

Abstract
The cluster [(Tp)MoFe3S4(PEt3)3]1+ containing the cubane-type [MoFe3(μ3-S)4]2+ reduced core undergoes facile ligand substitution reactions at the iron sites leading to an extensive set of mono- and disubstituted species [(Tp)MoFe3S4(PEt3)3-nLn]1-n with L = halide, N3-, PhS-, PhSe-, R3SiO-, and R3SiS- and n = 1 and 2. Structures of 10 members of the set are reported. For two representative clusters, Curie behavior at 2-20 K indicates a spin-quintet ground state. Zero-field Mössbauer spectra consist of two doublets in a 2:1 intensity ratio. 57Fe isomer shifts are consistent with the mean oxidation state Fe32.33+ arising from electron delocalization of the mixed-valence oxidation state description [Mo3+Fe3+Fe2+2]. Reaction of [(Tp)MoFe3S4(PEt3)2Cl] with (Me3Si)2S affords [(Tp)MoFe3S4(PEt3)2(SSiMe3)], a likely first intermediate in the formation of the tricluster compound {[(Tp)MoFe3S4(PEt)2]3S}(BPh4) from the reaction of [(Tp)MoFe3S4(PEt3)3](BPh4) and NaSSiMe3 in THF. The tricluster consists of three cluster units bound to a central μ3-S atom in a species of overall C3 symmetry. Relatively few clusters in the [MoFe3S4]2+ oxidation state have been prepared compared to the abundance of clusters in the oxidized [MoFe3S4]3+ state. This work is the first comprehensive study of the [MoFe3S4]2+ state, one conspicuous feature of which is its ability to bind hard and soft σ-donors and strong to weak π-acid ligands. (Tp = tris(pyrazolyl)hydroborate(1-))
Introduction
Heterometal clusters containing cubane-type [MFe3(μ3-S)4]z core units with M = Ni,1-4 Mo,5-10 and V5,11-14 continue to occupy a principal role in synthetic experiments directed toward attainment of the catalytic centers in enzymes such as carbon monoxide dehydrogenase and nitrogenase.15-18 The newer generation of M = Mo and V clusters prepared in this laboratory features a heterometal site protected by coordination of the facial tridentate ligand tris(pyrazolyl)hydroborate (Tp) such that reactivity is directed toward the tetrahedral iron sites. This allows a number of significant transformations,5,6,8 including the simultaneous reduction and substitution of [(Tp)MoFe3S4Cl3]1- to form [(Tp)MoFe3S4(PEt3)3]1+, which is a precursor of other cluster types. The phosphine cluster can be fully substituted with anionic ligands such as thiolate to give [(Tp)MoFe3S4(SR)3]2-, reduced with loss of phosphine to the edge-bridged double cubane [(Tp)2Mo2Fe6S8(PEt)4], and transformed by sulfide incorporation to [(Tp)2Mo2Fe6S9(SH)2]3-, whose core has the topology of the PN cluster of nitrogenase. A similar reaction sequence has been accomplished with vanadium-containing clusters, culminating in the PN-type cluster [(Tp)2V2Fe6S9(SH)2]3-. 12-14
The foregoing transformations, as well as those of cubane-type [Fe4S4]1+,0 clusters,19,20 are enabled by the lability of tertiary phosphine binding. For the oxidized case, this has allowed the formation of 3:1 site-differentiated clusters with a wide variety of anionic ligands by substitution reaction 1, and by reductive cleavage of disulfides, diselenides, or iodine in reaction 2 utilizing the indicated double cubane (or the tetracubane [Fe16S16(PPri3)8]).20 Reactions are usually performed in, and the neutral products are always crystallized from, THF or benzene to avoid ligand scrambling via charged clusters whose formation is disfavored by low dielectric solvents.
| (1) |
| (2) |
The present investigation was undertaken to determine if neutral mixed-ligand clusters with the heterometal [MoFe3S4]2+ core are accessible by the same or similar means. Suitably substituted clusters are potentially amenable to coupling reactions leading to the formation of covalently bridged [MoFe3S4]2 and [MoFe3S4]/[Fe4S4] double cubanes. These clusters are of much interest in their own right and also have the attractive feature of approaching the MoFe7S9X core nuclearity and composition of the FeMo-cofactor cluster of nitrogenase.21,22 As is widely recognized, the MoFe3S3 fragment of these clusters closely resembles a significant portion of the molybdenum coordination environment in the iron-molybdenum cofactor of nitrogenase.17
The clusters noted above with M2Fe6S8 and M2Fe6S9 cores (M = Mo, V) provide prima facie evidence for the existence of at least two structural types of octanuclear heterometal clusters. Here we report methods affording the clusters [(Tp)MoFe3S4(PEt3)2L] and related species, their structures, representative electronic properties, and an example of cluster coupling.
Experimental Section
Preparation of Compounds
All reactions and manipulations were performed under a pure dinitrogen atmosphere using either Schlenk techniques or an inert atmosphere box. Solvents were passed through an Innovative Technology solvent purification system prior to use. Benzene was distilled from sodium benzophenone ketyl. All solvents were further deoxygenated before use. Solvent removal steps were performed in vacuo; filtrations were through Celite. Extraction and crystallization steps were performed in the presence of excess PEt3 in order to promote cluster stability, apparently by repressing dissociation of bound phosphine. All new compounds were identified by combinations of 1H NMR spectroscopy, X-ray crystal structure determinations, and elemental analyses (Midwest Microlab). Neutral compounds are soluble in non-polar or weakly polar solvents such as benzene, toluene, and THF and are extremely air-sensitive and must be handled accordingly.
[(Tp)MoFe3S4(PEt3)2F]
To a dark brown solution of 62 mg (0.049 mmol) of [(Tp)MoFe3S4(PEt3)3](BPh4)6 in 4 mL of THF was added 16 mg (0.051 mmol) of Bu4NF·3H2O in 1 mL of THF. The reaction mixture was stirred for 10 min, filtered, and the brown filtrate was reduced to dryness. The residue was extracted with 5 mL of THF (containing 10 mg (0.085 mmol) of PEt3) and layered with hexanes. The product was obtained as 30 mg (72%) of black block-like crystals. 1H NMR (THF-d8): δ 20.08 (2), 19.0 (br, 2), 9.88 (1), 8.68 (1), 5.22 (2), 3.10 (6), 2.50 (24). The presence of fluoride in this compound was demonstrated by an NMR method (see below).
[(Tp)MoFe3S4(PEt3)2Cl]
To a dark brown solution of 258 mg (0.20 mmol) of [(Tp)MoFe3S4(PEt3)3](BPh4) in 5 mL of THF was added a solution of 79 mg (0.21 mmol) of Ph4PCl in 5 mL of acetonitrile. The reaction mixture was stirred for 2 h, filtered to remove a white solid, and the brown filtrate was reduced to dryness. The residue was extracted with 10 mL of THF (containing 20 mg (0.17 mmol) of PEt3) and layered with hexanes. The product was isolated as 130 mg (74%) of black needles. 1H NMR (THF-d8): δ 19.30 (2), 18.5 (br, 2), 9.74 (1), 9.20 (1), 5.63 (2), 3.09 (6), 2.74 (18), 2.54 (6). Anal. Calcd. for C21H40BClFe3MoN6P2S4: C, 28.78; H, 4.60; N, 9.59. Found: C, 31.39; H, 4.74; N, 9.35.
[(Tp)MoFe3S4(PEt3)2Br]
The preceeding procedure with 113 mg (0.088 mmol) of [(Tp)MoFe3S4(PEt3)3](BPh4) and 41 mg (0.10 mmol) of Ph4PBr was employed. The residue was extracted with 20 mL of THF (containing 10 mg (0.085 mmol) of PEt3). The product was isolated as 53 mg (65%) of black needles. 1H NMR (THF-d8): δ 19.05 (2), 17.9 (br, 2), 9.89 (1) 9.39 (1), 5.67 (2) 3.91 (6), 3.27 (6), 2.87 (18). Anal. Calcd. for C21H40BBrFe3MoN6P2S4: C, 27.39; H, 4.38; N, 9.13. Found: C, 27.74; H, 4.29; N, 9.10.
[(Tp)MoFe3S4(PEt3)2I]
To a suspension of 75 mg (0.044 mmol) of [(Tp)2Mo2Fe6S8(PEt3)4]6 in 5 mL of THF was added 16 mg (0.063 mmol) of I2 and 10 mg of PEt3. The dark brown reaction mixture was stirred overnight, filtered, and the filtrate was layered with hexanes. The product was obtained as 58 mg (68%) of black plate-like crystals. 1H NMR (THF-d8): δ 18.84 (2), 17.5 (br, 2), 10.13 (1), 9.78 (1), 5.70 (2), 4.35 (6), 3.03 (24). Anal. Calcd. for C21H40BFe3IMoN6P2S4: C, 26.06; H, 4.17; N, 8.68. Found: C, 26.42: H, 4.03; N, 8.38.
[(Tp)MoFe3S4(PEt3)2(N3)]
To a solution of 75 mg (0.059 mmol) of [(Tp)MoFe3S4(PEt3)3](BPh4) in 5 mL of THF was added a solution of 16 mg (0.056 mmol) of (Bu4N)(N3) in 1 mL of THF. The reaction mixture was stirred overnight, filtered, and the filtrate reduced to dryness. The residue was extracted with benzene (with 10 mg of PEt3). Solvent removal followed by crystallization of the residue from THF/hexanes afforded the product as 30 mg (53%) of black plate-like crystals. 1H NMR (C6D6): δ 18.7 (br, 2), 18.59 (2), 9.53 (1), 8.19 (1), 5.37 (2), 3.45 (6), 2.55 (24). Anal. Calcd. for C21H40BFe3MoN9P2S4: C, 28.56; H, 4.57; N, 14.27. Found: C, 29.81; H, 4.61; N, 14.80.
[(Tp)MoFe3S4(PEt3)2(OSiMe3)]
To a solution of 110 mg (0.086 mmol) of [(Tp)MoFe3S4(PEt3)3](BPh4) in 5 mL of THF was added 10 mg (0.089 mmol) of NaOSiMe3 in 1 mL of acetonitrile. The reaction mixture was stirred for 4 h, filtered, and the dark brown filtrate was reduced to dryness. The residue was extracted with benzene (with 10 mg of PEt3) and the solvent was removed to give the product as 59 mg (73%) of a brown solid. 1H NMR (C6D6): δ 19.07 (2), 18.6 (br, 2), 9.09 (1), 8.41 (1), 8.17 (9), 4.86 (2), 2.75 (6), 2.13 (18), 1.95 (6).
[(Tp)MoFe3S4(PEt3)2(OSiPh3)]
To a solution of 53 mg (0.042 mmol) of [(Tp)MoFe3S4-(PEt3)3](BPh4) in 5 mL of THF was added a solution of 14 mg (0.047 mmol) of NaOSiPh3 in 1 mL of THF. The reaction mixture was stirred overnight, filtered, and the dark brown filtrate was reduced to dryness. The residue was extracted with 10 mL of benzene (containing 10 mg of PEt3) and solvent was removed to give the product as 27 mg (58%) of brown solid. 1H NMR (C6D6): δ 19.29 (2), 18.9 (br, 2), 10.9 (br, 6), 9.22 (1), 8.95 (6), 8.15 (1), 7.64 (3), 4.89 (2), 2.55 (6), 2.21 (18), 1.70 (6). Anal. Calcd. for C39H55BFe3MoN6OP2S4Si: C, 41.96; H, 4.97; N, 7.53. Found: C, 42.01; H, 4.93; N, 7.55.
[(Tp)MoFe3S4(PEt3)2(SPh)]
Method A
To a solution of 43 mg (0.034 mmol) of [(Tp)MoFe3S4(PEt3)3](BPh4) in 5 mL of THF was added 9.0 mg (0.038 mmol) of (Et4N)(SPh) in 1 mL of acetonitrile. The reaction mixture was stirred overnight and filtered. The dark brown filtrate was reduced to dryness and the residue was extracted with benzene (containing 10 mg of PEt3). Volatiles were removed to afford the product as 23 mg (70%) of brown solid. 1H NMR (C6D6): δ 27.38 (2), 18.0 (br, 2), 16.98 (2), 8.85 (1), 8.35 (1), 5.46 (2), 3.77 (12), 2.38 (18), -20.47 (2), -23.39 (1).
Method B
To a dark brown solution of 68 mg (0.040 mmol) of [(Tp)2Mo2Fe6S8(PEt3)4]6 in 8 mL of THF was added 10 mg (0.046 mmol) of PhSSPh and 10 mg of PEt3. The reaction mixture was stirred overnight and filtered. The dark brown filtered was layered with hexanes. The product was collected as 32 mg (42%) of black plate-like crystals; the 1H NMR spectrum of this material is identical to that of the product from Method A.
[(Tp)MoFe3S4(PEt3)2(SePh)]
Method B for the benzenethiolate cluster was followed on a 0.043 mmol scale with use of PhSeSePh. The product was isolated as 70 mg (82%) of brown crystalline solid. 1H NMR (C6D6): δ 23.67 (2), 17.8 (br, 2), 16.82 (2), 8.94 (1), 8.47(1), 5.47 (2), 3.93 (12), 2.46 (18), -8.47 (2), -14.54(1). Anal. Calcd. for C27H45BFe3MoN6P2S4Se: C, 32.52; H, 4.55; N, 8.43. Found: C, 32.33; H, 4.38; N, 8.50.
[(Tp)MoFe3S4(PEt3)2(SSiMe3)]
To a solution of 20 mg (0.023 mmol) of [(Tp)MoFe3S4(PEt3)2Cl] in 1 mL of THF was added 39 mg (0.32 mmol) of (Me3Si)2S in 3 mL benzene. The reaction mixture was stirred overnight, filtered, and the filtrate was taken to dryness. The residue was extracted with benzene (with 10 mg of PEt3), volatiles were removed, and the solid was crystallized from THF/hexanes to afford the product as 15 mg (70%) of black needles. 1H NMR (C6D6): δ 17.7 (br, 2), 17.28 (2), 8.86 (1), 8.69 (1), 7.75 (9), 5.38 (2), 3.22 (6), 2.31(24).
[(Tp)MoFe3S4(PEt3)2(SSiPri3)]
A solution of 90 mg (0.070 mmol) of [(Tp)MoFe3S4(PEt3)3](BPh4) in 5 mL of THF was added 18 mg (0.085 mmol) of NaSSiPri3 in 1 mL of THF. The reaction mixture was stirred overnight, filtered, and the filtrate reduced to dryness. The residue was extracted with 10 mL of benzene (containing 10 mg of PEt3) and the extract was taken to dryness. The residue was recrystallized from THF/hexanes to give the product as 36 mg (50%) of a black needle-like solid. 1H NMR (THF-d8): δ 17.96 (2), 17.7 (br, 2), 14.8 (br, 3), 9.24 (1), 9.12 (1) 6.81 (18), 5.62 (2), 3.38 (12), 2.46 (18).
[(Tp)MoFe3S4(PEt3)2(SSiPh3)]
The preceding method was used with 58 mg (0.045 mmol) of [(Tp)MoFe3S4(PEt3)3](BPh4), 20 mg (0.047 mmol) of (Et4N)(SSiPh3) in 1 mL of acetonitrile, and a reaction time of 4 h. The product was obtained as 26 mg (50%) of brown solid. 1H NMR (C6D6): δ 17.8 (br, 2), 17.37 (2), 10.39 (6), 8.95 (1), 8.70 (6), 8.58 (1), 7.67 (3) 5.41 (2), 2.94 (6), 2.54 (6), 2.36 (18).
(Ph4P)[(Tp)MoFe3S4(PEt3)Cl2]
The procedure for the monochloride cluster was used with 43 mg (0.034 mmol) of [(Tp)MoFe3S4(PEt3)3](BPh4), 26 mg (0.068 mmol) of Ph4PCl, and a reaction time of 4 h. The residue was washed with 3 mL of THF and extracted with 10 mL of acetonitrile. The residue from solvent removal was recrystallized from acetonitrile/ether to afford the product as 14 mg (36%) of black needles. Anal. Calcd. for C39H45BCl2Fe3MoN6P2S4: C, 41.34; H, 4.00; N, 7.42. Found: C, 41.60; H, 4.04; N, 7.82.
(Ph4P)[(Tp)MoFe3S4(PEt3)Br2]
To a solution of 73 mg (0.057 mmol) of [(Tp)MoFe3S4(PEt3)3](BPh4) in 3 mL of THF was added 23 mg (0.056 mmol) of Ph4PBr in 3 mL of acetonitrile. The reaction mixture was stirred for 2 h, 23 mg of Ph4PBr in 3 mL of acetonitrile was added, and stirring was continued for 3 h. The mixture was filtered and the filtrate was taken to dryness. The residue was recrystallized from acetonitrile/ether to afford the product as 53 mg (76%) of black crystalline solid.
(Ph4P)[(Tp)MoFe3S4(PEt3)ClBr]
To a solution of 49 mg (0.055 mmol) of [(Tp)MoFe3S4(PEt3)2Cl] in 3 mL of THF was added a solution of 23 mg (0.055 mmol) of Ph4PBr in 3 mL of acetonitrile. The reaction mixture was stirred for 1 h, filtered, and the filtrate reduced to dryness. The residue was washed with THF and extracted with 10 mL of acetonitrile (containing 10 mg of PEt3). The residue from solvent removal of the extract was recrystallized from dichloromethane/hexanes to yield the product as 33 mg (50%) of brown needles. Calcd. for C39H45BBrClFe3MoN6P2S4: C, 39.78; H, 3.85; H, 7.14. Found: C, 39.23; H, 3.80: N, 7.42.
(Ph4P)2[(Tp)MoFe3S4Cl3]
A solution of 67 mg (0.052 mmol) of [(Tp)MoFe3S4(PEt3)3](BPh4) in 1 mL of THF was treated with a solution of 57 mg (0.15 mmol) of Ph4PCl in 5 mL of acetonitrile. A white precipitate formed immediately. The reaction mixture was stirred overnight, filtered, and the filtrate reduced to dryness. The black residue was washed with ether and recrystallized from acetonitrile/ether. The product was isolated as 35 mg (72%) of black needles. The 1H NMR spectrum of the anion in CD3CN (δ 19.03 (1), 18.1 (br, 1), 4.88 (1)) is identical to that of the Bu4N+ salt.5
{[(Tp)MoFe3S4(PEt3)2]3S}(BPh4)
To a solution of 31 mg (0.024 mmol) of [(Tp)MoFe3S4(PEt3)3](BPh4) in 3 mL of THF was added 2.5 mg (0.020 mmol) of NaSSiMe323 in 1 mL of THF. The reaction mixture was stirred for 3 h, filtered, and the filtrate was reduced to dryness. The residue was extracted with benzene (containing 10 mg of PEt3) and the solvent removed to give a brown solid. This material was twice recrytallized from THF/hexanes to produce hexagonally shaped black crystals (4.5 mg, 20%). 1H NMR (CD3CN): δ 17.2 (br), 13.70, 13.17, 7.39, 7.29, 7.01, 6.86, 3.28. Low solubility hampered accurate signal integration.
Fluorine Analysis
The use of a hydrated fluoride source in the preparation of the compound described as [(Tp)MoFe3S4(PEt3)2F] raises the possibility of hydroxide rather than fluoride ligation. Fluorine analysis has been performed using a previously described NMR method based on the minimal reaction Fe-F + Me3SiCl →Fe-Cl + Me3SiF.8 A solution of the 0.0070 mmol of the cluster and 1.98 mmol of Me3SiCl was allowed to react for 1 hr, 0.057 mmol of Ph3SiF was added, and the mixture was analyzed by 19F NMR integration of the peaks of the internal standard Ph3SiF (δ -171.1) and Me3SiF (δ -158.5). In two experiments the fluorine liberated from cluster was found to be 91% of that expected. We conclude that the cluster is correctly formulated.
In the following sections, clusters are numerically designated as in the Chart.
Chart. Abbreviations and Designation of Clusters.

X-ray Structure Determinations
The structures of the eleven compounds in Table 1 were determined. Diffraction-quality crystals were obtained by diffusion of hexanes into THF (2-6, 11-13, [18](BPh4)) or dichloromethane ((Ph4P)[16]) solutions or by ether diffusion into an acetonitrile solution ((Ph4P)[14]). Crystal mounting and data collections were performed as described26 on a Siemens (Bruker) SMART CCD diffractometer using Mo Kα radiation. Data integration was performed with SAINT, which corrects for Lorentz polarization and decay. Space groups were assigned unambiguously by analysis of symmetry and systematic absences determined by XPREP and were further checked by PLATON. All structures were solved and refined against all data in the 2θ ranges by full-matrix least-squares on F2 using SHELXTL. Hydrogen atoms at idealized positions were included in final refinements. Refinement details and explanations (wherever necessary) are included in individual CIF files. Crystallographic data and final agreement factors are given in Table 1.27
Table 1. Crystallographic Data for [MoFe3S4] Clusters at 100 K a.
| 2·½C4H8O | 3 | 4 | 5 | 6 | 11 | 12 | |
|---|---|---|---|---|---|---|---|
| formula | C23H44BFFe3MoN6O0.50P2S4 | C21H40BClFe3MoN6P2S4 | C21H40BBrFe3MoN6P2S4 | C21H40BFe3IMoN6P2S4 | C42H80B2Fe6Mo2N18P4S8 | C48H98B2Fe6Mo2N12P4S10Si2 | C30H61BFe3MoN6P2S5Si |
| formula weight | 896.12 | 876.52 | 920.98 | 967.97 | 1766.20 | 1892.64 | 1030.48 |
| crystal system | monoclinic | monoclinic | monoclinic | orthorhombic | triclinic | triclinic | triclinic |
| space group | C2/c | P21/n | P21/n | Pbca | P-1 | P-1 | P-1 |
| Z | 8 | 4 | 4 | 8 | 2 | 2 | 2 |
| a, (Å) | 35.518(6) | 14.760(3) | 14.585(6) | 15.2679(13) | 10.4374(5) | 11.683 (2) | 10.5481 (5) |
| b, (Å) | 9.8685(16) | 12.367(3) | 12.409(5) | 16.2173(13) | 15.5706(7) | 18.803 (3) | 11.1605 (5) |
| c, (Å) | 24.544(5) | 19.127(4) | 18.896(8) | 28.031(2) | 23.0073(10) | 22.516 (4) | 22.2512 (11) |
| α, deg | 90 | 90 | 90 | 90 | 75.923(1) | 65.571 (3) | 80.553 (3) |
| β, deg | 125.912(6) | 106.320(4) | 105.50(2) | 90 | 82.991(1) | 75.347 (4) | 84.709 (3) |
| γ, deg | 90 | 90 | 90 | 90 | 73.169(1) | 89.576 (4) | 61.897 (2) |
| V, Å3 | 6967(2) | 3350.6(13) | 3296(2) | 6940.6(10) | 3466.1(3) | 4330.2 (14) | 2278.96 (19) |
| dcalcd, g/cm3 | 1.709 | 1.738 | 1.856 | 1.853 | 1.692 | 1.452 | 1.502 |
| μ, mm-1 | 1.941 | 2.088 | 3.254 | 2.828 | 1.947 | 1.634 | 1.559 |
| 2θ range, deg | 3.34-51.42 | 3.10-51.74 | 3.98-52.34 | 3.94- 51.42 | 3.00- 51.44 | 3.62- 51.42 | 3.72- 51.92 |
| R1b (wR2c) | 0.0241 (0.0569) | 0.0558 (0.1757) | 0.0431 (0.1216) | 0.0318 (0.0728) | 0.0377 (0.0889) | 0.0686 (0.1406) | 0.0660 (0.2134) |
| GOF (F2) | 0.997 | 1.098 | 1.033 | 1.023 | 0.955 | 1.045 | 1.068 |
| 13 | (Ph4P)[14]·½C4H10O | (Ph4P)[16]·5CH2Cl2 | [18](BPh4) | ||||
|
| |||||||
| formula | C39H55BFe3MoN6P2S5Si | C41H45BCl2Fe3MoN6O0.50P2S4 | C83H100B2Br2Cl12Fe6Mo2N12P4S8 | C87H140B4Fe9Mo3N18P6S13 | |||
| formula weight | 1132.52 | 1165.21 | 2779.93 | 2874.48 | |||
| crystal system | triclinic | monoclinic | triclinic | hexagonal | |||
| space group | P-1 | P21/c | P-1 | P-3 | |||
| Z | 2 | 4 | 2 | 2 | |||
| a, (Å) | 10.6968 (4) | 17.361 (8) | 12.9300 (6) | 17.021(3) | |||
| b, (Å) | 10.9806 (4) | 26.451 (12) | 17.3950 (8) | 17.021(3) | |||
| c, (Å) | 23.6575 (10) | 12.831 (6) | 26.2891 (12) | 23.450 (5) | |||
| α, deg | 94.202 (2) | 90 | 90.582 (3) | 90 | |||
| β, deg | 99.012 (2) | 111.334 (6) | 92.433 (3) | 90 | |||
| γ, deg | 117.744 (1) | 90 | 111.480 (3) | 120 | |||
| V, Å3 | 2394.72 (16) | 5488 (4) | 5495.1 (4) | 5884 (2) | |||
| dcalcd, g/cm3 | 1.571 | 1.410 | 1.680 | 1.623 | |||
| μ, mm-1 | 1.492 | 1.342 | 2.265 | 1.742 | |||
| 2θ range, deg | 3.54-54.40 | 2.96-50.62 | 3.10-51.52 | 3.26-51.46 | |||
| R1b (wR2c) | 0.0573 (0.1488) | 0.0767 (0.2003) | 0.0898 (0.2113) | 0.0696 (0.1959) | |||
| GOF (F2) | 1.036 | 1.020 | 1.038 | 1.072 | |||
Mo Kα radiation (λ = 0.71073 Å).
R1 = Σ||Fo |− |Fc||/Σ|Fo|.
wR2 = {Σw(Fo2 - Fc2)2/ Σw(Fo2)2}1/2
Other Physical Measurements
1H NMR spectra were obtained with a Varian M400 spectrometer. 57Fe Mössbauer spectra were measured with a constant acceleration spectrometer. Data were analyzed with WMOSS software (WEB Research, Edina, MN); isomer shifts are referenced to iron metal at room temperature. Magnetic susceptibility data were collected with a Quantum Design SQUID magnetometer at 1 T and 2-300 K (MIT Center for Materials Science and Engineering).
Results and Discussion
Synthesis of Clusters
In the development of MoFe3S4 cluster chemistry, single cubane clusters have been nearly always isolated in the [MoFe3S4]3+ oxidation state stabilized by frequently employed thiolate or halide ligands at the iron sites.15,17,28 The potential E½ = -0.57 V vs. SCE for the couple [(Tp)MoFe3S4Cl3]1-/2- in acetonitrile, signifying facile oxidation of the reduced form, is one example of such stabilization.5 In several instances, chemical reduction of [MoFe3S4]3+ clusters has afforded isolable but oxidatively sensitive [MoFe3S4]2+ species. However, the initial preparation of the edge-bridged double cubane [(Cl4cat)2(Et3P)2Mo2Fe6S8(PEt3)4]29 followed by demonstration that tertiary phosphines are reductants of oxidized clusters25 has led to more direct syntheses of reduced clusters.6,7,10 These results show that stabililty of the [MoFe3S4]2+ oxidation state is clearly enhanced by phosphine coordination at the iron sites, irrespective of ligation at molybdenum.
The synthetic methods utilized in this investigation are summarized in Figure 1. The primary precursor is the tris(phosphine) cluster 1, readily obtainable from [(Tp)MoFe3S4Cl3]1- by reduction and ligand substitution with PEt3.6 Substitution reaction 3 (50-74%) and substrate reduction reaction 4 (42-82%) with the double cubane [(Tp)2Mo2Fe6S8(PEt3)4] afford monosubstituted [MoFe3S4]2+ clusters in the indicated yields, and find analogy with reactions 1 and 2 of [Fe4S4]1+ clusters. All reactions afford neutral clusters as brown or black air-sensitive solids that are soluble in low-polarity solvent media
Figure 1.
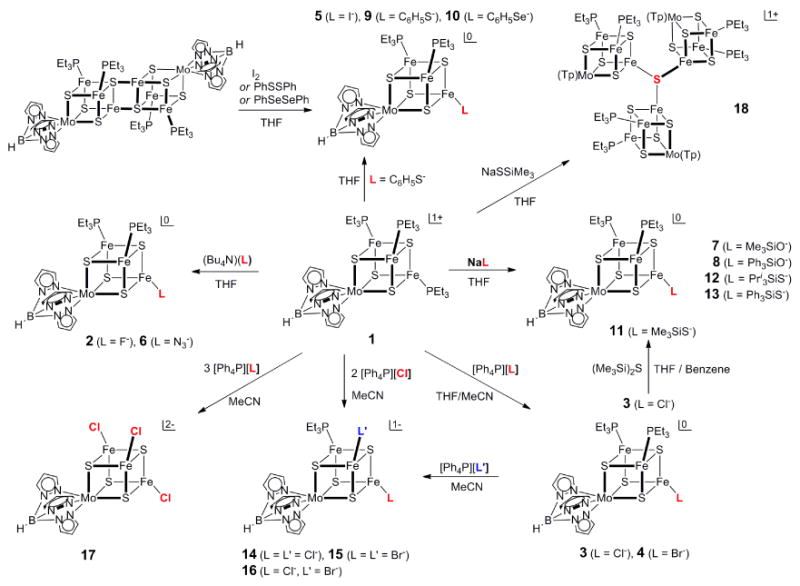
Scheme illustrating the synthesis of variously substituted single cubanes [(Tp)MoFe3S4(PEt3)3-n(L/L′)n]0/1- by monosubstitution (2-4, 6-8, 12,13), disubstitution (14-16), and trisubstitution (17) of cluster 1. Monosubstituted clusters are also formed by reductive substrate cleavage (5, 9, 10) with the double cubane [(Tp)2Mo2Fe6S8(PEt3)4]. Also shown is the formation of tricluster 18 from trimethylsilylthiolate cluster 11, which is accessible from 3 by reaction with (Me3Si)2S.
| (3) |
| (4) |
(THF, benzene) in which they do not undergo ligand exhange. These reactions are best conducted with excess phosphine to minimize phosphine dissociation and possible side reactions. Substitution reactions are readily followed by 1H NMR because cluster paramagnetism (see below) produces isotropically shifted pyrazolyl signals that are very sensitive to ligands at the iron sites. Thus in the complete substitution 1 → 17 in acetonitrile involving trigonally symmetric species, the three signals of the initial phosphine cluster (δ 7.35, 13.62, 17.1 (br)) are replaced by those of the product chloride cluster (δ 4.88, 18.1 (v br), 19.03). In monosubstitution reactions of 1 (Figure 1), the 2:1 inequivalence of pyrazolyl rings arising from idealized mirror symmetry of the product clusters is readily evident in 1H NMR spectra, as seen for the representative clusters 6 and 12 in Figure 2. Both show the three signals of the symmetry-related rings and two of the three signals of the ring in the mirror plane. The missing signal is either obscured by other resonances or broadened beyond detection. Also evident in the spectrum of 12 in particular is the diastereotopic splitting (0.47 ppm) of the phosphine methylene signals, an effect that is absent in the fully substituted clusters.
Figure 2.
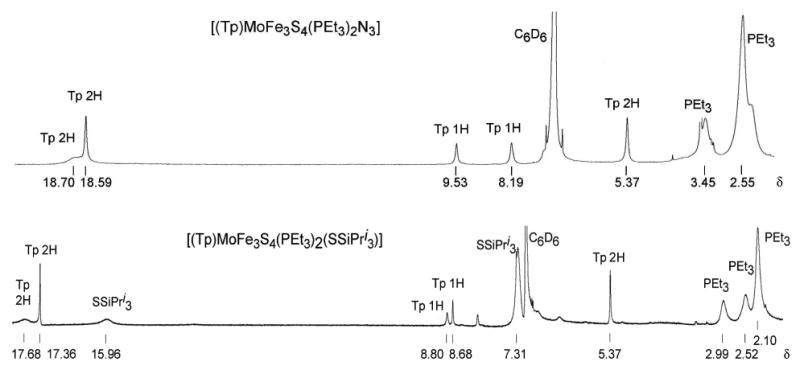
1H NMR spectra of representative single cubanes 6 and 12 in C6D6 solutions at ambient temperature; signal assignments are indicated.
Further cluster substitution has been achieved in the conversion of 1 to disubstituted 14-16 with two equivalents of ligand, and monosubstituted 3 and 4 to 14-16 with one equivalent of ligand. These anionic clusters have limited stability in acetonitrile but could be recrystallized to afford crystalline samples suitable for X-ray structure determinations.
Structural and Electronic Features
Structures of three monosubstituted clusters (3, 6, 11) are set out in Figure 3. Like other isoelectronic (Tp)MoFe3S4 clusters,5-7,9 they and clusters 4, 5, 12 and 13 display trigonally distorted octahedral coordination at molybdenum and distorted tetrahedral iron sites. The existence of disubstituted clusters with the same structural features is demonstrated by the structures of 14 and 16 in Figure 4. Core dimensions are practically invariant over the entire set of clusters. Bond distances are typified by the information for 3 and are collected in Table 2 together with terminal Fe-L structural data for the set.27 Cluster 16 has the singular property of three different ligands at the iron sites and therefore is chiral; the cluster as its Ph4P+ salt was, however, isolated as a racemate.
Figure 3.
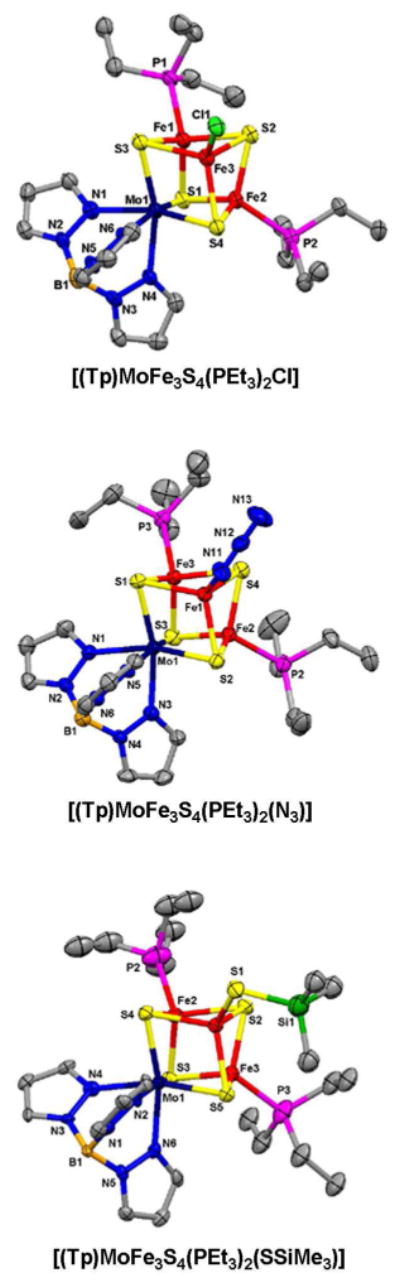
Structures of monosubstituted clusters [(Tp)MoFe3S4(PEt3)2L] with L = Cl-, N3-, and Me3SiS- shown with atom labeling schemes and 50% probability ellipsoids.
Figure 4.
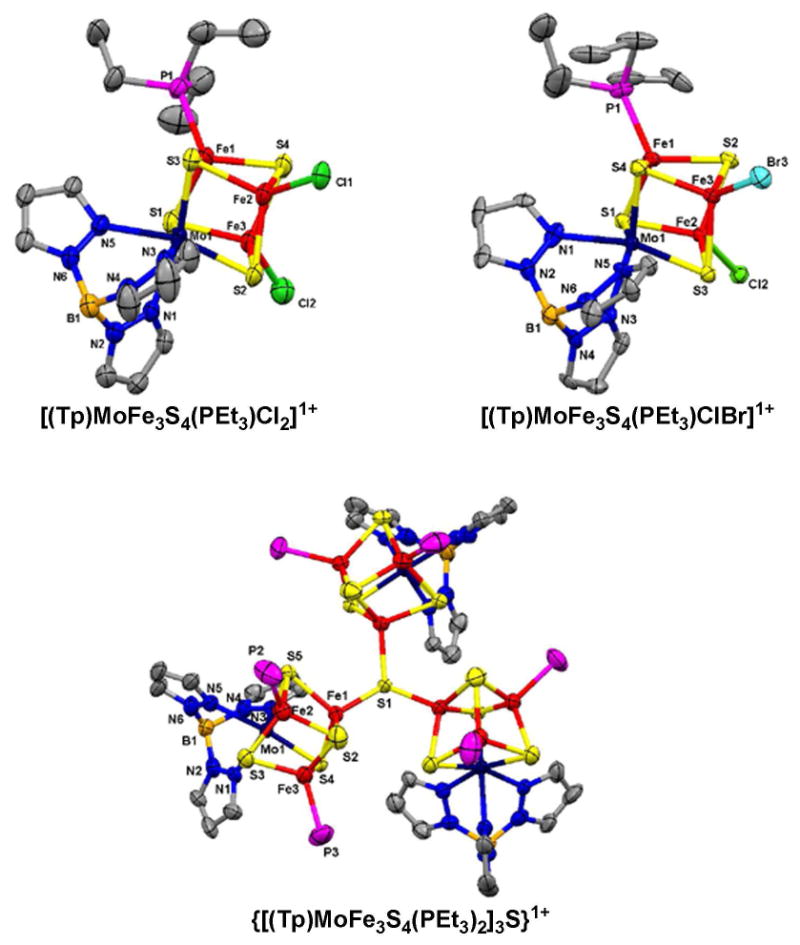
Structures of disubstituted clusters [(Tp)MoFe3S4(PEt3)LL′] with L = L′= Cl- and L = Cl-, L = Br- (upper) and the tricluster {[(Tp)MoFe3S4(PEt3)2]3S}1+ (lower) shown with atom labeling schemes and 50% probability ellipsoids. The tricluster has a crystallographically imposed C3 axis.
Table 2. Summary of Selected Structural Features of Clusters.
| cluster | bond distance (Å), angle (deg) |
|---|---|
| [(Tp)MoFe3S4(PEt3)2F] | Fe3-F1 1.853(2) |
| [(Tp)MoFe3S4(PEt3)2Cl]a | Mo-N 2.24[1], Mo-S 2,36[1], Mo-Fe 2,68[3], Fe-S 2.178(2)-2.318(2), Fe-Fe 2.61[3], Fe1-P1 2.350(2), Fe2-P2 2.276(2), Fe3-Cl1 2.243(2) |
| [(Tp)MoFe3S4(PEt3)2Br] | Fe3-Br1 2.352(1) |
| [(Tp)MoFe3S4(PEt3)2I] | Fe3-I1 2.568(1) |
| [(Tp)MoFe3S4(PEt3)2N3]b | Fe1-N11 1.943(4), Fe22-N21 1.945(4) |
| Fe1-N11-N12 133.0(3), Fe22-N21-N22 138.5(3) | |
| [(Tp)MoFe3S4(PEt3)2(SSiMe3)]b | Fe1-S1 2.263(3), Fe11-S11 2.275(3), Fe1-S1-Si1 105.4(1) Fe11-S11-Si11 105.5(2) |
| [(Tp)MoFe3S4(PEt3)2(SSiPri3)] | Fe3-S5 2.248(3), Fe3-S5-Si1 118.0(7) |
| [(Tp)MoFe3S4(PEt3)2(SSiPh3)] | Fe3-S5 2.277(2), Fe3-S5-Si1 111.7(1) |
| [(Tp)MoFe3S4(PEt3)Cl2]1- | Fe2-Cl1 2.249(3), Fe3-Cl2 2.230(3) |
| {[(Tp)MoFe3S4(PEt3)2]3S}1+ c | Fe1-S1 2.248(2), Fe1-S1-Fe1A 114.94(9), 0.52d |
Core dimensions are typical for the set of clusters; see Fig.3 for core atom numbering scheme.
Two independent clusters.
S1 = μ3-S.
Perpendicular displacement of S1 from Fe1-Fe1A-Fe1B plane.
Magnetic and Mössbauer spectroscopic properties for four representative polycrystalline clusters and two others reported earlier24,25 are provided in Figure 5 and Table 3. Clusters 3 and 4 exhibit Curie-type behavior at 2-20 K with Curie constants close to C = 3 emu K/mol for an S = 2 ground state (g = 2). These results are consistent with the only prior magnetic measurement of a [MoFe3S4]2+ single-cubane cluster, cluster 19, which also has a spin quintet ground state. The Mössbauer spectra of 4, 6, and 8 consist of two quadrupole doublets in an intensity ratio of 2:1, with the less intense doublet having the smaller isomer shift (δ) and larger quadrupole splitting (ΔEQ). This same pattern was found earlier for 19 and 20. The isomer shifts do not correspond to values expected for localized Fe2+ or Fe3+ sites, which have not been observed in any type of cubane Fe-S cluster. The similar spectrum of 19, in which the iron sites have identical ligands, is a further argument again assigning the majority doublet to the two FeS3P sites and the minority doublet to the unique site in monosubstituted clusters. At this stage, we do not have sufficient information to describe the spin coupling interactions leading to the two doublets and the S = 2 ground state. However, an interpretation of oxidation states can be made.
Figure 5.
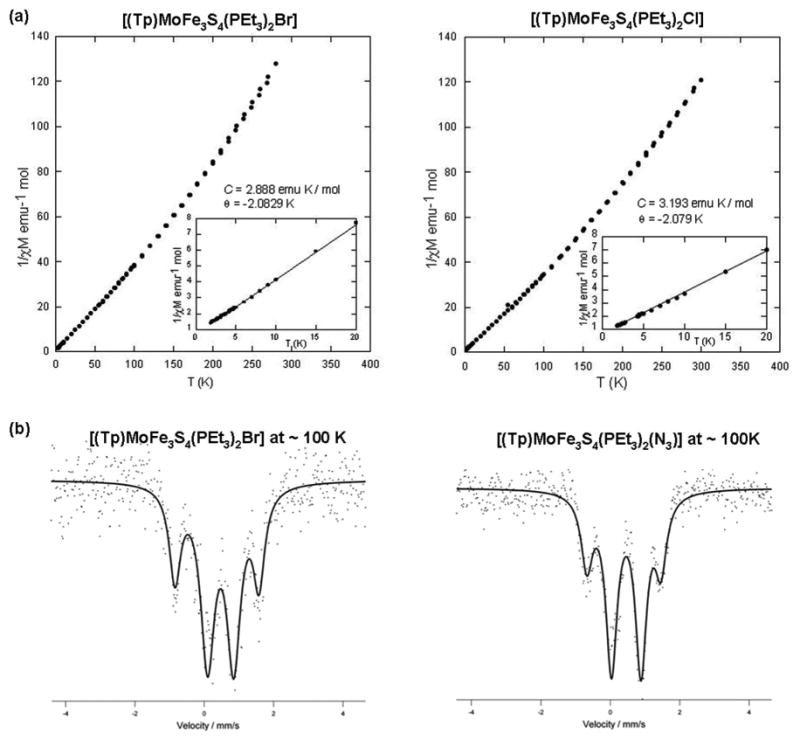
(a) Temperature dependence of the reciprocal molar susceptibilities of 3 and 4 at H = 1 T: inset: Curie plots at 2-20 K with Curie and Weiss constants indicated. (b) Zero-field Mossbauer spectra of 4 and 6 at 100 K; the solid lines are fits to the data using the parameters in Table 3. The correlation coefficient r = 0.9987 for both fits.
Table 3. Magnetic and Mössbauer Properties of [(Tp)MoFe3S4(PEt3)2L] (L = Cl-, Br-) and Related Clusters.
The mean isomer shifts of 4, 6, and 8 (0.42-0.44 mm/s) are the same and very close to that of phosphine-ligated 20 (0.46 mm/s), but less than that for thiolate cluster 19 (0.53 mm/s). This is consistent with previous observations that substitution of thiolate by phosphine decreases the isomer shift at constant oxidation state.12,30 For 19, application of the empirical relationship δ = 1.51 - 0.41s between isomer shift and (mean) oxidation state s for tetrahedral FeS431 gives Fe2.39+. This relationship cannot be strictly applied to FeS3P sites. However, the isomer shifts of phosphine clusters do not approach ≥0.6 mm/s expected for Fe2+3. Thus of the two oxidation state descriptions, c and Mo4+Fe2+3, the former (Fe32.33+) is the more consistent with the isomer shifts of the phosphine clusters. Lastly, these shifts are close to those of the clusters [Fe4S4(PR3)4]1+ (R = alkyl, 0.46-0.51 mm/s)30 and [Fe4S4(PPri3)3(SSiPri3)] (0.51 mm/s),20 which by their composition require the Fe2+3Fe3+ = Fe42.25+ description.
Tricluster Formation
The possibility of bridged cluster formation has thus far been investigated in the equimolar reaction mixture [1](BPh4)/NaSSiMe3 in THF. After 3 h reaction time and workup, the system afforded the tricluster salt [18](BPh4) (20%), which was identified by an X-ray structure determination (Figure 4). The formation of the tricluster can be interpreted by the suggested reaction scheme 5a-5c (formulas abbreviated) whose sum is the overall reaction 6. The scheme conveys the lability of the Me3Si-S bond previously exploited in Fe-S cluster formation.23,32-35 Substitution reaction 5a has been independently verified. In reaction 5b, Me3SiS- acts as a nucleophile toward silicon in a second cluster with bridge formation and release of the silylsufide. In reaction 5c, bridging sulfide attacks an iron site in another cluster with displacement of the silylthiolate and formation of a third Fe-S bridge. The modest yield is not unexpected for a three-step cluster assembly. Overall reaction 6 has been directly verified by the reaction of one equiv of 1 and two equiv of 11 in acetonitrile overnight. The sparingly soluble product was isolated and shown to be [18](BPh4) by 1H-NMR; yields were 20-30%. The scheme is a current working hypothesis, appropriate alteration of which will be directed toward the formation of bridged diclusters rather than triclusters.
| (5a) |
| (5b) |
| (5c) |
| (6) |
Structure proof of tricluster 18 is contained in Figure 4. Three (Tp)MoFe3S4(PEt3)2 units are bridged by a μ3-S atom which is displaced by 0.52 Å from the plane of the three iron atoms to which it is bonded. The cation has crystallographically imposed C3 symmetry with Fe-S-Fe bond angles of 115°; other metric features are unexceptional. Two other Mo-Fe-S triclusters have been prepared.36,37 These lack a μ3-S atom; instead components are linked in large rings through bridges between iron atoms in different clusters.
Summary
This work is the most comprehensive investigation of the [MoFe3S4]2+ oxidation state currently available. The following are the principal results and conclusions of this investigation.
The cluster [(Tp)MoFe3S4(PEt3)3]1+ readily undergoes phosphine substitution reactions to generate an extensive new family of clusters [(Tp)MoFe3S4(PEt3)3-nLn]1-n (n = 1, 2) with L = halide, N3-, PhS-, PhSe-, R3SiO- and R3SiS-. Monosubstituted clusters with L = PhS-, PhSe- and I- are also accessible by cleavage of oxidized precursors L2 with the all-ferrous double cubane [(Tp)2Mo2Fe6S8(PEt3)4]. The cubane-type stereochemistry of these clusters is demonstrated by X-ray structure determinations.
The neutral clusters (n = 1) in (1) are stable (no ligand exchange) in low dielectric solvents such as benzene and THF, in contrast to the usual behavior of charged clusters in more polar solvents.
Unlike the great majority of known MoFe3S4 clusters, which contain the [MoFe3S4]3+ (S = 3/2) core, the clusters in (1) are isolated in the reduced [MoFe3S4]2+ oxidation state. The clusters have a spin-quintet (S = 2) ground state and their formal electron distribution is best interpreted in terms of Mo3+Fe2+2Fe3+ from 57Fe isomer shifts, corresponding to a delocalized Fe32.33+ description.
A significant feature of the [MoFe3S4]2+ core is its ability to bind a variety of ligand types, ranging from hard (halide, azide) to softer (thiolate, selenolate) σ-domors to strong (phosphine) to weaker (cyanide9) π-acid ligands. Substitution reactions of single [MoFe3S4] clusters have been limited to mainly chloride displacement by RS- or ArO- in [MoFe3S4]3+ species,5, 38-40 which contain the oxidation state preferentially stabilized by ligands of this type.
Equimolar reaction of [(Tp)MoFe3S4(PEt3)3]1+ and NaSSiMe3 in THF affords the tricluster {[(Tp)MoFe3S4(PEt3)2]3S}1+ with a unique structure in which three cubane units are bridged by a central μ3-S atom.
Supplementary Material
Acknowledgments
This research was supported by NIH Grant 28856. We thank Drs. D. Harris, W. Lo, and S. Zheng for experimental assistance and helpful discussions. This work made use of the MRSEC Shared Experimental Facilities at MIT, supported by the National Science Foundation under award number DMR-08-19762.
Footnotes
Supporting Information Available. X-ray crystallographic files in CIF form for all compounds in Table 1. This material is available free of charge via the Internet at http://pubs.acs.org.
Reference List
- 1.Ciurli S, Ross PK, Scott MJ, Yu SB, Holm RH. J Am Chem Soc. 1992;114:5415–5423. [Google Scholar]
- 2.Panda R, Zhang Y, McLauchlan CC, Rao PV, Tiago de Oliveira FA, Münck E, Holm RH. J Am Chem Soc. 2004;126:6448–6459. doi: 10.1021/ja030627s. [DOI] [PubMed] [Google Scholar]
- 3.Panda R, Berlinguette CP, Zhang Y, Holm RH. J Am Chem Soc. 2005;127:11092–11101. doi: 10.1021/ja052381s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun J, Tessier C, Holm RH. Inorg Chem. 2007;46:2691–2699. doi: 10.1021/ic062362z. [DOI] [PubMed] [Google Scholar]
- 5.Fomitchev DV, McLauchlan CC, Holm RH. Inorg Chem. 2002;41:958–966. doi: 10.1021/ic011106d. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Holm RH. J Am Chem Soc. 2003;125:3910–3920. doi: 10.1021/ja0214633. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Holm RH. Inorg Chem. 2004;43:674–682. doi: 10.1021/ic030259t. [DOI] [PubMed] [Google Scholar]
- 8.Berlinguette CP, Miyaji T, Zhang Y, Holm RH. Inorg Chem. 2006;45:1997–2007. doi: 10.1021/ic051770k. [DOI] [PubMed] [Google Scholar]
- 9.Pesavento RP, Berlinguette CP, Holm RH. Inorg Chem. 2007;46:510–516. doi: 10.1021/ic061704y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koutmos M, Georgakaki IP, Tsiolis P, Coucouvanis D. Z Anorg Allgem Chem. 2008;634:255–261. [Google Scholar]
- 11.Malinak SM, Demadis KD, Coucouvanis D. J Am Chem Soc. 1995;117:3126–3133. [Google Scholar]
- 12.Hauser C, Bill E, Holm RH. Inorg Chem. 2002;41:1615–1624. doi: 10.1021/ic011011b. [DOI] [PubMed] [Google Scholar]
- 13.Zuo JL, Zhou HC, Holm RH. Inorg Chem. 2003:4624–4631. doi: 10.1021/ic0301369. [DOI] [PubMed] [Google Scholar]
- 14.Scott TA, Holm RH. Inorg Chem. 2008;47:3426–3432. doi: 10.1021/ic702372f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malinak SM, Coucouvanis D. Prog Inorg Chem. 2001;49:599–662. [Google Scholar]
- 16.Lee SC, Holm RH. Proc Natl Acad Sci USA. 2003;100:3595–3600. doi: 10.1073/pnas.0630028100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee SC, Holm RH. Chem Rev. 2004;104:1135–1157. doi: 10.1021/cr0206216. [DOI] [PubMed] [Google Scholar]
- 18.Groysman S, Holm RH. Biochemistry. 2009;48:2310–2320. doi: 10.1021/bi900044e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng L, Holm RH. J Am Chem Soc. 2008;130:9878–9886. doi: 10.1021/ja802111w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng L, Majumdar A, Lo W, Holm RH. Inorg Chem. 2010;49:11118–11126. doi: 10.1021/ic101702b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Einsle O, Tezcan FA, Andrade SLA, Schmid B, Yoshida M, Howard JB, Rees DC. Science. 2002;297:1696–1700. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 22.Fay AW, Blank MA, Lee CC, Hu Y, Hodgson KO, Hedman B, Ribbe MW. J Am Chem Soc. 2010;132:12612–12618. doi: 10.1021/ja1019657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Do Y, Simhon ED, Holm RH. Inorg Chem. 1983;22:3809–3812. [Google Scholar]
- 24.Mascharak PK, Papaefthymiou GC, Armstrong WH, Foner S, Frankel RB, Holm RH. Inorg Chem. 1983;22:2851–2858. [Google Scholar]
- 25.Osterloh F, Segal BM, Achim C, Holm RH. Inorg Chem. 2000;39:980–989. doi: 10.1021/ic991016x. [DOI] [PubMed] [Google Scholar]
- 26.Groysman S, Holm RH. Inorg Chem. 2007;46:4090–4102. doi: 10.1021/ic062441a. [DOI] [PubMed] [Google Scholar]
- 27.See paragraph at the end of this article for Supporting Information available.
- 28.Holm RH, Simhon ED. In: Molybdenum Enzymes. Spiro TG, editor. Wiley; New York: 1985. pp. 1–87. [Google Scholar]
- 29.Demadis KD, Campana CF, Coucouvanis D. J Am Chem Soc. 1995;117:7832–7833. [Google Scholar]
- 30.Goh C, Segal BM, Huang J, Long JR, Holm RH. J Am Chem Soc. 1996;118:11844–11853. [Google Scholar]
- 31.Rao PV, Holm RH. Chem Rev. 2004;104:527–559. doi: 10.1021/cr020615+. [DOI] [PubMed] [Google Scholar]
- 32.Noda I, Snyder BS, Holm RH. Inorg Chem. 1986;25:3851–3853. [Google Scholar]
- 33.Snyder BS, Holm RH. Inorg Chem. 1988;27:2339–2347. [Google Scholar]
- 34.Cen W, MacDonnell FM, Scott MJ, Holm RH. Inorg Chem. 1994;33:5809–5818. [Google Scholar]
- 35.Zhou C, Cai L, Holm RH. Inorg Chem. 1996;35:2767–2772. [Google Scholar]
- 36.Kawaguchi H, Yamada K, Ohnishi S, Tatsumi K. J Am Chem Soc. 1997;119:10871–10872. [Google Scholar]
- 37.Koutmos M, Coucouvanis D. Angew Chem Int Ed. 2004;43:5023–5025. doi: 10.1002/anie.200460154. [DOI] [PubMed] [Google Scholar]
- 38.Palermo RE, Holm RH. J Am Chem Soc. 1983;105:4310–4318. [Google Scholar]
- 39.Huang J, Mukerjee S, Segal BM, Akashi H, Zhou J, Holm RH. J Am Chem Soc. 1997;119:8662–8674. [Google Scholar]
- 40.Huang J, Holm RH. Inorg Chem. 1998;37:2247–2254. doi: 10.1021/ic971353a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.