

Abstract
Ionotropic GABA receptors (GABAA and GABAC) belong to the Cys-loop receptor family of ligand-gated ion channels. GABAC receptors are highly expressed in the retina, mainly localized at the axon terminals of bipolar cells. Ascorbic acid, an endogenous redox agent, modulates the function of diverse proteins, and basal levels of ascorbic acid in the retina are very high. However, the effect of ascorbic acid on retinal GABA receptors has not been studied. Here we show that the function of GABAC and GABAA receptors is regulated by ascorbic acid. Patch-clamp recordings from bipolar cell terminals in goldfish retinal slices revealed that GABAC receptor-mediated currents activated by tonic background levels of extracellular GABA, and GABAC currents elicited by local GABA puffs, are both significantly enhanced by ascorbic acid. In addition, a significant rundown of GABA puff-evoked currents was observed in the absence of ascorbic acid. GABA-evoked Cl− currents mediated by homomeric ρ1 GABAC receptors expressed in Xenopus laevis oocytes were also potentiated by ascorbic acid in a concentration-dependent, stereo-specific, reversible, and voltage-independent manner. Studies involving the chemical modification of sulfhydryl groups showed that the two Cys-loop cysteines and histidine 141, all located in the ρ1 subunit extracellular domain, each play a key role in the modulation of GABAC receptors by ascorbic acid. Additionally, we show that retinal GABAA IPSCs and heterologously expressed GABAA receptor currents are similarly augmented by ascorbic acid. Our results suggest that ascorbic acid may act as an endogenous agent capable of potentiating GABAergic neurotransmission in the CNS.
Introduction
The ionotropic GABA receptors, members of the Cys-loop receptor family, are GABA-gated pentameric chloride (Cl−) channels (Moss and Smart, 2001; Zhang et al., 2001). They are usually divided into two classes, namely, GABAA and GABAC receptors. GABAC receptors are composed of rho subunits, which are highly expressed in retinal bipolar cells (Wässle et al., 1998; McCall et al., 2002). They play an important role in the control of axon terminal excitability by mediating reciprocal synapses with amacrine cells (Matthews et al., 1994; Dong and Werblin, 1998; Protti and Llano, 1998; Hartveit, 1999). GABAC receptors also mediate tonic inhibitory currents, which arise in response to extracellular GABA concentrations controlled locally by GABA transporter 1 (GAT-1) transporters located on amacrine cells (Hull et al., 2006; Jones and Palmer, 2009). Unlike GABAA receptors, GABAC receptors display both high affinity for GABA and a lack of desensitization (Johnston, 1996). These two properties allow them to mediate several modes of inhibitory signaling in the retina (Zhang and Slaughter, 1995; Lukasiewicz et al., 2004), including inhibition of vesicle recycling (Hull and von Gersdorff, 2004).
Ascorbic acid (vitamin C), an endogenous redox agent highly concentrated in the retina and other regions of the CNS, is accumulated in neurons and glial cells by specific transporters (Rebec and Pierce, 1994; Rice, 2000; Harrison and May, 2009). In the retina, ascorbic acid levels rise above 100 times the concentration found in blood plasma (Rose and Bode, 1991; Hediger, 2002). Interestingly, the extracellular concentration of ascorbic acid can transiently undergo substantial increases during neuronal activity (Bigelow et al., 1984; Grünewald, 1993). In retinal neurons, the ascorbate transporter SVCT2 (sodium-dependent vitamin C transporter 2) mediates extensive sodium-dependent ascorbic acid extrusion through a mechanism regulated by neuronal depolarization and glutamate concentration (Portugal et al., 2009). In addition, ascorbic acid has been shown to modulate the activity of NMDA glutamate receptors and voltage-activated ion channels (Majewska et al., 1990; Fan and Yazulla, 1999; Alshuaib and Mathew, 2006; Nelson et al., 2007). Remarkably, its effects on Cys-loop receptors have not been previously studied in the retina.
We analyzed here whether GABAC receptor function can be regulated by ascorbic acid. GABAC receptor-mediated currents were measured from mixed rod-cone ON-type (Mb) bipolar cell terminals in goldfish retinal slices and from Xenopus laevis oocytes expressing recombinant homomeric ρ1 GABAC receptors. We show that the function of native retinal and recombinant GABAC receptors is significantly enhanced in the presence of ascorbic acid. Experiments involving the chemical modification of sulfhydryl groups and site-directed mutagenesis of the ρ1 subunit indicate that cysteines 177 and 191 forming the Cys-loop and histidine 141 (H141), all located in the N-terminal extracellular domain, are critical for the modulation of GABAC receptors by ascorbic acid. Additionally, we found that GABAA receptor-mediated miniature IPSCs (mIPSCs) at Mb retinal bipolar cell terminals, and ionic currents mediated by GABAA receptors heterologously expressed in HEK293 cells, are potentiated by ascorbic acid. Together, our results suggest that ascorbic acid can be a powerful endogenous modulator of GABAergic neurotransmission.
Materials and Methods
Retinal slice preparation and electrophysiology.
Retinal slices were prepared from pieces of retina, taken from goldfish (Carassius auratus; 8–16 cm) of either sex, according to procedures described previously (Palmer et al., 2003). The slices (250 μm thick) were constantly perfused at 4–5 ml/min with 2.5 mm Ca2+ Ringer's solution for patch-clamp recording. Bipolar cell terminals with severed axons (axotomized) were identified in the inner plexiform layer based on (1) single-exponential membrane time constant, (2) the presence of an L-type Ca2+ current and ΔCm jump, and (3) Mb-shaped (bulbous) terminal morphology (Palmer et al., 2003).
Axotomized bipolar cell terminals in retinal slices were voltage-clamped in the whole-cell mode using an HEKA Elektronik EPC-9 patch-clamp amplifier in conjunction with Pulse software running the xChart extension (Pulse version 8.53). The Sine+DC technique was used for real-time measurements of membrane capacitance. Briefly, a 30 mV peak-to-peak 1 kHz sine wave was superimposed on the holding potential of the cells (−60 mV) and was used by on-line analysis software to calculate time-resolved membrane capacitance. Standard external recording solutions contained the following (in mm): 100 NaCl, 2.5 KCl, 2.5 CaCl2, 1.0 MgCl2, 25 NaHCO3, and 12 glucose, pH 7.45 (osmolarity, 260–265 mOsm). In some cases, 3 mm ascorbic acid was added to recording solutions, which were then rebubbled with 95% O2/5% CO2. In these cases, pH and osmolarity were readjusted following bubbling. Patch pipettes (6–8 MΩ) were pulled from borosilicate capillary glass and coated with dental wax to reduce pipette capacitance. Internal pipette solutions contained the following solutions (in mm): 60 Cs gluconate, 40 CsCl, 10 TEA-Cl, 25 HEPES, 3 Mg-ATP, 0.5 Na-GTP, and 2 EGTA. Some internal solutions contained 3 mm ascorbic acid. All internals were set to pH 7.2 with CsOH, and osmolarity was adjusted to 250 mOsm. dl-APV, NBQX, TPMPA [(1,2,5,6-tetrahydropyridin-4-yl)methylphosphinic acid], and SR95531 (gabazine) were obtained from Tocris Bioscience. All other chemicals were obtained from Sigma.
For puff applications, GABA was dissolved in Ringer's solution along with, in some cases, 3 mm ascorbic acid and/or 25 μm SR95531. The pH was corrected to 7.45, and osmolarity was adjusted to 260 mOsm. This solution was then loaded in a patch pipette (see above) and puffed at 5 psi for 25 ms onto the axotomized Mb terminal at a distance of ∼10–30 μm using a Picospritzer III (Parker Instrumentation) driven by 100% N2 gas. Miniature GABAA currents (mIPSCs) were measured in recording solution containing 150 μm TPMPA, 1 μm TTX, 10 μm NBQX, and 50 μm dl-APV. The holding potential was −60 mV, and data were acquired in a series of successive 60 s sweeps in continuous recording mode. mIPSCs were analyzed using a custom Igor procedure that used template matching and thresholding techniques. In some cases, noise distributions were determined by analysis of sweeps following addition of 25 μm SR95531. Standing GABAC leak current was measured using the Pulse xChart extension (16 Hz acquisition rate, up to 50 point averaging) in the presence of 25 μm SR95531. Holding potential was set at −60 mV, and the minimum period for determination of stable leak current, as determined by recording chamber volume replacement rate, was 18 s.
Statistical analyses were performed using Prism (version 4; GraphPad Software) with one-way repeated-measures ANOVA, Dunnett's or Tukey's post hoc tests, and two-tailed unpaired or paired Student's t tests for comparing datasets. One-sample t tests were used to compare normalized data to a theoretical mean of zero. F tests were used to compare fitted slopes from linear regression either with a theoretical value of zero or between datasets. Data are reported as mean ± SEM. Statistics were performed on averaged traces where noted. mIPSC analysis was performed with Igor Pro v.5.04b (WaveMetrics).
RNA preparation, oocyte isolation, injection, and electrophysiology.
A human cDNA encoding the ρ1 GABAC receptor subunit cloned in the vector suitable for in vitro transcription pGEM was used as a template to synthesize cRNAs in vitro (mMessage mMachine kit, Ambion). Site-directed mutagenesis was achieved by the PCR overlap extension method using QuickChange Site-Directed Mutagenesis Kit (Stratagene). cRNA solutions (0.1–0.3 ng/nl) were prepared in RNase-free H2O and stored at −70°C. Oocytes, at stages V and VI, were used for expression of exogenous cRNAs. Isolation and maintenance of cells were performed as previously described (Miledi et al., 1989). Briefly, female X. laevis frogs (Nasco) were anesthetized with 3-aminobenzoic-acid ethylester (∼1 mg/ml), and ovaries were surgically removed. Ovaries were incubated with 200 U/ml type I or type II collagenase (Worthington) for 30 min at room temperature (RT) and isolated oocytes maintained in an incubator at 17°C in Barth's medium as follows (in mm): 88 NaCl; 0.33 Ca(NO3)2; 0.41 CaCl2; 1 KCl; 0.82 MgSO4; 2.4 NaHCO3; 10 HEPES and 0.1 mg/ml gentamicin; pH 7.4. After 1 d, each oocyte was manually microinjected (microinjector Drummond Scientific) with 50 nl of a solution containing 50 ng of cRNA. Two-electrode voltage-clamp recordings were performed 3–7 d after oocyte injection with an Axoclamp 2B amplifier (Molecular Devices). Standard glass recording electrodes were made in a PB-7 puller (Narishige Scientific Instrument Laboratory) and filled with 3 m KCl. Pipette resistance values were ∼1 MΩ. The holding potential was set to −70 mV, and current traces were acquired in a PC through Labmaster TL-1 DMA interface (Scientific Solutions) using pClamp software (Molecular Devices). Cells were placed in a chamber (volume 100 μl) continuously superfused (12 ml min−1) with frog Ringer's solution (in mm: 115 NaCl; 2 KCl; 1.8 CaCl2; 5 HEPES; pH 7.0). Drugs were applied through the perfusion system, and all the experiments were performed at RT (23–24°C). The agonist and all the drug and salts, HEPES, ascorbate analogs, 3-aminobenzoic-acid ethylester, and RNase-free H2O, were purchased from Sigma-Aldrich. Ascorbic acid, l-dehydroascorbic acid (DHA), calcium ascorbate [Ca(Asc)2], and d-isoascorbic acid (d-iso Asc) solutions were prepared freshly each day in normal Ringer's solution, and the pH was always adjusted to 7.0 with NaOH (1 m). Data were analyzed with Prism v. 6.0 (MicroCal). Statistical analysis was performed using one-way ANOVA and Tukey's or Dunnett's test. Dose–response curves (D–R) for GABA were fit with the expression of the mentioned logistic equation: Imax = B{1 − 1/[1 + (A/EC50)n]}, where A is the agonist concentration, B is the maximal response, EC50 is the concentration of agonist that elicits half-maximal responses, and n is the Hill coefficient.
Cell culture, transfection, and electrophysiology.
HEK293 cells were cultured using standard methodologies (Yevenes et al., 2006). Transfections were performed using Lipofectamine 2000 (Invitrogen) with 2 μg of DNA for each plasmid studied per well. cDNA constructs encoding the α1, β2, and γ2 rat GABAA was subcloned in the pRK5 vector (BD Pharmingen). Whole-cell recordings, at a holding potential of −60 mV, were performed as previously described 18–36 h after transfection. Patch electrodes were filled with the following (in mm): 140 CsCl, 10 1,2-bis-(2-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid, 10 HEPES, pH 7.4, 4 MgCl2, 2 ATP, and 0.5 GTP. The external solution contained the following (in mm): 150 NaCl, 10 KCl, 2.0 CaCl2, 1.0 MgCl2, 10 HEPES, pH 7.4, and 10 glucose. The amplitude of the GABA current was assayed using a short (1–2 s) pulse of 30–40 μm GABA every 60 s. Bicuculline (1 μm) blocked all of the current mediated by GABAA receptors (data not shown). GABA was unable to produce any response in >50 nontransfected cells. Statistical analyses were performed using ANOVA, and results were expressed as the arithmetic means ± SE; values of p < 0.05 were considered statistically significant. For all of the statistical analysis and plots, Origin 6.0 (MicroCal) software was used.
Results
GABAC currents in Mb retinal bipolar cell terminals are enhanced by ascorbic acid
Previous work has shown that both GABAA and GABAC receptors mediate chloride currents at Mb bipolar cell presynaptic terminals in goldfish retinal slices (Vigh and von Gersdorff, 2005). GABAC receptors at the bipolar cell terminals mediate a tonic current, which is activated by background levels of extracellular GABA. This current corresponds to a standing leak current sensitive to the selective GABAC receptor antagonist TPMPA and resistant to the GABAA receptor antagonist bicuculline-methiodide and SR-95531. Here we studied the effects of ascorbic acid on GABAC receptor-mediated responses using patch-clamp recording from axotomized Mb bipolar cell presynaptic terminals in the presence of 25 μm SR-95531 to block GABAA responses. Figure 1A, left, illustrates a representative trace showing the effects of ascorbic acid on a GABAC tonic current recorded at −60 mV. Overall, the GABAC-mediated standing leak current was significantly enhanced by bath application of 3 mm ascorbic acid [0 mm Asc = −49.17 ± 6.72 pA; 3 mm Asc = −61.44 ± 7.64 pA; wash (0 mm Asc) = −48.66 ± 6.80 pA, n = 11; repeated-measures ANOVA, p < 0.01; Dunnett's multiple comparison test, p < 0.01; paired t test, p = 0.0053). During washout, the leak current was significantly reduced to nearly baseline levels (Fig. 1A, right, summary data) (Dunnett's test, p < 0.01; paired t test, p < 0.001). Normalized measures of enhancement of leak current and washout were also significant (3 mm Asc = 30.43 ± 9.61%; 0 mm Asc = 0.24 ± 5.54%; one-sample t test, hypothetical value = 0; 3 mm Asc: p < 0.05; 0 mm Asc: p = 0.01). Similar highly significant enhancement [0 mm Asc = −235.3 ± 57.65 pA; 3 mm Asc = −302.4 ± 55.79 pA; wash (0 mm Asc) = −272.70 ± 45.40 pA; n = 4; repeated-measures ANOVA, p < 0.01; Dunnett's test, p < 0.01; paired t test, p = 0.0073] of leak current by ascorbic acid was obtained when the GABAC tonic current was recorded in the presence of the GAT-1 selective blocker NO-711 [example trace: 3 μm NO-711 (Fig. 1B, left); summary data: 1 or 3 μm NO-711, n = 1,3, data collapsed (Fig. 1B, right)], which increased the basal extracellular GABA concentration by inhibiting GABA uptake. Washout of the effect of ascorbic was not significant in the presence of NO-711 (paired t test, p = 0.058) (Fig. 1B, right), which may be due to increased variability of extracellular GABA concentration and difficulty establishing stable baseline leak current in the absence of functioning GAT-1 transporters (Hull et al., 2006). Normalized changes in leak current in the presence of NO-711 also revealed significant ascorbic acid-mediated enhancement of leak current (3 mm Asc = +33.63 ± 8.78%; 0 mm Asc = +21.97 ± 8.97%; one-sample t test, hypothetical value = 0, 3 mm Asc: p < 0.05; 0 mm Asc: p > 0.05). Bath application of 300 μm TPMPA in the presence of 25 μm SR-95531 (Fig. 1C, left, example trace) significantly reduced leak current (control: −31.5 ± 3.8 pA; TPMPA: − 14.2 ± 3.0 pA; n = 4, paired t test, p = 0.015) (Fig. 1C, right) in axotomized Mb terminals, indicating the presence of a small, non-GABAC-mediated leak current. Subtraction of this non-GABAC leak current from recordings in the absence or presence of NO-711 yielded estimated ascorbic acid potentiation of pure GABAC leak current of +65.0 ± 30.2% and +36.5 ± 9.9%, respectively. This is consistent with enhanced ascorbic acid-mediated potentiation of GABAC currents at low GABA concentrations.
Figure 1.

Ascorbic acid reversibly enhances GABAC-mediated standing leak current in axotomized Mb bipolar cell presynaptic terminals. A, GABAC leak current is enhanced by bath application of Asc. Whole-cell voltage-clamp recording of an Mb terminal in the presence of 25 μm SR-95531 shows a GABAC-mediated leak current of −52.5 pA at −60 mV (left). The leak current was enhanced by 8 pA following bath application of 3 mm ascorbic acid. During wash (0 mm Asc), the leak current was reduced nearly to its original level. Ascorbic acid significantly and reversibly enhanced GABAC-mediated leak current in the absence of NO-711 (n = 11; right). The mean for all cells with SE is shown in dark gray (circles) and is connected by a solid line; data from each single cell are shown in light gray (“x”) and are connected by a dashed line. B, Whole-cell voltage-clamp recording of an Mb terminal in the presence of 25 μm SR-95531 and 3 μm NO-711 shows a GABAC-mediated leak current of −400.6 pA at −60 mV (left). The leak current was enhanced by 55 pA following bath application of 3 mm Asc. During wash (0 mm Asc, 3 μm NO-711), the leak current was reduced to its original level. Ascorbic acid significantly enhanced GABAC-mediated leak current in the presence of 1–3 μm NO-711 (n = 4; right). Washout was nearly significant. C, Standing leak current consists of both TPMPA-sensitive and TPMPA-insensitive components. Whole-cell recording (left) of an axotomized Mb1 terminal voltage-clamped at −60 mV shows a leak current of −34.7 pA that is stably reduced to −16.4 pA by bath application of 300 μm TPMPA in the presence of 0 mm ascorbic acid. Addition of 300 μm TPMPA in the presence of 25 μm SR95531 and 0 mm ascorbic acid produced a statistically significant reduction in leak current (n = 4; right). *p < 0.05; **p < 0.01; ***p < 0.001; n.s., p > 0.05; all statistical tests reflect a repeated-measures design; all errors are expressed as ±SE, unless otherwise noted.
We also evoked GABAC responses by GABA pressure ejecting (puffing) GABA directly onto presynaptic terminals of bipolar cells (Fig. 2A) [25 ms puff, 30 s interstimulus interval (ISI)]. The amplitude of GABAC current responses to puff application of GABA exhibited significant rundown (∼30%) over 15 min in the absence of ascorbic acid [example: Fig. 2A left, summary data; B, 0 mm Asc rundown linear regression, −2.31 ± 0.08% per minute; slope significantly nonzero (p < 0.0001)]. Perfusion solution contained 25 μm SR-95531 in all cases. Some experiments were performed with 25 μm SR-95531 in the puff pipette, but showed no change in rundown kinetics (data not shown). Including 3 mm ascorbic acid in the bath solution, intracellular solution, and puff pipette significantly slowed and/or blocked this rundown over the same time period [example: Fig. 2A, right; B, 3 mm Asc linear regression: −0.16 ± 0.11% per minute; slope not significantly nonzero (p > 0.05)]. This effect on rundown slope was highly significant (Fig. 2B) [F = 28.7, degrees of freedom for numerator variance (DFn) = 1; degrees of freedom for denominator variance (DFd) = 306; p < 0.0001]. The two conditions were compared at individual time points, revealing significant block of GABAC current rundown by 3 mm ascorbic acid (in the puff pipette, patch pipette, and perfusion solution) at 5 min (unpaired t test, n = 7,7; p = 0.0036) and 10 min (p = 0.0031), but not at 14.5 min (p = 0.0936). Addition of 3 mm ascorbic acid to the bath solution alone appeared to slow rundown during the first 5 min of puff application, but this effect did not reach significance over the entire 15 min period (data not shown). Summary data were collapsed across conditions where either 200 μm (n = 4) or 1 mm GABA (n = 3) was used for puff application, as analysis showed no difference in rundown slope between these conditions from 0 to 15 min (n = 4, 3; F = 0.40; DFn = 1; DFd = 112; p = 0.53; data not shown).
Figure 2.
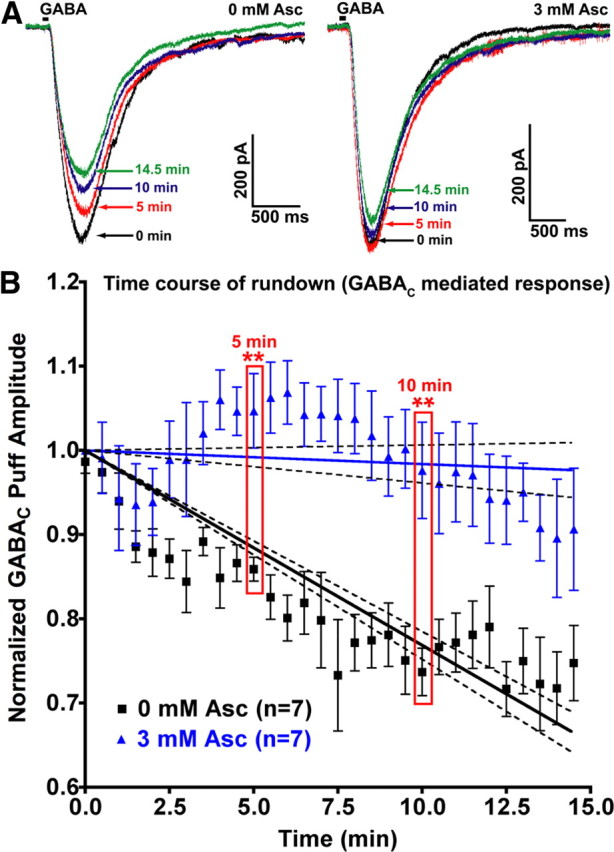
Rundown of GABAC-mediated responses to puff application of GABA in axotomized Mb bipolar cell presynaptic terminals is significantly slowed and/or prevented by the presence of intracellular and extracellular ascorbic acid. A, Puff application of GABA (25 ms duration, indicated by black bar) was performed every 30 s for 15 min to assess the degree of rundown of the resulting GABAC-mediated currents. Perfusion solution contained 25 μm SR-95531 (a GABAA antagonist) in all cases. Some experiments were performed with 25 μm SR-95531 in the puff pipette, with no change in rundown kinetics (data not shown). An individual example of GABAC current rundown shows that, in the absence of ascorbic acid either in the puff pipette, patch-clamp pipette, or perfusion solution, 25 ms puff application of 200 μm GABA with a 30 s (ISI) resulted in a 27.3% rundown in current amplitude over 14.5 min (left). Right, Same as left, except that in this example 3 mm ascorbic acid was included in the puff pipette, patch-clamp pipette, and perfusion solution. Under these conditions, rundown of GABAC current amplitude over 14.5 min was 8.7%. Over the first 5 min, GABAC current amplitude increased by 3.7%. B, The GABA puff experiments from A were repeated in the presence (n = 7) or absence (n = 7) of 3 mm ascorbic acid in the puff pipette, patch pipette, and perfusion solution, with 25 μm SR-95531 in the perfusion solution (and, in some cases, in the puff pipette). Shown are the mean normalized GABAC current amplitudes for each 30 s interval at which GABA was puff applied for 25 ms. Error bars indicate mean ± SEM. The slope of rundown for each condition was fit with linear regression (solid lines). Dashed lines indicate 95% confidence interval boundaries for each condition. The effect of ascorbic acid on rundown was highly significant at 5 and 10 min. **p < 0.01; all errors expressed as ±SE, unless otherwise noted.
We also quantified the amplitude of GABAC responses to puff application of GABA before (control), during, and after (wash) bath application of 3 mm ascorbic acid (Fig. 3A,B). Puff applications of GABA, collapsed across 1 mm (n = 7) and 200 μm (n = 1), produced large TPMPA-sensitive (data not shown) GABAC receptor-mediated inward currents at −60 mV (Fig. 3A). Ascorbic acid application significantly and reversibly [0 mm Asc = 661.2 ± 176.0 pA; 3 mm Asc = 729.8 ± 174.9 pA; wash (0 mm Asc) = 579.7 ± 153.8 pA; repeated-measures ANOVA (n = 8, p = 0.01); Tukey's multiple comparison test (0 mm Asc vs 3 mm Asc: p < 0.05; 3 mm Asc vs Wash (0 mm Asc): p < 0.05)] increased GABAC puff response amplitude by ∼16% (Fig. 3A, bottom). Analysis of responses normalized to control amplitudes also showed a significant increase in 3 mm ascorbic acid (+16.29 ± 5.37%; one-sample t test, p = 0.019), while normalized amplitudes during washout were not significantly nonzero (−9.11 ± 6.98%, one-sample t test, p = 0.233). Correction of averaged traces for GABAC rundown (based on linear regressions shown in Fig. 2), revealed a large effect of ascorbic acid that was nearly reversible during washout (Fig. 3B, bottom). Potentiation of GABAC-mediated responses to puff application of GABA was highly significant following correction for rundown, although this potentiation was not significantly reversed during washout (Fig. 3B, bottom) [0 mm Asc = 675.5 ± 178.3 pA; 3 mm Asc = 826.6 ± 198.0 pA; wash (0 mm Asc) = 746.8 ± 196.1 pA; repeated-measures ANOVA (n = 8, p = 0.01); Tukey's multiple comparison test (0 mm Asc vs 3 mm Asc: p < 0.01, 3 mm Asc vs Wash: p > 0.05)]. The lack of significant washout for corrected data is probably due to increased variability in rundown for data acquired >10 min after break-in (Fig. 2B). Analysis of corrected responses normalized to control amplitudes showed a highly significant increase in 3 mm ascorbic acid (+28.29 ± 6.73%; one-sample t test, p = 0.004), while normalized amplitudes during washout were not significantly nonzero (14.18 ± 9.17%; one-sample t test, p = 0.166). It is worth noting that, even though we know the exact concentration of GABA inside the pipette, it is difficult to quantitatively control the local concentration of GABA at bipolar terminal presynaptic receptors due to effects of transporters and extracellular diffusion. Thus, low concentrations of GABA applied to the bath or locally puff applied often do not produce a robust, stable response. Despite these issues, a significant dose dependency of ascorbic acid-mediated potentiation on the concentration of puffed GABA was observed between 1 and 10 mm (1 mm GABA: +16.31 ± 6.20%; 10 mm GABA: − 4.65 ± 6.70%; n = 7,5; unpaired t test, p = 0.047; data not shown). No difference in potentiation was seen between puff applications of 200 μm and 1 mm GABA (n = 1, 7; data not shown). Together with the effect of ascorbic acid on standing leak current, these results demonstrate that endogenous GABAC receptor function is strongly modulated by ascorbic acid.
Figure 3.

Response to puffing GABA directly onto axotomized Mb bipolar cell presynaptic terminals is enhanced by bath application of ascorbic acid. A, Puff application of 1 mm GABA (25 ms duration, indicated by black bar) directly onto an axotomized Mb bipolar cell presynaptic terminal in the presence of 25 μm SR-95531 produced (upper) a large GABAC-mediated inward current (black trace). Bath application of 3 mm ascorbic acid (red trace) increased the amplitude of this current in a reversible manner (washout; blue trace). Each trace is the average of three successive stimulations with an intertrace interval of 20 s. Summary data (bottom; n = 8) showed a significant and reversible increase in puff response amplitude caused by 3 mm ascorbic acid. Mean for all cells with SE is shown in green (circles); data from each single cell (“x”) are connected by gray dashed line, control data points (0 mm Asc) are shown in black, 3 mm Asc points are shown in red, wash (0 mm Asc) points are shown in blue. B, Rundown normalization of GABAC currents from cell in A, calculated using the mean time after initial GABA puff, and the linear rundown regressions from Figure 2B, show potentiation of nearly 25% in the presence of 3 mm ascorbic acid, with a nearly complete washout in 0 mm Asc (top). Summary of corrected data (bottom; n = 8) showed a significant increase in puff response amplitude caused by bath application of 3 mm ascorbic acid. Individual repeated-measures data are shown in gray (filled circles), and mean values are shown in black (“x”; bottom). *p < 0.05; **p < 0.01; n.s., p > 0.05; all statistical tests reflect a repeated-measures design; all errors are expressed as ±SE, unless otherwise noted.
Responses mediated by homomeric ρ1 GABAC receptors are enhanced by ascorbic acid
To further characterize the actions of ascorbic acid on GABAC receptors, we heterologously expressed recombinant GABAC receptors in frog oocytes. GABA applications to oocytes expressing homomeric ρ1 GABAC receptors induced large inward Cl− currents displaying all of the features of retinal GABAC receptor-mediated responses. For example, they are bicuculline insensitive, TPMPA and picrotoxin sensitive, non-desensitizing, and display the same pharmacological profile for agonists (Kusama et al., 1993; Woodward et al., 1993). In addition, this experimental model allowed us to control more precisely the extracellular concentration of ascorbic acid and the GABA concentration sensed by the GABAC receptors; thus, a more quantitative approach was possible. Figure 4A shows GABAρ1 responses measured in normal frog Ringer's solution (control, 0.3 μm GABA) or immediately after the application of ascorbic acid to the recording solution. Current amplitude was significantly enhanced (∼+160%) in the presence of 3 mm ascorbic acid. Similar effects were produced by brief exposures of the oocytes to a lower concentration of ascorbic acid (700 μm) during sustained GABA applications (0.3, 1, or 10 μm) (Fig. 4B). Ascorbic acid actions were strongly dependent on GABA concentration, and were very rapid, stable, and completely reversible after washout (Fig. 4B). No use-dependent effects were observed, and no changes in activation, deactivation, or desensitization of the GABAρ1 responses were detected during ascorbic acid applications (data not shown). D–R curves for GABA were shifted to the left and slightly but significantly increased in their maximal values in the presence of ascorbic acid (Fig. 5A, Table 1). Figure 5B illustrates the effects of increasing concentrations of ascorbic acid on GABAρ1 responses elicited by 0.3 and 1 μm GABA. Maximal values of potentiation obtained during applications of high concentrations of ascorbic acid (30 mm) were substantial (∼750%) (Fig. 5C). No significant differences were observed in either the degree of potentiation at different membrane potentials (reflected by the change in I–V curves slopes) or in the reversal potentials of Cl− currents in the presence of ascorbic acid (Fig. 5D). The fast onset, fast offset, and reversible character of ascorbic acid effects on GABAC receptors all suggest that these effects are not due to its entry into the cells. Based on this evidence, an extracellular superficial site of action is more likely. This is in agreement with the fact that short (0–10 s) or persistent (up to 30 min) applications of ascorbic acid produced identical results in oocytes expressing GABAρ1 receptors (data not shown), and with data reporting that oocytes do not express ascorbic acid transporters (SCVT1 and SCVT2) (Dyer et al., 1994). I–V curves showing that modulation is independent of the membrane potential also indicate regulatory sites located outside of the channel pore.
Figure 4.

Homomeric ρ1 GABAC receptor function is enhanced by ascorbic acid. Representative traces of ionic (Cl−) currents elicited by different GABA concentrations in oocytes expressing homomeric GABAρ1 receptors. Calibration: current amplitude (y-axis), time (x-axis). For this and the subsequent figures, oocytes were voltage-clamped at −70 mV. A, GABAρ1 responses recorded before (left, control) and after the application of 3 mm ascorbic acid (right). Potentiation of GABAρ1 responses by ascorbic acid was 162 ± 22.6% (n = 3). B, Ionic currents elicited by increasing concentrations of GABA (0.3, 1, and 10 μm) were also enhanced during concurrent applications of 700 μm ascorbic acid.
Figure 5.

Analysis of ascorbic acid effects on GABAρ1 receptor function. A, D–R curves for GABA performed in the presence or absence of ascorbic acid. Response amplitudes were expressed as a fraction of maximal GABA-evoked currents (30 μm GABA). B, Dose–effects curves for ascorbic acid acting on GABAρ1 responses. Data were normalized to control values, obtained in the absence of ascorbic acid, for two different GABA concentrations. All the points tested >100 μm were significantly different from control. C, Degree of potentiation of the GABAρ1 responses by ascorbic acid for increasing concentrations of GABA. D, I–V relationships of the GABAρ1 responses measured in the presence or absence of ascorbic acid.
Table 1.
Dose-response curve parameters
| EC50 (μm) | n (Hill coefficient) | n | |
|---|---|---|---|
| Control | 0.74 ± 0.01 | 1.58 ± 0.04 | 5–10 |
| 3 mm Asc | 0.49 ± 0.01* | 1.88 ± 0.07 | 5–10 |
| Control | 1.52 ± 0.04 | 2.24 ± 0.05 | 4 |
| 30 μm NEM | 1.29 ± 0.02 | 2.24 ± 0.10 | 4 |
| 30 μm NEM + 3 mm Asc | 0.75 ± 0.01 | 2.44 ± 0.19 | 4 |
| Control | 0.86 ± 0.10 | 2.58 ± 0.30 | 3 |
| 30 μm NEM | 0.78 ± 0.09 | 2.32 ± 0.10 | 3 |
| 30 μm NEM + 2 mm DTT | 0.74 ± 0.06 | 2.04 ± 0.08 | 3 |
| H141D | 1.76 ± 0.21 | 3.41 ± 0.61 | 10 |
| H141D + 3 mm Asc | 2.07 ± 0.32 | 2.94 ± 0.38 | 5 |
| H141D + 30 μm NEM | 1.40 ± 0.16 | 3.24 ± 0.16 | 4 |
| H141D + 30 μm NEM + 3 mm Asc | 1.46 ± 0.24 | 3.48 ± 0.28 | 4 |
Parameters of the D–R curves for GABA, performed for GABA ρ1 wt and GABA ρ1 H141D in the absence or presence of the different agents.
*p < 0.03.
The specificity of ascorbic acid actions on GABAρ1 responses was analyzed by testing structurally related analogs (Fig. 6A). DHA (3 mm) and Ca(Asc)2 (1.5 mm), two forms of ascorbate that lack antioxidizing activity, were unable to produce changes in currents elicited by 0.3 μm GABA (Fig. 6B, Table 2). In contrast, the stereoisomer d-iso-Asc (3 mm), which shows the same antioxidizing power as ascorbic acid (Organisciak et al., 1992), had a much lower efficacy (Fig. 6B, Table 2), indicating that structural determinants are also essential for modulation. Kinetic analyses of the GABAρ1 responses elicited by 0.3 μm GABA, recorded in normal frog Ringer's solution or in the presence of ascorbic acid, Ca(Asc)2, DHA, or d-iso-Asc showed time courses of activation (τact) and deactivation (τdeact) well fitted to single exponential functions. No kinetic changes were observed in the presence of 3 mm ascorbic acid, 1.5 mm Ca(Asc)2, 3 mm DHA, or 3 mm d-iso-Asc (Table 2). Desensitization values were negligible, thus τdeact was not included for analysis.
Figure 6.

Effects of different ascorbic acid analogs on GABAρ1 receptor function. A, Chemical structures of the diverse compounds used. B, Histogram summarizing the values obtained for GABAρ1 responses evoked by 0.3 μm GABA recorded before (control, dotted line) and after exposure to the different ascorbic acid analogs (n = 4).
Table 2.
Effects of ascorbic acid and its analogs on GABAρ1 responses
| Compound | % Potentiation | n | τact (s) | τdeact (s) | n |
|---|---|---|---|---|---|
| Control (GABA 0.3 μm) | 6 | 24.8 ± 1.6 | 23.5 ± 3.8 | 6 | |
| 3 mm Asc | 139.5 ± 87.5** | 7 | 22.6 ± 2.6 | 28.1 ± 3.3 | 6 |
| 3 mm D-isoAsc | 33.5 ± 12.5* | 4 | 28.5 ± 2.5 | 33.4 ± 1.9 | 6 |
| 3 mm DHA | −1.6 ± 4.2 | 3 | 26.1 ± 2.8 | 32.0 ± 2.8 | 6 |
| 1.5 mm Asc(Ca)2 | 7.4 ± 4.8 | 6 | 22.5 ± 2.5 | 27.4 ± 4.3 | 4 |
Potentiation (%) produced by ascorbic acid and its different analogs on GABAρ1 responses and values obtained for kinetic parameters in the presence of the different compounds.
*p < 0.04,
**p < 0.01.
Involvement of cysteines forming the Cys-loop and histidine 141 in the modulation of GABAρ1 receptors by ascorbic acid
We studied whether ascorbic acid effects on GABAρ1 responses could be due entirely or in part to a direct interaction of this agent with specific aminoacidic residues at the receptor subunits. Previous studies have suggested that cysteine residues are commonly involved in the redox modulation of various ion channels (Ruiz-Ǵomez et al., 1991; Ruppersberg et al., 1991; Sullivan et al., 1994; Lipton et al., 2002; Chu et al., 2006). GABAC receptor ρ1 subunits carry only two cysteines (C177 and C191) at the extracellular domain forming the characteristic Cys-loop. Mutations of these cysteines are precluded because they render receptors nonfunctional (Amin et al., 1994; Sedelnikova et al., 2005). Thus, to determine whether C177 and C191 are involved in the modulation of the homomeric ρ1 GABAC receptors by ascorbic acid, we used the irreversible thiol alkylating agent N-ethylmaleimide (NEM), which forms covalent bonds with the free sulfhydryl groups preventing any further chemical reaction at these sites. NEM concentration was kept as low as possible, and incubation periods were kept very short to prevent nonspecific effects. As shown in Figure 7A, NEM alone produced, on GABAρ1 receptors expressed in oocytes, effects similar to those observed for ascorbic acid (Fig. 5A). D–R curves for GABA after NEM (30 μm) treatment were shifted to the left, and their maximal values were significantly increased. As expected for a specific sulfhydryl reagent, NEM completely abolished dithiothreitol (DTT; 2 mm) potentiation of the GABAρ1 responses (Fig. 7B). Next, we studied the actions of ascorbic acid on GABAρ1 receptors previously treated with 30 μm NEM. D–R curves showed that the increase produced by 3 mm ascorbic acid in GABAρ1 maximal responses (evidenced in Fig. 5A) was entirely prevented after NEM treatment, whereas the leftward shift was partially resistant to NEM (Fig. 7C). These results indicate that the extracellular cysteines forming the Cys-loop of the ρ1 subunits are involved in the modulation of GABAC receptors by ascorbic acid. Effects of ascorbic acid may also be partially due to redox modulation of C177 and C191, so additional mechanisms may also be involved.
Figure 7.
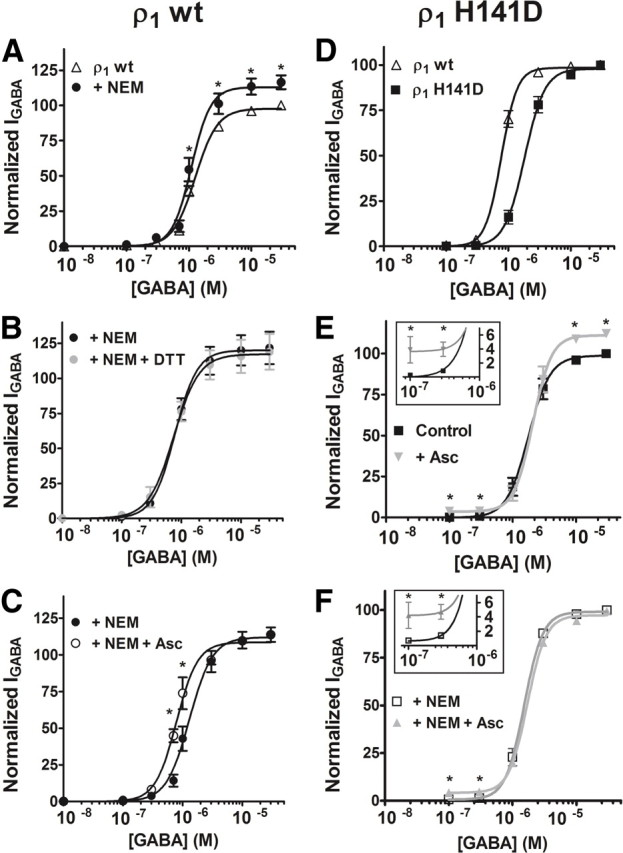
Identification of amino acid residues involved in the modulation of homomeric ρ1 GABAC receptors by ascorbic acid. A, The irreversible methylation of C177 and C191 at the Cys-loop, by pretreatment of oocytes with 30 μm NEM, produced similar effects to those shown by ascorbic acid on D–R curves for GABA. B, NEM pretreatment completely abolished the potentiation of GABAρ1 responses induced by 2 mm DTT. C, NEM pretreatment partially prevented the potentiation of GABAρ1 responses by 3 mm ascorbic acid. D, D–R curves for GABA performed on wild-type GABAρ1 and mutant GABAρ1H141D receptors. E, Mutation of H141 to D at the ρ1 subunit partially prevented the potentiating actions of ascorbic acid. Effects of 3 mm ascorbic acid on D–R curves for GABA were performed on GABAρ1H141D receptors. Ascorbic acid significantly enhanced maximal responses and currents evoked by very low GABA concentrations (inset), but failed to induce a leftward shift in the D–R curve (as that observed in C), indicating that the H141 is critical for ascorbic acid modulatory actions. F, NEM pretreatment abolished the potentiation induced by ascorbic acid on maximal responses mediated by GABAρ1H141D receptors, indicating that they are more likely mediated by C177 and C191, but effects at very low GABA concentrations were not prevented by NEM (inset). Response amplitudes were expressed as a fraction of the 30 μm GABA-evoked currents (maximal response). Each point represents the mean and SEM of the responses obtained from four to six oocytes. Notice that many of the SE bars are hidden by the symbols.
Previous studies have demonstrated that H141, located near the ρ1 subunit extracellular Cys-loop, is essential for allosteric modulation of homomeric GABAρ1 receptors (Wang et al., 1995; Zhang et al., 2001; Sedelnikova et al., 2005). Thus, we used site-directed mutagenesis to study whether this amino acid residue can also participate in the modulation of GABAρ1 receptors by ascorbic acid. H141 was replaced with an aspartic acid to generate mutant homomeric GABAρ1H141D receptors. When expressed in oocytes, these receptors had typical responses to GABA (bicuculline insensitive and TPMPA sensitive) and showed EC50 values for GABA slightly higher than wild-type receptors (Fig. 7D) (Wang et al., 1995). The effects of ascorbic acid on mutant GABAρ1H141D receptors were analyzed in D–R curves. Similarly to wild-type receptors, maximal responses to GABA were significantly increased in the presence of 3 mm ascorbic acid (Fig. 7E). However, the shift to the left produced by ascorbic acid on D–R curves performed on wild-type GABAρ1 receptors (Fig. 5A) was completely abolished in receptors carrying the mutation H141D (Fig. 7E). In addition, ascorbic acid remained capable of potentiating responses evoked by low GABA concentrations (Fig. 7E, inset). We also analyzed the specific contribution of C177 and C191 to the effects induced by ascorbic acid on GABAρ1H141D receptors in oocytes previously treated with NEM. Ascorbic acid-induced increases in maximal responses evoked by GABA on wild-type receptors was completely prevented by NEM treatment, but potentiating effects of ascorbic acid on GABAρ1 responses induced by low GABA concentrations on the mutant GABAρ1H141D receptors remained unaltered (Fig. 7F, inset). Ascorbic acid might exert pro-oxidant effects on membrane proteins through a selective metal-catalyzed oxidation involving the modification of histidines (Stadtman, 1991, 1993; Nelson et al., 2007). However, this possibility was dismissed because ascorbic acid effects on GABAρ1 responses were identical in the presence of the metal-chelating agents EDTA and tricine (data not shown). In addition, several oxidizing agents antagonize GABAρ1 responses (Calero and Calvo, 2008), and Zn2+ interaction with H141 also causes a reversible inhibition (Wang et al., 1995). In contrast, we found that H141 is critically involved in potentiation of GABAρ1 receptor function by ascorbic acid. A proton transfer from ascorbic acid to H141 was also disregarded because pH (7.4) was constant during experiments. Moreover, critical residues for pH sensitivity are located within the first 60 N-terminal amino acids in ρ1 subunits (Rivera et al., 2000). Thus, H141 would be involved in an independent conformational change, allosterically induced by ascorbic acid at or near the Cys-loop of GABAρ1 receptors, which might take place simultaneously with redox modification of C177 and C191 at ρ1 subunits. All of these events would occur near the extracellular agonist-binding site.
Modulation of neuronal and recombinant GABAA receptors by ascorbic acid
Based on our findings, we decided to test whether ascorbic acid could produce similar effects on GABAA receptors. Whole-cell patch-clamp recordings were performed on both presynaptic terminals of retinal bipolar cells and transfected cells expressing two types of recombinant GABAA receptors. Figure 8A shows an example recording demonstrating a reversible increase in the amplitude of miniature GABAA receptor-mediated IPSCs (i.e., mIPSCs) following extracellular application of 3 mm ascorbic acid. The mean mIPSC waveforms for this example recording (Fig. 8B, left) show a distinct, reversible increase in mIPSC amplitude. This effect can also be seen as a reversible rightward shift in the cumulative probability curve for mIPSC amplitude (same example cell) following addition of ascorbic acid (Fig. 8B, right). Although ascorbic acid appeared to increase mIPSC frequency (2.44 Hz control vs 5.34 Hz Asc) and preferentially affect larger mIPSCs (>12 pA) in this example cell (Fig. 8B, right), there was no overall significant effect of ascorbic acid on mIPSC frequency [control (0 mm Asc) = 3.01 ± 1.26 Hz; 3 mm Asc = 3.80 ± 1.45 Hz; wash (0 mm Asc) = 3.58 ± 1.39 Hz; repeated-measures ANOVA, n = 5, p = 0.28] (Fig. 8C, right). It is possible that in some cells, ascorbic acid potentiation of mIPSCs raised their amplitude above our detection threshold and thus caused an apparent increase in the frequency of small mIPSCs. However, examination of the mIPSC amplitude distribution (Fig. 8B, right) showed a clear shift in cumulative probability over the whole range of amplitudes, indicating that the effect of ascorbic acid was not specific for a small subset of mIPSCs (Fig. 8C, right). In fact, both the enhancement of mIPSC amplitude by bath application of 3 mm ascorbic acid, and the reversibility of this effect upon washout, were statistically significant [control (0 mm Asc) = 15.42 ± 2.51 pA; 3 mm Asc = 18.45 ± 3.23 pA; wash (0 mm Asc) = 15.62 ± 2.48 pA; repeated-measures ANOVA, n = 5, p = 0.014; Tukey's multiple comparison test, control (0 mm) vs 3 mm Asc, p < 0.05; 3 mm Asc vs wash (0 mm Asc), p < 0.05] (Fig. 8C, left). These results show that ascorbic acid potentiation is effective at local GABA concentrations in the synaptic cleft seen under physiological conditions.
Figure 8.
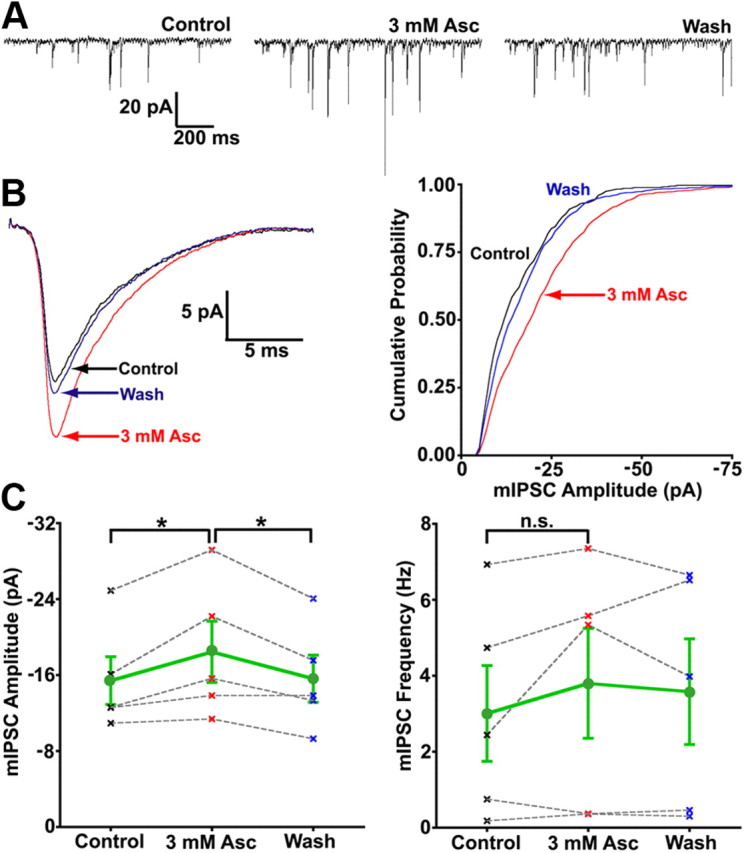
The amplitude of GABAA mIPSCs is enhanced by bath application of ascorbic acid. A, Excerpts (1.3 s each) from continuous records of mIPSCs from a single axotomized Mb1 terminal (at least 120 s per condition) in the presence of 150 μm TPMPA (GABAC antagonist), 10 μm NBQX, 50 μm dl-APV, and 1 μm TTX show that 3 mm ascorbic acid reversibly increased the amplitude of GABAA mIPSCs without significantly increasing mIPSC frequency. B, The amplitude of the mean IPSC waveform of GABAA mIPSCs was reversibly enhanced by nearly 30% (left) following bath application of 3 mm ascorbic acid (same cell as in A). The mIPSC amplitude cumulative probability distribution (bin = 1 pA) for control (0 mm Asc, count = 353), 3 mm ascorbic acid (count = 770), and wash (0 mm Asc, count = 574) conditions (right) showed a distinct, reversible rightward shift (larger amplitudes) in cumulative probability in the presence of 3 mm ascorbic acid. C, Summary of data for all cells (n = 5) shows a significant, reversible increase in GABAA mIPSC amplitude (left), with no significant effect on mIPSC frequency (right), following bath application of 3 mm ascorbic acid. Mean for all cells with SE shown in green (circles), data from each single cell (“x”) connected by gray dashed line, control data points (0 mm Asc) shown in black, 3 mm Asc shown in red, wash (0 mm Asc) shown in blue. *p < 0.05; n.s, p > 0.05; all statistical tests reflect a repeated-measures design; all errors are expressed as ±SE, unless otherwise noted.
Native and recombinant GABAA receptors can be modulated by a number of redox agents whose effects depend on receptor subunit composition. In particular, the presence of the γ2 subunit was shown to reduce the sensitivity of GABAA receptors to exogenous and naturally occurring redox reagents (Amato et al., 1999; Pan et al., 2000). Thus, redox modulation at GABAergic synapses is predicted to vary throughout the CNS, depending on the GABAA receptor subtypes expressed. To examine the ascorbic acid sensitivity of GABAA receptors with known subunit compositions, two combinations of recombinant GABAA receptors were transfected in HEK cells, namely, α1β2γ2 and α1β2 receptors. In fact, expression of α1, α2, α3, β1, β2, β3, γ2, and δ subunits has been previously reported in retinal neurons (Wässle et al., 1998; Koulen, 1999; Zhang et al., 2003). GABAA receptor-mediated inward currents recorded in HEK cells at −60 mV were significantly enhanced by extracellular application of ascorbic acid over the entire range of concentrations tested (0.5–10 mm). Responses mediated by GABAA receptors lacking the γ2 subunit were strongly potentiated by ascorbic acid (up to a ∼600% by 10 mm), whereas responses mediated by α1β2γ2 receptors were also effectively increased by ascorbic acid, but to a lesser extent (up to ∼200% by 10 mm). Data were summarized in Figure 9. These results indicate that ascorbic acid can produce on GABAA receptors effects similar to those exerted by other redox agents, and that GABAA receptor subtypes show a differential sensitivity to ascorbic acid modulation.
Figure 9.
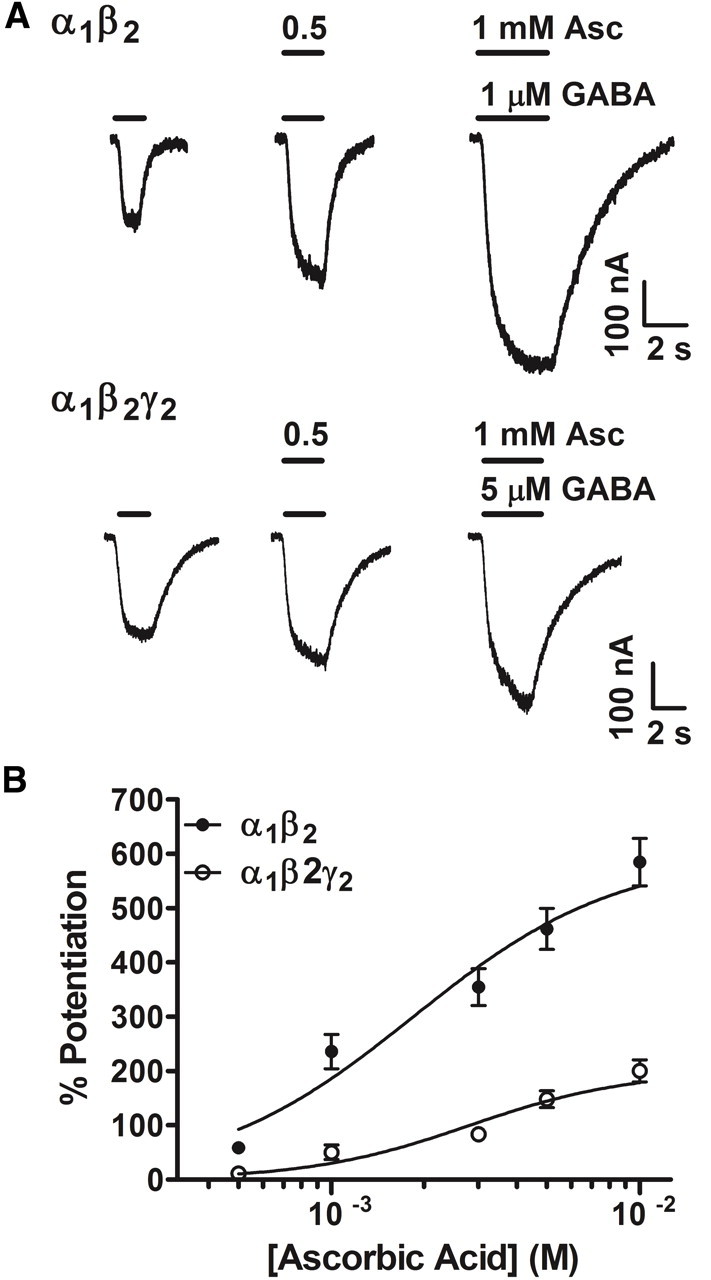
Potentiation of the α1β2 and α1β2γ2 GABAA receptor function by ascorbic acid. A, Representative traces of ionic (Cl−) currents mediated by α1β2 and α1β2γ2 GABAA receptors expressed in HEK cells. Inward GABAA currents were elicited by bath applications of 1 or 5 μm GABA (the corresponding EC50 values for GABA showed by α1β2 and α1β2γ2 GABAA receptors, respectively) and recorded by whole-cell voltage-clamp at −60 mV. GABAA responses mediated by both α1β2 and α1β2γ2 GABAA receptors were significantly enhanced in the presence of 0.5 and 1 mm ascorbic acid. B, Dose–effect curves for ascorbic acid actions on α1β2 and α1β2γ2 GABAA receptors (n = 3; curves performed at the corresponding EC50 values). Data were normalized to control values, obtained in the absence of ascorbic acid.
Discussion
We have shown a remarkably potent modulation of retinal and heterologous GABAC and GABAA receptors by physiologically relevant concentrations of ascorbic acid. Surprisingly, we believe that the present findings are the first to demonstrate that this ubiquitous and endogenous redox agent can regulate the function of Cys-loop receptors.
Effects of ascorbic acid on GABAC receptors
The potentiating effects of ascorbic acid on native and recombinant GABAC receptors were similar to those shown by many reducing agents acting on different ionotropic GABA receptors and other Cys-loop receptors (Bouzat et al., 1991; Ruiz-Gómez et al., 1991; Pan et al., 1995, 2000; Amato et al., 1999; Calero and Calvo, 2008). Efficacy of ascorbic acid on retinal GABAC responses appears to be relatively lower than on responses mediated by GABAρ1 receptors, probably because experiments in the retina required higher concentrations of GABA to counteract the action of GABA transporters and extracellular diffusion. Additionally, differences may be due to variations in the sensitivity to ascorbic acid among receptors from different species (fish vs human), the possible existence of heteromeric receptors in the retina (e.g., GABAC receptors containing ρ2 or ρ3 subunits), the interaction of native receptors with accessory subunits or regulatory proteins not present in oocytes, or endogenous redox buffers that could be absent or altered in heterologous systems.
Mechanisms underlying the modulation of GABAC receptors by ascorbic acid
We showed that the Cys-loop residues C177 and C191, along with H141, all located near the extracellular agonist-binding site, take part in the modulation of GABAC receptors by ascorbic acid. The shift to the left produced by ascorbic acid on the GABAρ1 receptor D–R curve (Fig. 5A) can be explained by a decrease in the energy barrier for GABA activation induced by this agent. Ascorbic acid could bind to or modify one or more sites capable of allosterically modulating single-channel properties, for example, by inducing an increase in GABA affinity that involves H141. In addition, to describe the simultaneous increase observed in the maximal response, which is more likely mediated by a redox modification of C177 and C191, a possibility is that ascorbic acid acts to favor the conversion from the last GABA-bound closed state to the open state (Goutman et al., 2005). Alternatively, ascorbic acid could induce the channels to transition toward additional open states in which the receptor adopts lower energy conformations with higher open probabilities. We found that GABAρ1 channel deactivation from open states was not altered in the presence of ascorbic acid (Table 2), which suggests that the first alternative is more likely. However, a combination of different mechanisms could also occur. Further analysis, including binding experiments, single-channel recording, and kinetic studies, is necessary to address these questions.
We also showed that the intracellular application of ascorbic acid prevents rundown of GABAC receptor-mediated inward currents at the Mb presynaptic terminals. There are many intracellular targets of redox modulation, and mitigation of general oxidative stress is likely to also prolong the viability of a cell following dialyzation by the patch pipette. Rundown of GABAC currents during the course of a 15–20 min patch-clamp recording has been shown to depend on PKC phosphorylation and to be inhibited by addition of phosphatases to the intracellular solution (Feigenspan and Bormann, 1994). Thus, a candidate could be the potential interaction of ascorbic acid at these signaling pathways. Interestingly, recent studies showed that the generation of reactive oxygen species (ROS) can induce rundown of currents mediated by neuronal acetylcholine receptors (nAChRs) (Campanucci et al., 2010). This rundown is triggered by ROS-induced oxidation of C239, an intracellular cysteine residue conserved in many nAChR subunits (e.g: α3, α4, β2, and β4) that forms a ring located at the M1-M2 linker near the inner mouth of the channel pore. Based on these data, and on the fact that ρ subunits in GABAC receptors also carry an intracellular cysteine (C364) at the M3-M4 linker, the prevention of GABAC current rundown exerted by ascorbic acid might act as a protective mechanism against ROS actions.
Effects of ascorbic acid on GABAA receptors
Redox modulation of GABAA receptors has been demonstrated in heterologous systems and in different areas of the CNS, including the retina (Pan et al., 1995; Amato et al., 1999). GABAA receptors from retinal ganglion cells are significantly inhibited by oxidants and are potentiated by reducing agents (Pan et al., 1995), but studies in retinal bipolar cells were lacking. We show that GABAA receptors located on the presynaptic terminals of Mb bipolar cells, which likely contain the γ2 subunit (Wässle et al., 1998), can be modulated by ascorbic acid. This is a significant finding, because it directly shows that ascorbic acid can potentiate synaptic GABAA-mediated currents in intact retinal tissue. Additionally, experiments performed on recombinant α1β2γ2 and α1β2 GABAA receptors showed that ascorbic acid actions depend on subunit composition. GABAA receptors containing the γ2 subunit had a decreased sensitivity to ascorbic acid (Fig. 9), in agreement with the reduced sensitivity to redox agents exhibited by hippocampal, cerebellar, and ganglionic GABAA receptors (Amato et al., 1999).
Potential physiological relevance of the modulation of GABAC receptors by ascorbic acid
Ascorbic acid is highly concentrated in the retina, both in extracellular (200–500 μm) and intracellular (≤10 mm) compartments (Rose and Bode, 1991; Rice, 2000; Castro et al., 2001; Hediger, 2002; Hosoya et al., 2004). During and after neuronal activity, local levels of ascorbic acid surrounding retinal bipolar neurons can reach millimolar values due to SVCT2-mediated sodium-dependent extrusion (Portugal et al., 2009). GABAρ1 receptor potentiation by ascorbic acid can be significant at extracellular concentrations as low as 0.3 mm, with effects becoming substantial at >0.7 mm (Fig. 4B). Meanwhile, the activity of retinal GABAC receptors was considerably increased by extracellular ascorbic acid in the low-millimolar range (3 mm) (Figs. 1, 2). Ascorbic acid effects on GABAC receptors strongly depended on GABA concentration. As GABAC receptors show relatively high affinity for GABA (compared with GABAA receptors), ascorbic acid modulation at low GABA concentrations could be ideally located within the dynamic range of ion channel activation. If so, the regulation of retinal GABAC receptors would be highly dependent on local ascorbic acid release and instantaneous extracellular GABA concentration. In particular, extrasynaptic receptors, which would presumably see lower concentrations of GABA, might be more powerfully modulated by ascorbic acid.
Tonic inhibition provides a constant shunt to the Mb terminal, which likely acts to prevent release in the absence of strong, high-frequency inputs, and activation of the standing leak current is closely controlled by the GABA transporter GAT1 (Hull et al., 2006). Reciprocal inhibition, which occurs when release of glutamate from the Mb terminal triggers direct GABA feedback from local amacrine cell presynaptic boutons, is comprised of a fast GABAA component and a slow, sustained GABAC component (Vigh and von Gersdorff, 2005). Reciprocal inhibition directly controls the duration and intensity of glutamate release from the Mb terminal. Finally, lateral inhibition, elicited when stimulation of a parallel bipolar cell pathway triggers unidirectional GABA release from a local amacrine cell bouton onto both GABAA and GABAC receptors on the Mb terminal, may act to shape the offset of bipolar cell light responses (Chávez et al., 2010). Strong modulation of tonic, reciprocal, and lateral inhibition at Mb terminals by ascorbic acid is likely to shape ganglion cell responses via control of glutamate release. However, given the previously described effects of ascorbic acid on T-type Ca2+ channels (Nelson et al., 2007) and K+ channels (Fan and Yazulla, 1999), mechanisms of modulation of retinal physiological responses by this agent might be more complex.
We showed here that ascorbic acid enhanced standing leak currents and responses activated by local GABA puffs in the presence of gabazine, both of which are mediated by GABAC receptors at the presynaptic terminals of retinal Mb bipolar cells. This suggests that ascorbic acid modulation can occur during either acute (similar to reciprocal or lateral synaptic inhibition), sustained (tonic standing leak current), or strong (under conditions of a high rate of GABA release) activation of GABAC receptors. Additionally, ascorbic acid could be involved in a “negative feedback” phenomenon following strong bipolar cell depolarization. This depolarization would cause strong glutamate release, which in turn would stimulate release of ascorbic acid into the extracellular space (Portugal et al., 2009). Increased concentrations of extracellular ascorbic acid can potentiate GABAC- and GABAA-mediated currents, enhancing local inhibition. In this context, ascorbic acid effects on both GABAA and GABAC responses would be consistent with the effects of glutamate on SVCT2-mediated ascorbic acid extrusion. Unfortunately, the subcellular localization of SVCT2 in neurons or glial cells still is unknown (Castro et al., 2001; Harrison and May, 2009). In conclusion, although the precise physiological role that ascorbate plays in the regulation of GABAergic neurotransmission needs to be further investigated, our results clearly indicate that ascorbic acid can act as an important endogenous modulator of ionotropic GABAC and GABAA receptors in the retina and probably also in other areas of the CNS.
Footnotes
This work was funded by Fondo para la Investigación Científica y Tecnológica and Consejo Nacional de Investigaciones Científicas y Técnicas. We also thank the Pew Foundation and International Brain Research Organization for support. H.v.G. was supported by Grant RO1 (EY14043) from the National Eye Institute of the NIH. We thank Javier Gasulla and Paul Brehm for helpful discussions, and Marcela Lipovsek and Guido Corallo for technical assistance.
References
- Alshuaib WB, Mathew MV. Vitamins C and E modulate neuronal potassium currents. J Membr Biol. 2006;210:193–198. doi: 10.1007/s00232-006-0001-x. [DOI] [PubMed] [Google Scholar]
- Amato A, Connolly CN, Moss SJ, Smart TG. Modulation of neuronal and recombinant GABAA receptors by redox reagents. J Physiol. 1999;517:35–50. doi: 10.1111/j.1469-7793.1999.0035z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin J, Dickerson IM, Weiss DS. The agonist binding site of the gamma-aminobutyric acid type A channel is not formed by the extracellular cysteine loop. Mol Pharmacol. 1994;45:317–323. [PubMed] [Google Scholar]
- Bigelow JC, Brown DS, Wightman RM. Gamma-aminobutyric acid stimulates the release of endogenous ascorbic acid from rat striatal tissue. J Neurochem. 1984;42:412–419. doi: 10.1111/j.1471-4159.1984.tb02693.x. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Barrantes FJ, Sigworth FJ. Changes in channel properties of acetylcholine receptors during the time course of thiol chemical modifications. Pflugers Arch. 1991;418:51–61. doi: 10.1007/BF00370451. [DOI] [PubMed] [Google Scholar]
- Calero CI, Calvo DJ. Redox modulation of homomeric rho1 GABA receptors. J Neurochem. 2008;105:2367–2374. doi: 10.1111/j.1471-4159.2008.05319.x. [DOI] [PubMed] [Google Scholar]
- Campanucci V, Krishnaswamy A, Cooper E. Diabetes depresses synaptic transmission in sympathetic ganglia by inactivating nAChRs through a conserved intracellular cysteine residue. Neuron. 2010;66:827–834. doi: 10.1016/j.neuron.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Castro M, Caprile T, Astuya A, Millán C, Reinicke K, Vera JC, Vásquez O, Aguayo LG, Nualart F. High-affinity sodium-vitamin C co-transporters (SVCT) expression in embryonic mouse neurons. J Neurochem. 2001;78:815–823. doi: 10.1046/j.1471-4159.2001.00461.x. [DOI] [PubMed] [Google Scholar]
- Chávez AE, Grimes WN, Diamond JS. Mechanisms underlying lateral GABAergic feedback onto rod bipolar cells in rat retina. J Neurosci. 2010;30:2330–2339. doi: 10.1523/JNEUROSCI.5574-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu XP, Close N, Saugstad JA, Xiong ZG. ASIC1a-specific modulation of acid-sensing ion channels in mouse cortical neurons by redox reagents. J Neurosci. 2006;26:5329–5339. doi: 10.1523/JNEUROSCI.0938-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong CJ, Werblin FS. Temporal contrast enhancement via GABAC feedback at bipolar terminals in the tiger salamander retina. J Neurophysiol. 1998;79:2171–2180. doi: 10.1152/jn.1998.79.4.2171. [DOI] [PubMed] [Google Scholar]
- Dyer DL, Kanai Y, Hediger MA, Rubin SA, Said HM. Expression of a rabbit renal ascorbic acid transporter in Xenopus laevis oocytes. Am J Physiol. 1994;267:C301–C306. doi: 10.1152/ajpcell.1994.267.1.C301. [DOI] [PubMed] [Google Scholar]
- Fan SF, Yazulla S. Suppression of voltage-dependent K+ currents in retinal bipolar cells by ascorbate. Vis Neurosci. 1999;16:141–148. doi: 10.1017/s0952523899161091. [DOI] [PubMed] [Google Scholar]
- Feigenspan A, Bormann J. Modulation of GABAC receptors in rat retinal bipolar cells by protein kinase C. J Physiol. 1994;481:325–330. doi: 10.1113/jphysiol.1994.sp020442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutman JD, Escobar AL, Calvo DJ. Analysis of macroscopic ionic currents mediated by GABA rho1 receptors during lanthanide modulation predicts novel states controlling channel gating. Br J Pharmacol. 2005;146:1000–1009. doi: 10.1038/sj.bjp.0706411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünewald RA. Ascorbic acid in the brain. Brain Res Brain Res Rev. 1993;18:123–133. doi: 10.1016/0165-0173(93)90010-w. [DOI] [PubMed] [Google Scholar]
- Harrison FE, May JM. Vitamin C function in the brain: vital role of the ascorbate transporter SVCT2. Free Radic Biol Med. 2009;46:719–730. doi: 10.1016/j.freeradbiomed.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartveit E. Reciprocal synaptic interactions between rod bipolar cells and amacrine cells in the rat retina. J Neurophysiol. 1999;81:2923–2936. doi: 10.1152/jn.1999.81.6.2923. [DOI] [PubMed] [Google Scholar]
- Hediger MA. New view at C. Nat Med. 2002;8:445–446. doi: 10.1038/nm0502-445. [DOI] [PubMed] [Google Scholar]
- Hosoya K, Minamizono A, Katayama K, Terasaki T, Tomi M. Vitamin C transport in oxidized form across the rat blood-retinal barrier. Invest Ophthalmol Vis Sci. 2004;45:1232–1239. doi: 10.1167/iovs.03-0505. [DOI] [PubMed] [Google Scholar]
- Hull C, von Gersdorff H. Fast endocytosis is inhibited by GABA-mediated chloride influx at a presynaptic terminal. Neuron. 2004;44:469–482. doi: 10.1016/j.neuron.2004.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull C, Li GL, von Gersdorff H. GABA transporters regulate a standing GABAC receptor-mediated current at a retinal presynaptic terminal. J Neurosci. 2006;26:6979–6984. doi: 10.1523/JNEUROSCI.1386-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston GA. GABAC receptors: relatively simple transmitter-gated ion channels? Trends Pharmacol Sci. 1996;17:319–323. [PubMed] [Google Scholar]
- Jones SM, Palmer MJ. Activation of the tonic GABAC receptor current in retinal bipolar cell terminals by nonvesicular GABA release. J Neurophysiol. 2009;102:691–699. doi: 10.1152/jn.00285.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulen P. Postnatal development of GABAA receptor beta1, beta2/3, and gamma2 immunoreactivity in the rat retina. J Neurosci Res. 1999;57:185–194. doi: 10.1002/(SICI)1097-4547(19990715)57:2<185::AID-JNR4>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Kusama T, Spivak CE, Whiting P, Dawson VL, Schaeffer JC, Uhl GR. Pharmacology of GABA rho 1 and GABA alpha/beta receptors expressed in Xenopus oocytes and COS cells. Br J Pharmacol. 1993;109:200–206. doi: 10.1111/j.1476-5381.1993.tb13554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Choi YB, Takahashi H, Zhang D, Li W, Godzik A, Bankston LA. Cysteine regulation of protein function as exemplified by NMDA-receptor modulation. Trends Neurosci. 2002;25:474–480. doi: 10.1016/s0166-2236(02)02245-2. [DOI] [PubMed] [Google Scholar]
- Lukasiewicz PD, Eggers ED, Sagdullaev BT, McCall MA. GABAC receptor-mediated inhibition in the retina. Vision Res. 2004;44:3289–3296. doi: 10.1016/j.visres.2004.07.023. [DOI] [PubMed] [Google Scholar]
- Majewska MD, Bell JA, London ED. Regulation of the NMDA receptor by redox phenomena: inhibitory role of ascorbate. Brain Res. 1990;537:328–332. doi: 10.1016/0006-8993(90)90379-p. [DOI] [PubMed] [Google Scholar]
- Matthews G, Ayoub GS, Heidelberger R. Presynaptic inhibition by GABA is mediated via two distinct GABA receptors with novel pharmacology. J Neurosci. 1994;14:1079–1090. doi: 10.1523/JNEUROSCI.14-03-01079.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall MA, Lukasiewicz PD, Gregg RG, Peachey NS. Elimination of the rho1 subunit abolishes GABAC receptor expression and alters visual processing in the mouse retina. J Neurosci. 2002;22:4163–4174. doi: 10.1523/JNEUROSCI.22-10-04163.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miledi R, Parker I, Sumikawa K. Transplanting receptors from brain into oocytes. In: Smith JJ, editor. Fidia Research Foundation Neuroscience Award Lecture Series. Vol 3. New York: Raven Press; 1989. pp. 57–90. [Google Scholar]
- Moss SJ, Smart TG. Constructing inhibitory synapses. Nat Rev Neurosci. 2001;2:240–250. doi: 10.1038/35067500. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Joksovic PM, Su P, Kang HW, Van Deusen A, Baumgart JP, David LS, Snutch TP, Barrett PQ, Lee JH, Zorumski CF, Perez-Reyes E, Todorovic SM. Molecular mechanisms of subtype-specific inhibition of neuronal T-type calcium channels by ascorbate. J Neurosci. 2007;27:12577–12583. doi: 10.1523/JNEUROSCI.2206-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organisciak DT, Bicknell IR, Darrow RM. The effects of L- and D-ascorbic acid administration on retinal tissue levels and light damage in rats. Curr Eye Res. 1992;11:231–241. doi: 10.3109/02713689209001774. [DOI] [PubMed] [Google Scholar]
- Palmer MJ, Taschenberger H, Hull C, Tremere L, von Gersdorff H. Synaptic activation of presynaptic glutamate transporter currents in nerve terminals. J Neurosci. 2003;23:4831–4841. doi: 10.1523/JNEUROSCI.23-12-04831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan ZH, Bähring R, Grantyn R, Lipton SA. Differential modulation by sulfhydryl redox agents and glutathione of GABA- and glycine-evoked currents in rat retinal ganglion cells. J Neurosci. 1995;15:1384–1391. doi: 10.1523/JNEUROSCI.15-02-01384.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan ZH, Zhang X, Lipton SA. Redox modulation of recombinant human GABAA receptors. Neuroscience. 2000;98:333–338. doi: 10.1016/s0306-4522(00)00114-7. [DOI] [PubMed] [Google Scholar]
- Portugal CC, Miya VS, Calaza Kda C, Santos RA, Paes-de-Carvalho R. Glutamate receptors modulate sodium-dependent and calcium-independent vitamin C bidirectional transport in cultured avian retinal cells. J Neurochem. 2009;108:507–520. doi: 10.1111/j.1471-4159.2008.05786.x. [DOI] [PubMed] [Google Scholar]
- Protti DA, Llano I. Calcium currents and calcium signaling in rod bipolar cells of rat retinal slices. J Neurosci. 1998;18:3715–3724. doi: 10.1523/JNEUROSCI.18-10-03715.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebec GV, Pierce RC. A vitamin as neuromodulator: ascorbate release into the extracellular fluid of the brain regulates dopaminergic and glutamatergic transmission. Prog Neurobiol. 1994;43:537–565. doi: 10.1016/0301-0082(94)90052-3. [DOI] [PubMed] [Google Scholar]
- Rice ME. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000;23:209–216. doi: 10.1016/s0166-2236(99)01543-x. [DOI] [PubMed] [Google Scholar]
- Rivera C, Wegelius K, Reeben M, Kaila K, Michael P. Different sensitivities of human and rat rho1 GABA receptors to extracellular pH. Neuropharmacology. 2000;39:977–989. doi: 10.1016/s0028-3908(99)00208-7. [DOI] [PubMed] [Google Scholar]
- Rose RC, Bode AM. Ocular ascorbate transport and metabolism. Comp Biochem Physiol A. 1991;100:273–285. doi: 10.1016/0300-9629(91)90470-w. [DOI] [PubMed] [Google Scholar]
- Ruiz-Gómez A, Fernández-Shaw C, Morato E, Marvizón JC, Vázquez J, Valdivieso F, Mayor F., Jr Sulfhydryl groups modulate the allosteric interaction between glycine binding sites at the inhibitory glycine receptor. J Neurochem. 1991;56:1690–1697. doi: 10.1111/j.1471-4159.1991.tb02069.x. [DOI] [PubMed] [Google Scholar]
- Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Frank R, Koenen M. Regulation of fast inactivation of cloned mammalian IK(A) channels by cysteine oxidation. Nature. 1991;352:711–714. doi: 10.1038/352711a0. [DOI] [PubMed] [Google Scholar]
- Sedelnikova A, Smith CD, Zakharkin SO, Davis D, Weiss DS, Chang Y. Mapping the rho1 GABAC receptor agonist binding pocket. Constructing a complete model. J Biol Chem. 2005;280:1535–1542. doi: 10.1074/jbc.M409908200. [DOI] [PubMed] [Google Scholar]
- Stadtman ER. Ascorbic acid and oxidative inactivation of proteins. Am J Clin Nutr. 1991;54:1125S–1128S. doi: 10.1093/ajcn/54.6.1125s. [DOI] [PubMed] [Google Scholar]
- Stadtman ER. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu Rev Biochem. 1993;62:797–821. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- Sullivan JM, Traynelis SF, Chen HS, Escobar W, Heinemann SF, Lipton SA. Identification of two cysteine residues that are required for redox modulation of the NMDA subtype of glutamate receptor. Neuron. 1994;13:929–936. doi: 10.1016/0896-6273(94)90258-5. [DOI] [PubMed] [Google Scholar]
- Vigh J, von Gersdorff H. Prolonged reciprocal signaling via NMDA and GABA receptors at a retinal ribbon synapse. J Neurosci. 2005;25:11412–11423. doi: 10.1523/JNEUROSCI.2203-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TL, Hackam A, Guggino WB, Cutting GR. A single histidine residue is essential for zinc inhibition of GABA rho 1 receptors. J Neurosci. 1995;15:7684–7691. doi: 10.1523/JNEUROSCI.15-11-07684.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wässle H, Koulen P, Brandstätter JH, Fletcher EL, Becker CM. Glycine and GABA receptors in the mammalian retina. Vision Res. 1998;38:1411–1430. doi: 10.1016/s0042-6989(97)00300-3. [DOI] [PubMed] [Google Scholar]
- Woodward RM, Polenzani L, Miledi R. Characterization of bicuculline/baclofen-insensitive (rho-like) gamma-aminobutyric acid receptors expressed in Xenopus oocytes. II. Pharmacology of gamma-aminobutyric acid A and gamma-aminobutyric acid B receptor agonists and antagonists. Mol Pharmacol. 1993;43:609–625. [PubMed] [Google Scholar]
- Yevenes GE, Moraga-Cid G, Guzmán L, Haeger S, Oliveira L, Olate J, Schmalzing G, Aguayo LG. Molecular determinants for G protein betagamma modulation of ionotropic glycine receptors. J Biol Chem. 2006;281:39300–39307. doi: 10.1074/jbc.M608272200. [DOI] [PubMed] [Google Scholar]
- Zhang D, Pan ZH, Awobuluyi M, Lipton SA. Structure and function of GABAC receptors: a comparison of native versus recombinant receptors. Trends Pharmacol Sci. 2001;22:121–132. doi: 10.1016/s0165-6147(00)01625-4. [DOI] [PubMed] [Google Scholar]
- Zhang J, Slaughter MM. Preferential suppression of the ON pathway by GABAC receptors in the amphibian retina. J Neurophysiol. 1995;74:1583–1592. doi: 10.1152/jn.1995.74.4.1583. [DOI] [PubMed] [Google Scholar]
- Zhang J, De Blas AL, Miralles CP, Yang CY. Localization of GABAA receptor subunits alpha 1, alpha 3, beta 1, beta 2/3, gamma 1, and gamma 2 in the salamander retina. J Comp Neurol. 2003;459:440–453. doi: 10.1002/cne.10626. [DOI] [PubMed] [Google Scholar]