that these two types of phase II metabolisms should be responsible for the intensive first-pass metabolism of Ba. In addition, BS formation is much less than that of BG in the liver. As shown in our previous study (14), the metabolic capacity of UGTs in the rat liver microsome was higher than that of SULTs in the rat liver cytosol (Vmax of Glu versus that of Sul = 34 versus 7 nmol min−1 mg−1). Moreover, sulfation of Ba experienced a significant substrate inhibition. Therefore, although Clint of sulfation is higher than that of glucuronidation, it is probable that SULTs were more readily saturated and inhibited at a relatively high concentration of Ba, which resulted in less formation of BS than BG in the liver. Despite the difference in anatomical structures, the expression levels, and numbers of metabolic enzymes and transporters between the liver and intestine, the two organs share some common manners for metabolism and disposition of Ba. As proposed in Fig. 8, Ba delivered from portal vein would be ready to diffuse into the hepatic cell due to its good permeability or lipophilicity, as demonstrated by its high hepatic ER as well as our previous observations in Caco-2 cell and rat in situ intestinal perfusion models. The Ba that diffused into hepatic cells was then subjected to intensive phase II metabolism, which was evidenced by the predominant phase II metabolites in both the plasma and bile. Nevertheless, transporters should be involved in the efficient removal of intracellular conjugates and their relocation into blood circulation and the bile, given the poor membrane permeability of the metabolites, as suggested in Fig. 8. It is suggested that membrane transporters such as MRPs and BCRP, etc. may take an important role in the disposition of the intracellular conjugates of Ba. Our previous study using inside-out cell membrane of the corresponding MRPs 1, 2, 3, and BCRP transfected cell lines demonstrated that BG, one of the major conjugates of Ba, is the substrate of these transporters. Given the location of these transporters in hepatic cells, MRP2 and BCRP at the apical side may be responsible for the biliary excretion of the conjugates of Ba. To demonstrate the in vivo significance of these processes, we studied the biliary secretion of BG in the presence or absence of MK 571, a potent Mrp inhibitor. The presence of MK 571 could remarkably reduce the biliary secretion of BG (Table III), which confirms our previous finding using an in vitro model (15). On the basolateral side, a transporter-mediated efflux (such as by Mrp3 and other unidentified transporters) may play important roles in the relocation of Ba conjugates into the systemic circulation. As for the hepatic disposition of conjugates already existing in the circulation, the hepatic uptake of the metabolites is critical due to their difficulty to traverse the basolateral cell membrane. Probenecid, one of the substrates of OATP, was used to compete with BG on the hepatic uptake and did significantly reduce the CLbile of BG. However, probenecid was not only the substrate of OATP but also the substrate of MRP (22). Thus, we tried estrone sulfate, another substrate of Oatp, and found that it could successfully inhibit the basal to apical transport of BG in Caco-2 cells (unpublished data), but fail to inhibit the biliary secretion of BG in vivo. Such discrepancies between in vitro and in vivo may be ascribed to the difference in the expression level of OATPs and the local concentration of inhibitors between the in vitro and in vivo situations. In order to provide straightforward information about the interaction of BG with OATPs, the uptake study was performed in a much simpler systems, i.e., various OATP isoform transfected cell lines. As shown in Table IV, OATP 1B3 and especially OATP 2B1 may contribute to the hepatic uptake of conjugates. It was surprising to discover that an increase of the BG concentration from 10 to 100 μM did not proportionally increase the inhibition effect on the uptake of Fluo3 by OATP 1B3, which could be due to the partial competitive inhibition effect of BG on OATP 1B3. A previous study by Wang et al. (23) showed interactions between OATP 1B1 and a series of flavonoid aglycones. In the present study, we demonstrated in both in vitro and in vivo systems that flavonoid conjugate metabolites are also likely eliminated in the liver through OATPs. In summary, it seems that the significant hepatic extraction, efficient phase II metabolism coupled with efflux transport, gave rise to the intensive first-pass effect and the rapid elimination of Ba in the body. On the other hand, since there was about 20% of BG that was excreted via the bile, around 80% of BG should be disposed by other pathways. Previously, we found that BG formed rapidly secreted to the gut lumen during intestinal perfusion of Ba (13), and OATP and MRP2 are expressed in both the liver and kidney. It is very likely that extrahepatic elimination of BG, such as eliminations in the intestine and kidney, could also play an important role. Inhibition effect by probenecid and MK571, which resulted in a decrease in both CLbile and total CL, should be due to their influence on both hepatic and extrahepatic elimination of BG.
Fig. 8.

Proposed diagram of hepatic metabolism and disposition of Ba
At a higher dose given through i.p.v. bolus administration, hepatic extraction was significantly reduced, as expected. However, it is necessary to address that intra-portal vein bolus with high dose of Ba may be less likely to follow physiological condition since intestinal absorption should be a process in which the introduction of substrates to the liver is slow and gradual. Therefore, we further conducted the i.p.v. and i.v. infusion study to investigate the hepatic metabolism and disposition of Ba at steady state. At both high and low doses, CssBa and AUCBa for i.v. administration were substantially higher than those for i.p.v. administration, demonstrating significant hepatic extraction of Ba. The plasma concentrations of glu/sul and AUCglu/sul for two different infusion pathways were basically similar, which further suggested that the capability of hepatic and extrahepatic glucuronidation and sulfation of Ba at steady state were comparable. The amount of glu/sul biliary secretion was similar for the two infusion pathways at high dose, but more metabolites were secreted via the bile at low doses than those at higher doses. Two possible mechanisms might be responsible for such observation. Firstly, glu/sul metabolites formed in hepatic cells were preferentially transported to the apical side (into the bile) at their low cellular concentrations. Such directional preference of BG and BS was also found in our previous in vitro study. During the permeation of Ba through Caco-2 cell monolayers, BG and BS formed in the cell were preferentially transported to the basal side and apical side, respectively. We also found that the preferential transport was dose-dependent (15). Although the underlying mechanism needs further investigation, it is suggested to be related to the different affinity and capacity of transporters to the metabolites between the apical and basal sides. Secondly, it also may be due to the competition of disposition of glu/sul among the liver, intestine, and kidney. We found that 55–65% of the dose of glu/sul was secreted via the bile after i.v. or i.p.v. bolus administration. Thus, an appreciable amount of glu/sul should be disposed via those organs such as the intestine and kidney. For example, Akao et al. (24) and our previous study using a rat intestinal perfusion model demonstrated the secretion of BG in the intestine. Kidney disposition of conjugates of Ba was also reported (25). Therefore, it is probable that different organs may show their preference to the uptake of conjugates, and the preference may also vary with different plasma concentrations of the conjugates.
In summary, the current study demonstrated a significant hepatic extraction of Ba, and an efficient phase II metabolism and transporter coupling in the liver substantially contribute to the first-pass effect of Ba after oral administration. Scutellariae radix containing an appreciable amount of Ba is popularly used in Asian countries as herbal medicine for remedying inflammatory and bacteria-related ailments. It is highly possible that Ba could be co-administrated with other anti-inflammatory and antibiotics. In such cases, there might be potential competition between Ba and non-steroidal anti-inflammatory drugs (NSAIDS) such as acetaminophen, ibuprofen or naproxen, etc. on phase II metabolic enzymes (26) or potential competition between Ba conjugates and NSAIDS conjugates on MRPs (27,28), as well as potential competition between Ba conjugates and antibiotics such as beta-lactam antibiotics on OATPs (29). All of those interactions may lead to the alteration of the clinical pharmacokinetic profiles of Western drug and should be taken into consideration in designing a therapeutic regimen.
Acknowledgment
The authors are grateful for CUHK 478607 from the Research Grants Council of the Hong Kong SAR, China.
References
- 1.Yang D, Hu H, Huang S, Chaumont JP, Millet J. Study on the inhibitory activity, in vitro, of baicalein and baicalin against skin fungi and bacteria. Zhongyaocai. 2000;23:272–4. [PubMed] [Google Scholar]
- 2.Wakabayashi I. Inhibitory effects of baicalein and wogonin on lipopolysaccharide-induced nitric oxide production in macrophages. Pharmacol Toxicol. 1999;84:288–91. doi: 10.1111/j.1600-0773.1999.tb01496.x. [DOI] [PubMed] [Google Scholar]
- 3.Hong T, Jin GB, Cho S, Cyong JC. Evaluation of the anti-inflammatory effect of baicalein on dextran sulfate sodium-induced colitis in mice. Planta Med. 2002;68:268–71. doi: 10.1055/s-2002-23143. [DOI] [PubMed] [Google Scholar]
- 4.Kimata M, Shichijo M, Miura T, Serizawa I, Inagaki N, Nagai H. Effects of luteolin, quercetin and baicalein on immunoglobulin E-mediated mediator release from human cultured mast cells. Clin Exp Allergy. 2000;30:501–8. doi: 10.1046/j.1365-2222.2000.00768.x. [DOI] [PubMed] [Google Scholar]
- 5.Shao ZH, Vanden Hoek TL, Qin Y, Becker LB, Schumacker PT, Li CQ, et al. Baicalein attenuates oxidant stress in cardiomyocytes. Am J Physiol. 2002;282:H999–1006. doi: 10.1152/ajpheart.00163.2001. [DOI] [PubMed] [Google Scholar]
- 6.Evers DL, Chao CF, Wang X, Zhang Z, Huong SM, Huang ES. Human cytomegalovirus-inhibitory flavonoids: studies on antiviral activity and mechanism of action. Antivir Res. 2005;68:124–34. doi: 10.1016/j.antiviral.2005.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brinkworth RI, Stoermer MJ, Fairlie DP. Flavones are inhibitors of HIV-1 proteinase. Biochem Biophys Res Commun. 1992;188:631–7. doi: 10.1016/0006-291X(92)91103-W. [DOI] [PubMed] [Google Scholar]
- 8.Ciesielska E, Gwardys A, Metodiewa D. Anticancer antiradical and antioxidative actions of novel antoksyd S and its major components, baicalin and baicalein. Anticancer Res. 2002;22:2885–91. [PubMed] [Google Scholar]
- 9.Lee JH, Li YC, Ip SW, Hsu SC, Chang NW, Tang NY, et al. The role of Ca2+ in baicalein-induced apoptosis in human breast MDA-MB-231 cancer cells through mitochondria- and caspase-3-dependent pathway. Anticancer Res. 2008;28:1701–11. [PubMed] [Google Scholar]
- 10.Li-Weber M. New therapeutic aspects of flavones: the anticancer properties of Scutellaria and its main active constituents Wogonin, Baicalein and Baicalin. Cancer Treat Rev. 2009;35:57–68. doi: 10.1016/j.ctrv.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 11.Akao T, Kawabata K, Yanagisawa E, Ishihara K, Mizuhara Y, Wakui Y, et al. Baicalin, the predominant flavone glucuronide of Scutellariae radix, is absorbed from the rat gastrointestinal tract as the aglycone and restored to its original form. J Pharm Pharmacol. 2000;52:1563–8. doi: 10.1211/0022357001777621. [DOI] [PubMed] [Google Scholar]
- 12.Lai MY, Hsiu SL, Tsai SY, Hou YC, Chao PD. Comparison of metabolic pharmacokinetics of baicalin and baicalein in rats. J Pharm Pharmacol. 2003;55:205–9. doi: 10.1211/002235702522. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L, Lin G, Chang Q, Zuo Z. Role of intestinal first-pass metabolism of baicalein in its absorption process. Pharm Res. 2005;22:1050–8. doi: 10.1007/s11095-005-5303-7. [DOI] [PubMed] [Google Scholar]
- 14.Zhang L, Lin G, Zuo Z. Involvement of UDP-glucuronosyltransferases in the extensive liver and intestinal first-pass metabolism of flavonoid baicalein. Pharm Res. 2007;24:81–9. doi: 10.1007/s11095-006-9126-y. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Lin G, Kovács B, Jani M, Krajcsi P, Zuo Z. Mechanistic study on the intestinal absorption and disposition of baicalein. Eur J Pharm Sci. 2007;31:221–31. doi: 10.1016/j.ejps.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Walle UK, Galijatovic A, Walle T. Transport of the flavonoid chrysin and its conjugated metabolites by the human intestinal cell line Caco-2. Biochem Pharmacol. 1999;58:431–8. doi: 10.1016/S0006-2952(99)00133-1. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Hu M. Absorption and metabolism of flavonoids in the Caco-2 cell culture model and a perused rat intestinal model. Drug Metab Dispos. 2002;30:370–7. doi: 10.1124/dmd.30.4.370. [DOI] [PubMed] [Google Scholar]
- 18.Chen J, Lin H, Hu M. Metabolism of flavonoids via enteric recycling: role of intestinal disposition. J Pharmacol Exp Ther. 2003;304:1228–35. doi: 10.1124/jpet.102.046409. [DOI] [PubMed] [Google Scholar]
- 19.Zhang L, Lin G, Zuo Z. High-performance liquid chromatographic method for simultaneous determination of baicalein and baicalein 7-glucuronide in rat plasma. J Pharm Biomed Anal. 2004;36:637–41. doi: 10.1016/j.jpba.2004.07.024. [DOI] [PubMed] [Google Scholar]
- 20.Leuthold S, Hagenbuch B, Mohebbi N, Wagner CA, Meier PJ, Stieger B. Mechanisms of pH-gradient driven transport mediated by organic anion polypeptide transporters. Am J Physiol Cell Physiol. 2009;296:C570–82. doi: 10.1152/ajpcell.00436.2008. [DOI] [PubMed] [Google Scholar]
- 21.Gui C, Miao Y, Thompson L, Wahlgren B, Mock M, Stieger B, et al. Effect of pregnane X receptor ligands on transport mediated by human OATP1B1 and OATP1B3. Eur J Pharmacol. 2008;584:57–65. doi: 10.1016/j.ejphar.2008.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerk PM, Vore M. Regulation of expression of the multidrug resistance-associated protein 2 (MRP2) and its role in drug disposition. J Pharmacol Exp Ther. 2002;302:407–15. doi: 10.1124/jpet.102.035014. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Wolkoff AW, Morris ME. Flavonoids as a novel class of human organic anion-transporting polypeptide OATP1B1 (OATP-C) modulators. Drug Metab Dispos. 2005;33:1666–72. doi: 10.1124/dmd.105.005926. [DOI] [PubMed] [Google Scholar]
- 24.Akao T, Sakashita Y, Hanada M, Goto H, Shimada Y, Terasawa K. Enteric excretion of baicalein, a flavone of Scutellariae radix, via glucuronidation in rat: involvement of multidrug resistance-associated protein 2. Pharm Res. 2004;21:2120–6. doi: 10.1023/B:PHAM.0000048205.02478.b5. [DOI] [PubMed] [Google Scholar]
- 25.Lai MY, Hsiu SL, Chen CC, Hou YC, Chao PD. Urinary pharmacokinetics of baicalein, wogonin and their glycosides after oral administration of scutellariae radix in humans. Biol Pharm Bull. 2003;26:79–83. doi: 10.1248/bpb.26.79. [DOI] [PubMed] [Google Scholar]
- 26.Kuehl GE, Lampe JW, Potter JD, Bigler J. Glucuronidation of nonsteroidal anti-inflammatory drugs: identifying the enzymes responsible in human liver microsomes. Drug Metab Dispos. 2005;33:1027–35. doi: 10.1124/dmd.104.002527. [DOI] [PubMed] [Google Scholar]
- 27.Xiong H, Turner KC, Ward ES, Jansen PL, Brouwer KL. Altered hepatobiliary disposition of acetaminophen glucuronide in isolated perfused livers from multidrug resistance-associated protein 2-deficient TR(−) rats. J Pharmacol Exp Ther. 2000;295:512–8. [PubMed] [Google Scholar]
- 28.Manautou JE, de Waart DR, Kunne C, Zelcer N, Goedken M, Borst P, Elferink RO. Altered disposition of acetaminophen in mice with a disruption of the Mrp3 gene. Hepatology. 2005;42:1091–8. doi: 10.1002/hep.20898. [DOI] [PubMed] [Google Scholar]
- 29.Nakakariya M, Shimada T, Irokawa M, Maeda T, Tamai I. Identification and species similarity of OATP transporters responsible for hepatic uptake of beta-lactam antibiotics. Drug Metab Pharmacokinet. 2008;23:347–55. doi: 10.2133/dmpk.23.347. [DOI] [PubMed] [Google Scholar]