

Abstract
Long chain fatty acids and pharmacologic ligands for the peroxisome proliferator activated receptor alpha (PPARα) activate expression of genes involved in fatty acid and glucose oxidation including carnitine palmitoyltransferase-1A (CPT-1A) and pyruvate dehydrogenase kinase 4 (PDK4). CPT-1A catalyzes the transfer of long chain fatty acids from acyl-CoA to carnitine for translocation across the mitochondrial membranes and is an initiating step in the mitochondrial oxidation of long chain fatty acids. PDK4 phosphorylates and inhibits the pyruvate dehydrogenase complex (PDC) which catalyzes the conversion of pyruvate to acetyl-CoA in the glucose oxidation pathway. The activity of CPT-1A is modulated both by transcriptional changes as well as by malonyl-CoA inhibition. In the liver, CPT-1A and PDK4 gene expression are induced by starvation, high fat diets and PPARα ligands. Here, we characterized a binding site for PPARα in the second intron of the rat CPT-1A gene. Our studies indicated that WY14643 and long chain fatty acids induce CPT-1A gene expression through this element. In addition, we found that mutation of the PPARα binding site reduced the expression of CPT-1A-luciferase vectors in the liver of fasted rats. We had demonstrated previously that CPT-1A was stimulated by the peroxisome proliferator activated receptor gamma coactivator (PGC-1α) via sequences in the first intron of the rat CPT-1A gene. Surprisingly, PGC-1α did not enhance CPT-1A transcription through the PPARα binding site in the second intron. Following knockdown of PGC-1α with short hairpin RNA, the CPT-1A and PDK4 genes remained responsive to WY14643. Overall, our studies indicated that PPARα and PGC-1α stimulate transcription of the CPT-1A gene through different regions of the CPT-1A gene.
Keywords: Carnitine palmitoyltransferase-1A (CPT-1A), pyruvate dehydrogenase kinase (PDK), PPARα, PGC-1α
Introduction
The expression of genes involved in mitochondrial fatty acid and glucose oxidation is modulated by long chain fatty acids and fibrate compounds (Chatelain et al., 1996; Harris et al., 2002; Sugden and Holness 2003). Carnitine palmitoyltransferase-1 (CPT-1) is an initiating step in the translocation of long chain fatty acids across the mitochondrial membranes for beta-oxidation (McGarry and Brown 1997; Park and Cook 1998). CPT-1 catalyzes the transfer of long chain fatty acids from acyl-CoA to carnitine to form acyl-carnitine which is moved across the mitochondrial membranes by carnitine acyl-carnitine translocase (CACT) (McGarry and Brown 1997). In the liver, CPT-1A is the primary isoform expressed while the CPT-1B and CPT-1C are highly expressed in other tissues. Previous studies from our laboratory and others have found that the oxidation of long chain fatty acids in the liver is elevated with high fat diets, fasting and streptozotocin induced diabetes (Park et al., 1995; Sugden et al., 2002; Howell et al., 2009). We have observed that the expression of the rat CPT-1A gene and CPT-1A activity are increased in these states (Cook et al., 2001). In addition, the sensitivity of the CPT-1A enzyme to malonyl-CoA inhibition is decreased with fasting and insulin deficient diabetes (Cook et al., 1980; Park et al., 1995). The pyruvate dehydrogenase complex (PDC) catalyzes the conversion of pyruvate to acetyl-CoA (Sugden and Holness 2003). PDC activity is inhibited by the dedicated pyruvate dehydrogenase kinases (PDK2 and PDK4) and phosphorylation of PDC will reduce the conversion of pyruvate to acetyl-CoA (Harris et al., 2002; Sugden and Holness 2003). Expression of the PDK4 gene is induced by high fat diets and long chain fatty acids (Sugden et al., 2002).
The peroxisome proliferator activated receptor α (PPARα) is a nuclear receptor which stimulates genes involved in mitochondrial fatty acid oxidation and increases expression of those modulating pyruvate oxidation. Long chain fatty acids and pharmacologic agonists such as gemfibrozol or WY14643 are ligands for PPARα (Desvergne and Wahli 1999). PPARα binds to the PPAR response element (PPRE) as a heterodimer with the retinoid X receptor (RXR) (Mangelsdorf and Evans 1995). In PPARα deficient mice, a number of genes involved in mitochondrial fatty acid oxidation are not induced during starvation (Kersten et al., 1999). However the role of PPARα in CPT-1A regulation is controversial. A PPRE was identified in the first intron of the human CPT-1A gene and this sequence was conserved between the rat, mouse and human genes (Napal et al., 2005). However, there have been several reports demonstrating that long chain fatty acids stimulate CPT-1A expression through a PPARα independent mechanism (Louet et al., 2001; Le May et al., 2005). PPARα ligands induce expression of the PDK4 gene (Huang et al., 2002; Sugden et al., 2002; Holness et al., 2003) and a PPRE has been described in the promoter of the human PDK4 gene (Degenhardt et al., 2007). In addition, PDK4 expression was not induced by fasting or WY14643 administration in PPARα deficient mice (Wu et al., 2001). Here, we have further examined the regulation of the CPT-1A and PDK4 genes by PPARα and selected coactivators.
Transcriptional coactivators are recruited to liganded nuclear receptors and function in part by stimulating gene expression, covalently modifying histones and mediating interactions between transcription factors (Rosenfeld and Glass 2001). The peroxisome proliferator activated receptor gamma coactivator (PGC-1α) was initially identified in brown adipose tissue as a protein that interacted with the peroxisome proliferator activated receptor gamma (PPARγ) (Puigserver et al., 1998). PGC-1α is highly expressed in tissues with high metabolic rates including heart, muscle and brown adipose tissue (Puigserver and Spiegelman 2003). Overexpression of PGC-1α in heart and muscle promotes mitochondrial biogenesis (Finck and Kelly 2006; Finck and Kelly 2007). In the heart, many genes involved in fatty acid oxidation such as medium chain acyl-CoA dehydrogenase (MCAD) and CPT-1B are stimulated by PGC-1α. The induction of CPT-1B occurs via direct interactions of PPARα and PGC-1α (Vega et al., 2000). In the liver, PGC-1α abundance is increased in fasting and diabetes (Yoon et al., 2001). PGC-1α promotes hepatic gluconeogenesis by stimulating the genes encoding phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6P) (Yoon et al., 2001). PGC-1α can interact with a variety of nuclear receptors including the thyroid hormone receptor (TR), hepatic nuclear factor 4 (HNF-4), estrogen related receptors (ERR), the glucocorticoid receptor (GR) and others through a leucine-X-X-leucine-leucine (LXXLL) motif in PGC-1α (Wu et al., 2002; Lin et al., 2005; Zhang et al., 2006). Overexpression of PGC-1α induced the expression of CPT-1A and PDK4 in the liver suggesting that PGC-1α stimulates hepatic fatty acid oxidation while decreasing glucose oxidation (Zhang et al., 2004; Ma et al., 2005). PGC-1α increased PDK4 expression via interactions with ERRα (Wende et al., 2005; Zhang et al., 2006). Previous studies from our laboratory have demonstrated that PGC-1α stimulates CPT-1A through the elements in the first intron (Song et al., 2004).
Here, we investigated the interrelationship of PGC-1α and PPARα in the induction of rat CPT-1A gene expression. We found that PPARα stimulates through a PPRE in the second intron of the rat CPT-1A gene. Although PGC-1α interacts with PPARα to induce many genes, our results suggest that PGC-1α does not stimulate the CPT-1A gene through the PPRE in the second intron. Our data provide the unexpected finding that PGC-1α does not induce CPT-1A via interactions with PPARα.
Materials and Methods
Transient transfection of luciferase vectors
CPT-1A-luciferase constructs (CPT-1A-luc) were transfected into HepG2 cells by the calcium phosphate method (Song et al., 2004). Transfections included 2 μg CPT-1A-luciferase along with expression vectors for CMV-PPARα, pSV-PGC-1α and TK-Renilla as indicated. Cells were transfected in Dulbecco's Modified Eagle's Medium (DMEM) containing 5% calf serum/5% fetal calf serum and incubated overnight at 37°C. The medium was replaced by DMEM containing no serum, and cells were treated with fatty acids conjugated to bovine serum albumin (BSA) as indicated or 1 μM WY14643 for 24 hrs (Howell et al., 2009). Luciferase and renilla activity were measured. Protein content in each lysate was determined by Bio-Rad Protein assay (Bio-Rad). Luciferase activity was corrected for both protein content and renilla activity to account for cell density and transfection efficiency, respectively (Zhang et al., 2006).
Construction of the -1653/+1240 CPT-1A-luciferase vector was described previously (Song et al., 2004). The second intron of the CPT-1A gene was obtained by PCR amplification and cloned in front of the luciferase reporter gene in pGL3 (Promega) to create -1779/+2502 CPT-1A-luc. The forward primers contained a Sac1 restriction site (atggagctcgatgaggaaggaaaatgag) and the reverse primer contained a BglII restriction site (ctaagatctagactctggcctacccgta). Mutations in the CPT-1A gene were introduced by site directed mutagenesis with the Quikchange site-directed mutagenesis kit (Stratagene). The following forward primers were used to introduce mutations into -1779/+2502 CPT-1A-luc: MutSp1(+2125) cgcgccagcacgggatatgggctcccgtaacc, MutPPRE ggcggggctcccgtagaatttcccctacttttctcagcc, MutSp1 (+2195) cgggtcggtggaaggaatagggcgggaagccgc, Mut(+2169) cttttctcagccagactgtgaccctggccgggtggg. The primer used to create the mutation in the - 1779/+2502 CPT-1A-luc Mut +686/+689 was ggtgtccagagactgctcaacagtgcgctgggacc. All mutations were confirmed by DNA sequencing at the University of Tennessee Molecular Resource Center.
Real-time PCR
For the preparation of cDNA, RNA was isolated from primary rat hepatocytes with RNA-Stat-60 (Tel-test) (Zhang et al., 2006). The RNA was treated with DNase I (2 units) at 37 °C for 1 h followed by addition of DNase Inactivation Reagent (Ambion). The concentration of each sample containing DNA free RNA was measured using a Nano drop machine (Thermo scientific). Two μg of DNA-free RNA extracted from different cells were converted to cDNA using Superscript reverse transcriptase III and random hexamers (Invitrogen). The concentration of primers in each well of the PCR plates was 0.1 μM, and 1.0 μl of a 1:20 dilution of each cDNA was used as a template. The parameters for real time PCR were as follows: 95 °C for 10 min, 40 cycles of 95 °C 30 s and 60 °C 1 min. Amplification of the target gene was detected by Sybr Green and analyzed by the ΔΔCt method (Howell et al., 2009). The 18S rRNA was used as the reference gene. The following forward (FP) and reverse primers (RP) were used for real time PCR to quantify mRNA abundance: PDK4 FP, ggattactgaccgcctctttagtt; PDK4 RP, gcattccgtgaattgtccatc; PGC-1α FP, atgaatgcagcggtcttagc; PGC-1α RP, aacaatggcagggtttgttc; CPT-1A FP, cggttcaagaatggcatcatc; CPT-1A RP, tcacacccaccaccacgat; 18S FP, cggctaccacatccaaggaa; 18S RP, ttttcgtcactacctccccg.
Adenoviral infection
Purified adenoviruses encoding shRNA specific for the PGC-1α (Ad-shPGC-1α) and control adenoviruses encoding non-template shRNA (Ad-NC) were amplified by Viraquest incorporation (Attia et al., 2010). Primary rat hepatocytes were prepared and plated at a density of 3 × 106 in a 60-mm dish in RPMI 1640 media as described previously (Howell et al., 2009). Five hours after plating the hepatocytes, media was aspirated and purified adenoviruses at an MOI of 10 were added to the cells. The media was changed 24 hours post transduction and the cells were treated with fatty acids or WY14643 for an additional 24 hours. The hepatocytes were harvested, RNA was isolated and real-time PCR was performed as described above.
In vivo electroporation and luciferase assays
Electroporation was performed as outlined previously with some modifications (Lagor et al., 2007). The rat liver was surgically exposed (Lagor et al., 2007). Fifty μl of sterile saline containing 10 μg of the wild type CPT-1A promoter linked to firefly luciferase and 0.01 μg of phRL-CMV renilla were introduced by subcapsular injection into different regions of the right lobe in duplicate. A six-needle hexagonal array electrode (0.5 cm in diameter) was placed over the area and 6 × 150 msec pulses of 150 V/cm with 150 msec rests between pulses were administered with a BTX T830 square-wave electroporator (Harvard Apparatus). Similarly, 10 μg of the CPT-1A promoter linked to firefly luciferase with mutations in the PPRE site and 0.01 μg of phRL-CMV were introduced in duplicate in the left lobe. Thus, wild type and mutant promoters were compared in duplicate in the same animal. After electroporation, the liver was placed back in the abdomen and the wound was closed with surgical staples. The rats were given a single subcutaneous injection of ketoprofen (5 mg/kg) for analgesia before surgery. The animals returned to normal activities in a few minutes. They were maintained on warm water blankets following surgery.
Eight male Sprague Dawley rats (Harlan) weighing 125 to 150 g were adapted to a reverse cycle light room (12 hrs of light followed by 12 hrs of darkness) for a week prior to experimentation. They were allowed free access to Teklad 22/5 rodent chow and water. Four of the rats were fasted for 18 hrs prior to surgery. The other four remained on chow diets. The wild type CPT-1 and CPT-1 PPRE mutant luciferase vectors were introduced into the liver by in vivo electroporation as described above (Attia et al., 2010). Twenty-four hrs after the introduction of the CPT-1 constructs the livers from the fasted (fasted another 24 hrs) and fed rats were harvested. The circular areas defined by six light needle scars were punched out with a 0.5 cm diameter cork borer. The firefly and renilla luciferase activity was determined using the Promega dual luciferase reporter kit and a Turner Designs 20/20 Luminometer. Luciferase activity was expressed as the ratio of firefly to renilla signal.
Real-time PCR of rat liver RNA
Liver samples of 200 μg from areas outside of the transfected spots were obtained from these same animals and used for isolation of total RNA using TRI Reagent from Molecular Research Center. Levels of CPT-1A mRNA were determined by RT-PCR in duplicate as previously described using SYBR green chemistry and a Bio-Rad Chromo4 DNA Engine thermal cycler (Boone et al., 2009). Levels of HMG-CoA reductase mRNA were also determined as a control. The CPT-1A primers were: forward 5′agaccgtgaggaactcaaacccat3′ and reverse 5′cacaacaatgtgcctgctgtcctt3′. The primers used for HMG-CoA reductase were those used previously (Boone et al., 2009).
Electrophoretic mobility shift
Electrophoretic mobility shift assays were conducted by labeling double-stranded oligonucleotides with Klenow enzyme and [α-32P]dCTP as previously reported (Jansen et al., 2000). Oligonucleotides contained sequences representing the various elements in the second intron and are shown in Table 1. The mutant oligomers contained the same sequence as was used in the site directed mutagenesis reactions. The protein-DNA binding mixtures contained labeled probe (30,000 cpm) and the purified protein in 80 mM KCl, 25 mM Tris-HCl (pH 7.4), 0.1 mM EDTA, 1 mM dithiothreitol, 10% glycerol and poly deoxyinosine-deoxycytidine as a nonspecific competitor. The binding reactions were incubated at room temperature for 20 min and then resolved on 5% non-denaturing acrylamide gels (80:1, acrylamide/bisacrylamide) in Tris-glycine running buffer (22 mM Tris and 190 mM glycine). The electrophoresis was carried out at 180 volts for 80 min at 4 °C.
Table 1.
Putative elements in the rat CPT-1A second intron. The sequence of the rat CPT-1A second intron between +2120 and +2100 is shown. The mutations introduced into the luciferase vectors are shown underneath. These are also the oligomer sequences use in the gel shift mobility assays. The altered nucleotides are underlined and placed in italics.
| Sp1(+2125) | PPRE | CREB/ATF | Sp1(+2195) |
| CACGGGGGCGGGGCTCCCGTAACCTTTCCCCTACTTTTCTCAGCCATCACGTGACCCTGGCCGGGTGGGTGGAAGGGGCGGGGCGGG | |||
| MutSp1(+2125) | Mut (+2167) | ||
| CACGGGATATGGGCTC | GCCAGACTGTGACCCTGG | ||
| Mut PPRE | MutSp1(+2195) | ||
| CCGTAGAATTTCCCCTACT | GGAAGGAATAGGGCGG | ||
Results
Our first experiments examined the regulation of the CPT-1A and PDK4 genes by two long chain fatty acids, linoleic acid (18:2) and docosahexaenoic acid (22:6 n-3, DHA), as well as the PPARα agonist WY14643. Primary rat hepatocytes were exposed overnight to these agents. CPT-1A was induced 2 to 3 fold by the addition of each PPARα ligand (Fig. 1A). In addition, we tested the ability of insulin to reduce the induction by fatty acids or WY14643. In the hepatocytes, insulin had a limited ability to reduce the stimulation by these compounds. Only the 35% reduction of the WY14643 CPT-1A mRNA levels achieved statistical significance. The PPARα agonists also increased expression of the PDK4 gene and insulin modestly suppressed this induction (Fig. 1B).
Figure 1. Long chain fatty acids and PPARα agonists induce CPT-1A and PDK4.
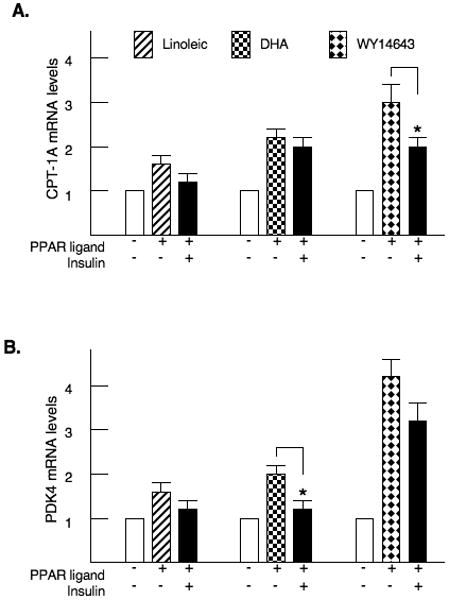
Primary rat hepatocytes were treated with 200μM linoleic acid, 40μM docosahexaenoic acid or 100μM WY14643. Insulin was added at a concentration of 100 nM. After 24 hrs, RNA was harvested and the abundance of CPT-1A (Fig. 1A) and PDK4 mRNA (Fig. 1B) were measured by real-time PCR. The p< 0.05 for the insulin inhibition is indicated by the * asterisk. These numbers are the average of four independent preparations of hepatocytes.
Our next studies were designed to identify a response element for PPARα in the rat CPT-1A gene as there have been varying reports regarding the location of the PPRE (Louet et al., 2001; Napal et al., 2005). We cloned the promoter, first intron and a portion of the CPT-1A second intron and ligated this genomic fragment in front of the luciferase reporter gene to create -1770/+2502 CPT-1A-luc (Fig. 2A). This reporter was transfected into HepG2 hepatoma cells. Cotransfection with an expression vector for PPARα increased the luciferase expression 3.4 fold (Fig. 2A). Addition of WY14643 further increased the activity of the luciferase reporter to 4.1 fold. The addition of insulin did not alter the activity of this reporter construct. Since PGC-1α can interact with PPARα (Lehman and Kelly 2002; Koo et al., 2004), we tested whether overexpression of PGC-1α would enhance the PPARα induction of CPT-1A. Cotransfection with an expression vector for PGC-1α stimulated the CPT-1A gene 2.1 fold. However with respect to stimulating the CPT-1A-luciferase gene, the combination of PGC-1α and PPARα was only additive (Fig 2A). We transfected the -1653/+1240 CPT-1A-luciferase vector that does not contain the second intron. This gene was strongly induced by PGC-1α but not by PPARα further confirming that these factors stimulate CPT-1A through different elements (Fig. 2B).
Figure 2. PPARα and PGC-1α induce CPT-1A through different elements in the gene.
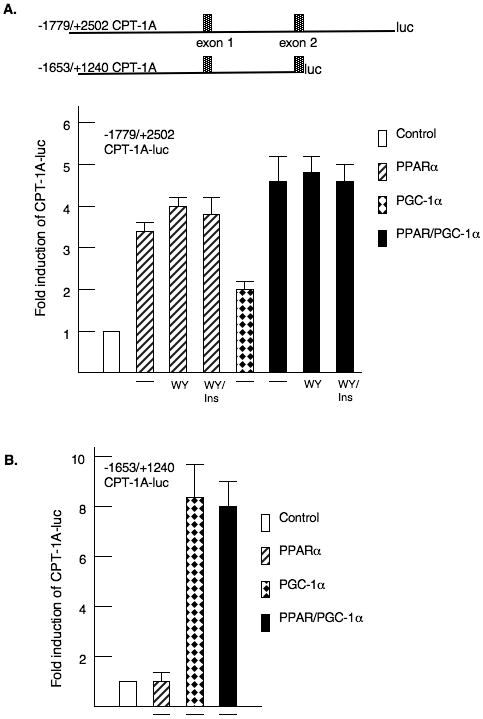
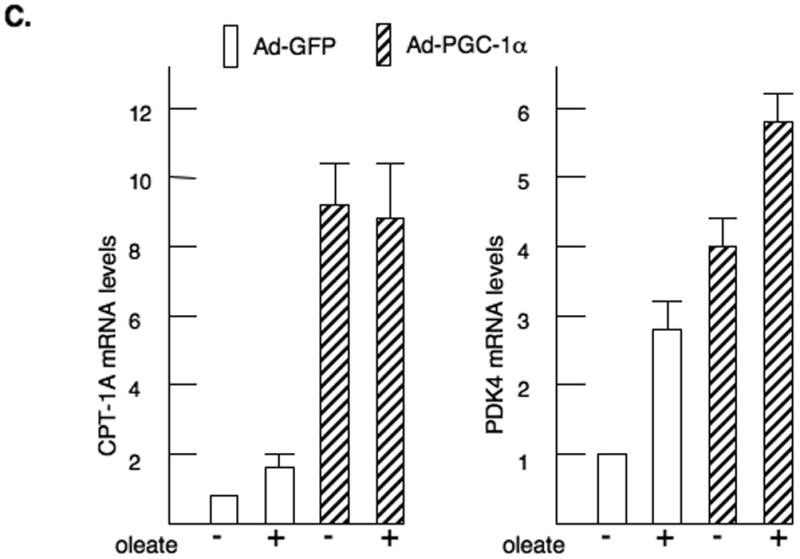
HepG2 hepatoma cells were transfected with CPT-1A-luciferase reporter genes and expression vectors for CMV-PPARα or pSV-PGC-1α as described in the materials and methods. A. A model of the CPT-1A luciferase genes is shown. The first and second exons are identified. The fold induction of the - 1770/+2502 CPT-1A-luciferase (luc) is shown. WY14643 and insulin were added at concentrations of 100μM and 100 nM respectively. All luciferase data was corrected for the protein content in the plate and renilla activity. All experiments were repeated four times in duplicate. B. The -1653/+1240 CPT-1A-luc was cotransfected with PPARα and PGC-1α expression vectors. The data were expressed as fold induction by the various factors. C. Primary rat hepatocytes were infected with adenoviruses expressing GFP (Ad-GFP) or PGC-1α (Ad-PGC-1α) as described previously (Ma et al., 2005). The cells were treated with 350 μM oleic acid for 24 hours prior to harvesting the RNA. CPT-1A and PDK4 mRNA abundance were measured by real-time PCR. The data are expressed as fold induction by either PGC-1α or long chain fatty acid.
Next, we over-expressed PGC-1α in primary rat hepatocytes by adenoviral infection (Zhang et al., 2004). In agreement with our previous reports, overexpression of PGC-1α induced CPT-1A mRNA abundance 9.0 fold (Fig. 2C) while the addition of the long chain fatty acid, oleate, stimulated 1.6 fold (Ma et al., 2005). When oleate was added in the presence of PGC-1α, the stimulation was not increased. These results are similar to the luciferase assays. The PDK4 gene was increased 4.0 fold by PGC-1α overexpression and 2.7 fold by oleate, but the combination of agents only elevated PDK4 expression 5.9 fold. The data from these overexpression approaches suggested that PPARα and PGC-1α do not act synergistically for these genes.
Using the MatInspector program, we identified a potential PPRE in the second intron of the rat CPT-1A gene at nucleotides -2139/-2150. This element had been described in the first intron of the human CPT-1A gene (Napal et al., 2005). First, a broad deletion was generated at the 3′ end of the second intron that removed the PPRE (-1779/+1990 CPT-1A-luc). Deletion of this region eliminated the induction by WY14643 (Fig 3A). In addition, site directed mutagenesis of the PPRE in the second intron (MutPPRE) blocked the induction by PPARα and WY14643. Furthermore, deletion of most of the first intron also attenuated the PPARα induction suggesting that proteins within the first intron are needed for the full WY14643 induction. Proteins bound in the first intron also participate in the T3 induction of CPT-1A (Jackson-Hayes et al., 2003). We tested whether linoleic acid would induce our CPT-1A-luciferase vectors through the PPRE in the second intron. Addition of linoleic acid increased the induction by 1.7 fold and linoleic acid further enhanced the induction by PPARα (Fig 3B). Mutation of the PPRE eliminated the induction by linoleic acid suggesting that fatty acids stimulate CPT-1A through this element. Finally, we used gel shift mobility assays to demonstrate that PPARα and RXRα bound to the PPRE in the CPT-1A promoter (Fig 3C). Mutation of the PPRE eliminated the binding of PPARα to this element consistent with the loss of PPARα responsiveness in the luciferase assays.
Figure 3. Identification of the PPARα response element (PPRE) in CPT-1A.
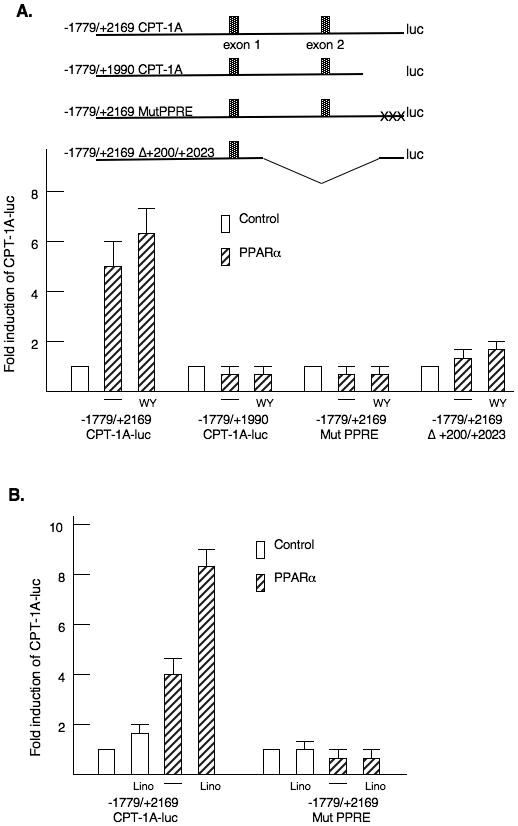
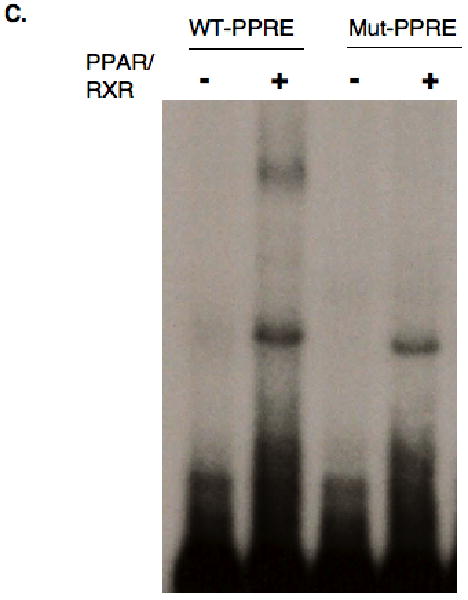
A. A schematic model of the four CPT-1A-luciferase vectors is shown. The CPT-1A-luciferase vectors were transfected with PPARα as described in the legend to figure 2. WY14643 was added at a concentration of 100μM. The results are expressed as the fold induction of luciferase activity. The changes in luciferase activity were corrected for the protein content and renilla activity. These experiments were repeated four times in duplicate. B. The CPT-1A-luciferase vectors were transfected with CMV-PPARα and linoleic acid (200μM) was added. All transfections were repeated four times in duplicate. C. Gel shift mobility assays were conducted with oligomers containing the wild type CPT-1A-PPRE or an oligomer with a mutation (Mut PPRE). The rapidly migrating unbound probe and a non-specific binding protein from the protein preparations from E. Coli are shown in the lower part of the gel. The sequences used in these assays are provided in the materials and methods section. Recombinant GST-PPARα and His-RXRα proteins were prepared as described in the materials and methods.
To determine if the PPRE contributed to CPT-1A regulation in vivo, we transfected the - 1770/+2502 CPT-1A-luciferase wild type and PPRE mutant reporters into rat livers. These vectors were electroporated into discreet regions of the liver of fed or fasted rats (Lagor et al., 2007). This approach allowed direct comparison of the expression of the wild type and mutant promoter in each animal. The data were expressed as the ratio of the expression from the wild type to mutant promoters in each rat. Our results indicate that the CPT-1A-luciferase gene with the disrupted PPRE is expressed at a considerably lower level than the wild type promoter resulting in a ratio of 2.8 (Fig 4A). Fasting results in a significantly increased expression of the wild type CPT-1A-luciferase gene as opposed to the mutant (Fig 4A). The expression of mutant promoter was not different between the fed and fasted animals. The data demonstrate that the PPRE contributes to the fasting induction of CPT-1A in vivo. In addition, we measured the mRNA abundance of CPT-1A in the fasted as opposed to fed rats and found that the CPT-1A mRNA levels were increased three fold with fasting (Fig 4B). Abundance of HMG-CoA reductase mRNA was decreased 75% by fasting (data not shown).
Figure 4. PPARα response element regulates CPT-1A gene expression in rat liver.
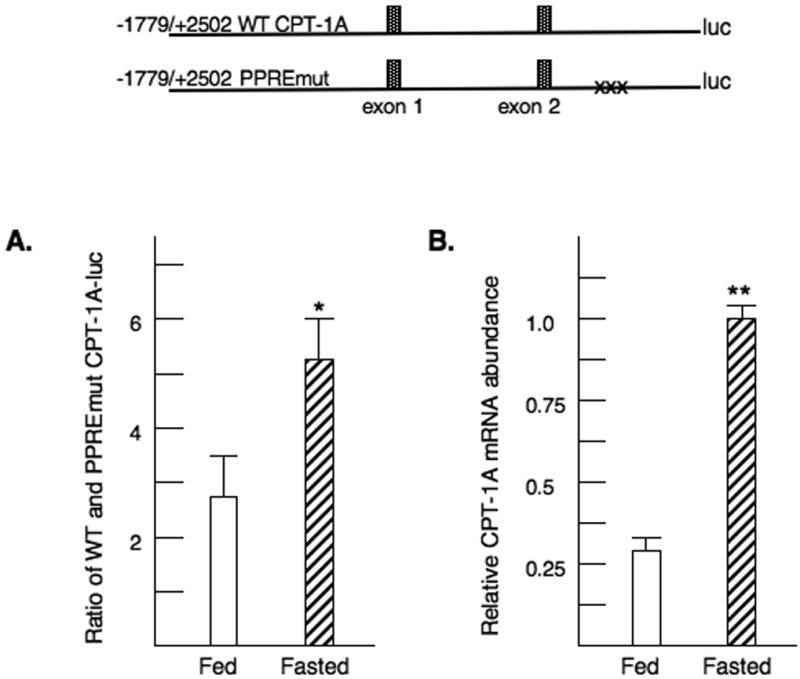
A. Rat livers were transfected by electroporation with the wild type or PPRE mutant CPT-1A-luciferase vectors as described in the materials and methods. Each rat received both constructs. Rats were fasted or fed overnight. Liver punches were removed for luciferase determinations the next day. For each rat, the ratio of luciferase activity from the wild type and mutant PPRE reporters was determined. The p< 0.05 for the relative induction of the WT to PPREmut CPT-1A-luc activity is indicated by the * asterisk. The values are the average of four fed and four fasted rats. B. Liver samples were also removed for isolation of RNA from the livers of these transfected rats. The levels of endogenous CPT-1A mRNA were assessed by real time PCR. The p< 0.01 for the relative CPT-1A mRNA abundance is indicated by the ** asterisks. The values are the average of four fed and four fasted rats.
Surrounding the PPRE are potential binding sites for Sp1 (Table 1). To test whether Sp1 contributed to the transcriptional activation by PPARα, we introduced mutations into each site in the context of the -1779/+2502 CPT-1A-luciferase gene. Only mutation of the PPRE reduced the induction by WY14643 (Fig 5A). Mutation of the 3′ Sp1 site called Sp1(+2195) and the adjacent conserved element, which is similar to a CREB/ATF binding site, decreased basal expression of the CPT-1A gene. We have reported that Sp1 sites in the proximal promoter stimulate basal expression of the CPT-1A gene (Steffen et al., 1999). However, the induction by PPARα and WY14643 were fully retained (Fig 5A). Our results suggest that the elements surrounding the PPRE do not participate in PPARα responsiveness. We also tested whether the induction by linoleic acid required the Sp1 sites. Linoleic acid alone induced the CPT-1A-luciferase vector modestly. As with WY14643, the surrounding Sp1 binding sites were not involved in the fatty acid induction (Fig 5B). Gel shift mobility assays were used to test whether Sp1 would bind to the GC rich regions surrounding the PPARα binding site (Fig 5C). Proteins isolated from rat liver nuclear extract bound the Sp1 elements and these complexes were disrupted by the addition of an antibody demonstrating that Sp1 can bind to these elements.
Figure 5. Characterization of binding sites surrounding the rat CPT-1A PPRE.
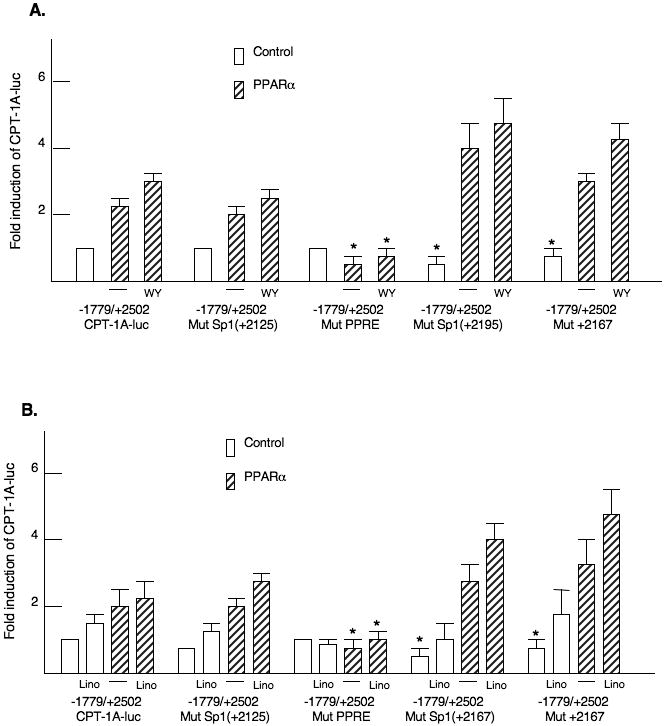
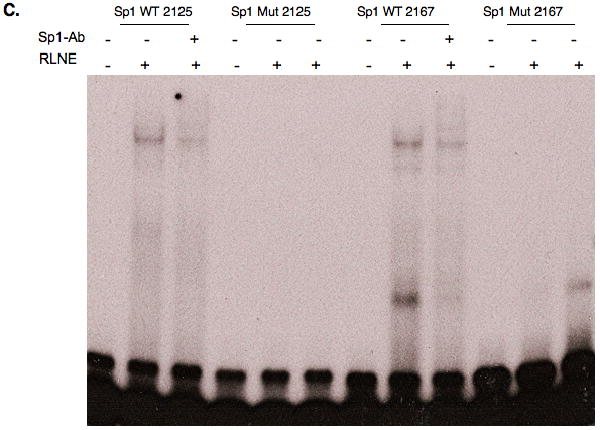
Mutations were introduced into the Sp1 and PPRE binding sites in the second intron of the -1770/+2502 CPT-1A-luciferase vector. A. The various luciferase vectors were transfected with or without PPARα as described in the legend to figure 2. WY14643 was added at a concentration of 100μM. The p< 0.05 for the relative WT to mutant CPT-1A-luc activity is indicated by the * asterisk. The transfection experiments were conducted four times in duplicate. B. The transfections were conducted in HepG2 cells exactly as described above except that linoleic acid was added at a concentration of 200μM. C. Gel shift mobility assays were conducted with nuclear proteins prepared from rat liver nuclei. Rapidly migrating DNA-protein complexes that do not contain Sp1 were observed with the Sp1 (2167) oligonucleotide. Antibodies were added for Sp1 as described in the materials and methods.
We had shown previously that elements in the first intron recruited PGC-1α to the CPT-1A promoter (Jackson-Hayes et al., 2003; Song et al., 2004). To further examine the role of the first intron in the CPT-1A gene, we disrupted one of the elements needed for PGC-1α responsiveness of the promoter. The transcription factor binding to this element is currently unknown. As expected, the stimulation by PGC-1α was reduced by this mutation. Disruption of this element decreased the induction by liganded PPARα from 4.8 ± 0.6 to 2.9 ± 0.2 suggesting that factors in the first intron are need for PPARα actions with respect to the CPT-1A gene (Fig 6). Overall these data in combination with the broad deletion in figure 3 suggest that proteins bound to the first intron are needed for the induction of CPT-1A by PPARα ligands.
Figure 6. Elements in the CPT-1A first intron contribute to the induction by PPARα.
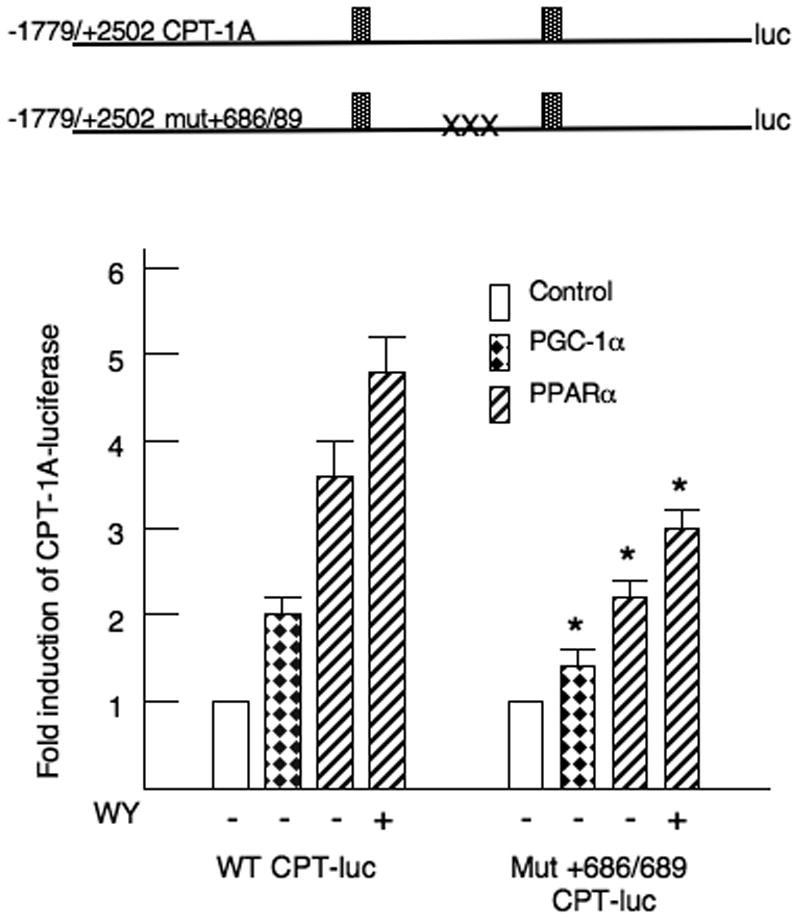
CPT-1A-luciferase vectors were transfected into HepG2 cells exactly as described in the legend to figure 2. The mutation of the nucleotides +686/+689 in the first intron is described in the materials and methods. The p< 0.05 for the fold induction of the WT to mutant CPT-1A-luc activity for each condition is indicated by the * asterisk. All transfections were repeated four times in duplicate.
We investigated the question of whether the coactivator PGC-1α participated in the PPARα induction of CPT-1A and PDK4 with shRNA directed against PGC-1α. To conduct these experiments, we infected primary rat hepatocytes with either a control adenovirus (Ad-NC) or a short hairpin RNA against PGC-1α (Ad-shPGC-1α) (Connaughton et al., 2010). We treated the cells with WY14643 as this agonist provided a larger and more consistent induction of CPT-1A. CPT-1A mRNA abundance was increased 5.9 ± 1.5 fold by the addition of WY 14,643. The induction of CPT-1A by WY14643 was attenuated to 3.2 ± 0.9 fold in the presence of the PGC-1α shRNA (Fig 7). However, this reduction was not statistically significant. In addition, the 10.9 ± 2.1 fold induction of the pyruvate dehydrogenase kinase 4 (PDK4) gene by WY 14,643 was decreased to 7.9 ± 2.6 by PGC-1α knockdown. We did not observe any stimulation of PGC-1α by WY14643 suggesting that PPARα does not induce PGC-1α expression at least in the liver. Overall, our results indicate that PGC-1α is not required for the induction of these genes by PPARα in the liver.
Figure 7. Induction of CPT-1A and PDK4 by WY14643 are independent of PGC-1α.
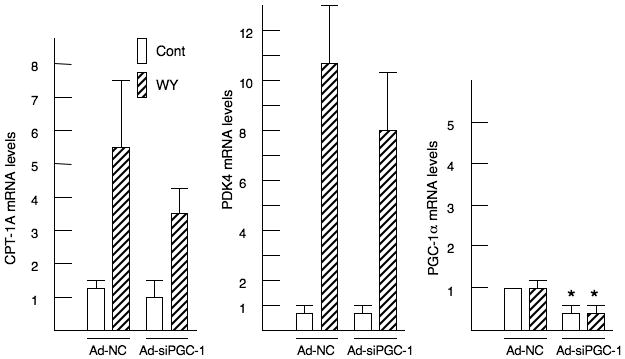
Primary rat hepatocytes were infected with either Ad-NC or Ad-shPGC-1α for 40 hrs. After 24 hrs of infection, the cells were exposed to 100 μM WY14643. RNA was isolated and the CPT-1A, PDK4 and PGC-1α mRNA abundance were measured by real time PCR. The p< 0.05 for the mRNA abundance of the shPGC-1α infected cells is indicated by the * asterisk. These experiments were repeated five times on independent preparations of hepatocytes.
Discussion
Previous studies from our laboratory and others determined that expression of the CPT-1A and PDK4 genes is induced by long chain fatty acids and PPARα agonists (Cook et al., 2001; Wu et al., 2001; Huang et al., 2002). Here, we report the characterization of a binding site for PPARα in the second intron of the rat CPT-1A gene. This element mediates the induction by PPARα ligands and contributes to the induction of CPT-1A gene expression in the liver of fasted rats. In addition, we investigated whether PGC-1α would directly coactivate PPARα on the rat CPT-1A gene. Our data indicate that PGC-1α stimulates the CPT-1A gene independently of PPARα through elements in the first intron and suggest that PPARα and PGC-1α do not synergistically stimulate CPT-1A expression.
PPARα knockout mice have diminished induction of numerous genes including CPT-1A and PDK4 following fasting or administration of pharmacologic ligands for PPARα (Louet et al., 2001; Wu et al., 2001). There have been varying reports regarding the mechanisms by which long chain fatty acids induce the CPT-1A gene. There is general agreement that pharmacologic PPARα agonists act through PPARα (Desvergne and Wahli 1999). For the rat CPT-1A gene, a PPRE consisting of a direct repeat separated by one nucleotide (DR1) was described in the promoter (Louet et al., 2001). This DR1 element was also found to bind hepatic nuclear factor 4 (HNF4) suggesting a complex pattern of transcription factor association with this element (Louet et al., 2002). While we did not find that the CPT-1A promoter responded to PPARα in our studies, these differences may reflect transfection of CPT-luciferase reporters into different cell types. In addition, there have been reports that long chain fatty acids induce CPT-1A via a PPARα independent mechanism (Le May et al., 2005). A fatty acid responsive region was described in the first intron, and it was shown that the CPT-1A gene is stimulated by long chain fatty acids in the presence of a dominant negative PPARα vector (Louet et al., 2001; Le May et al., 2005). To our knowledge, no specific proteins involved in the PPARα independent induction of CPT-1A have been identified. Our transfection data suggest that long chain fatty acids utilize PPARα to induce the CPT-1A gene. However, it is highly likely that long chain fatty acids modulate gene expression through other mechanisms and our experiments do not eliminate this possibility (Duplus and Forest 2002; Pegorier et al., 2004).
The human and rat CPT-1A genes are highly homologous in the exons encoding the protein but there is divergence in the promoter regions and the introns. Both the rat and human genes contain an intronic PPARα binding site (Napal et al., 2005). The human CPT-1A gene contains alternate first exons (exons 1a and 1b) while the rat gene has a single exon 1 (Park et al., 1998; Gobin et al., 2002; Napal et al., 2005). The rat gene contains a non-coding second exon that is not expressed in the human (Park et al., 1998; Gobin et al., 2002). The initiating ATG for protein synthesis is in the third exon of the rat and the second exon of the human genes. One salient difference in the regulation of the rat and human genes lies with the interaction of PPARα and PGC-1α. Using human CPT-1A-luciferase reporters, it was found that mutation of the PPRE completely eliminated the induction by both PGC-1α and PPARα (Napal et al., 2005). In our transfections with rat CPT-1A luciferase genes, PGC-1α induced the reporter through elements other than the PPRE. The element in the first intron through which PGC-1α stimulates is closely conserved in the rat (tgctcaaggtcgcgctg) and human (tgcccaaggtcgtgccg) genes. In addition, our PGC-1α knockdown experiments suggest that PPARα ligands can induce the rat CPT-1A gene in a PGC-1α independent manner. The reason for this different response in rat and human is not clear. Although we did not clone the intronic region of the human CPT-1A gene containing the PPRE, we have observed in luciferase assays that PGC-1α will also stimulate human CPT-1A gene expression through the promoter upstream of exon 1a (data not shown) suggesting that the induction of human CPT-1A by PGC-1α may involve several regions of the gene. Although there are differences in the architecture of the rat and human CPT-1A genes, PPARα and PGC-1α retain the ability to stimulate the CPT-1A gene in both species.
PPARα has been found to be an important mediator of the hepatic response to starvation, and PPARα knockout mice develop hepatic steatosis with fasting (Kersten et al., 1999; Leone et al., 1999). Our data indicate that the PPRE in the second intron is important for the CPT-1A induction in fasting. Although many hormonal changes occur in the fasted animal, the elevation of free fatty acids released from adipose tissue contributes to altered gene expression (Duplus and Forest 2002). Additional data from our laboratory suggested that the CPT-1A promoter and first intron were not sufficient for the induction of CPT-1A in the fasted animal. We had previously generated two lines of transgenic mice that expressed the -6870/+1240 region of the rat CPT-1A gene driving the luciferase reporter (Jackson-Hayes et al., 2003). This transgene possessed the first intron and a large promoter region but did not contain the second intron with the PPRE. While this transgene was responsive to thyroid hormone (Jackson-Hayes et al., 2003), the CPT-1A reporter gene was not induced when the animals were fasted suggesting that additional elements stimulate CPT-1A (data not shown). Our in vivo liver transfections of CPT-1A-luciferase vectors containing the second intron with a mutation in the PPRE demonstrated that the PPRE induces the CPT-1A gene in fasted animals.
Multiple hormones regulate expression of the CPT-1A gene including glucocorticoids, thyroid hormone and insulin (Mynatt et al., 1994; Park et al., 1995; Cook et al., 2001). We previously reported that insulin decreased expression of the CPT-1A gene in H4IIE hepatoma cells, but we were only able to observe an inhibition of the CPT-1A gene in the presence of glucocorticoids even though the induction of CPT-1A by dexamethasone was modest (Park et al., 1995). Here, we show that fed rats have lower levels of CPT-1A mRNA in the liver which is likely due in part to inhibition by insulin as well as the lower levels of free fatty acids. The PPRE mediates the stimulation of CPT-1A-luciferase in the livers of fasted rats. These data point to PPARα as contributing to the induction of CPT-1A in fasted rats, but the mechanism by which insulin reduces CPT-1A is not known. In our studies in primary hepatocytes, we have found only modest decreases in the WY14643 induction of the CPT-1A gene by insulin. Expression of the PDK4 gene is also inhibited by insulin (Huang et al., 2002). The insulin inhibition of PDK4 occurs primarily through the phosphorylation and cytosolic relocalization of FoxO1 (Kwon et al., 2004; Connaughton et al., 2010). To our knowledge, there is no evidence implicating FoxO1 in the regulation of the CPT-1A gene.
One issue raised by our studies is which of hormonal responses of the CPT-1A and PDK4 genes utilize PGC-1α. Thyroid hormone (T3) stimulates CPT-1A and PDK4 expression. PGC-1α is a crucial coactivator in T3 responsiveness as knockdown of PGC-1α decreased the ability of T3 to induce both these genes (Zhang et al., 2004; Attia et al., 2010). We used multiple approaches including overexpression, promoter mutagenesis and shRNA mediated knockdown to evaluate the role of PGC-1α in CPT-1A gene expression. While PGC-1α stimulates basal expression of the CPT-1A gene, the most unexpected aspect of this study was that we did not observe a synergistic induction between PPARα and PGC-1α for the CPT-1A gene. This synergism was anticipated in part because PPARα and PGC-1α can physically interact through the respective AF2 and LLXXL motifs of the respective proteins (Finck and Kelly 2006). Both factors induce overlapping metabolic pathways, and PGC-1α is elevated in the liver of fasted animals (Leone et al., 1999; Rhee et al., 2003). Following PGC-1α knockout, there is hepatic steatosis in the fasted mice clearly demonstrating a role for this coactivator in stimulating hepatic fatty acid oxidation (Lin et al., 2004; Leone et al., 2005). We did not observe that WY14643 directly increased PGC-1α abundance in cell culture experiments. In contrast to the CPT-1A gene, PPARα and PGC-1α directly coactivate the CPT-1B and MCAD genes in heart and skeletal muscle (Lehman and Kelly 2002). We have also observed that mutation of the PPRE in the CPT-1B gene decreases the stimulation by PPARα and PGC-1α (data not shown). There are likely to be numerous genes where direct interactions of these factors are an important regulatory mechanism (Finck and Kelly 2006). In summary, we have found that long chain fatty acids induce the rat CPT-1A gene through PPARα binding site in the second intron. PGC-1α stimulates the CPT-1A and PDK4 genes through other gene elements. PPARα responsiveness is retained following PGC-1α knockdown suggesting that for some lipid regulated genes the association of PGC-1α with the gene does not involve direct recruitment by PPARα.
Acknowledgments
This work is supported in part by grants DK059368 to EAP, DK075504 to MBE and DK075414 to GCN.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Attia RR, Connnaughton S, Boone LR, Wang F, Elam MB, Ness GC, Cook GA, Park EA. Regulation of pyruvate dehydrogenase kinase 4 (PDK4) by thyroid hormone: role of the peroxisome proliferator-activated receptor gamma coactivator (PGC-1 alpha) J Biol Chem. 2010;285:2375–85. doi: 10.1074/jbc.M109.039081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone LR, Niesen MI, Jaroszeski M, Ness GC. In vivo identification of promoter elements and transcription factors mediating activation of hepatic HMG-CoA reductase by T3. Biochem Biophys Res Commun. 2009;385:466–71. doi: 10.1016/j.bbrc.2009.05.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatelain F, Kohl C, Esser V, McGarry JD, Girard J, Pegorier JP. Cyclic AMP and fatty acids increase carnitine palmitoyltransferase I gene transcription in cultured fetal rat hepatocytes. Eur J Biochem. 1996;235:789–98. doi: 10.1111/j.1432-1033.1996.00789.x. [DOI] [PubMed] [Google Scholar]
- Connaughton S, Chowdhury F, Attia RR, Song S, Zhang Y, Elam MB, Cook GA, Park EA. Regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) gene expression by glucocorticoids and insulin. Mol Cell Endocrinol. 2010;315:159–67. doi: 10.1016/j.mce.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook GA, Edwards TL, Jansen MS, Bahouth SW, Wilcox HG, Park EA. Differential regulation of carnitine palmitoyltransferase-I gene isoforms (CPT-I alpha and CPT-I beta) in the rat heart. J Mol Cell Cardiol. 2001;33:317–29. doi: 10.1006/jmcc.2000.1304. [DOI] [PubMed] [Google Scholar]
- Cook GA, Otto DA, Cornell NW. Differential inhibition of ketogenesis by malonyl-CoA in mitochondria from fed and starved rats. Biochem J. 1980;192:955–8. doi: 10.1042/bj1920955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt T, Saramaki A, Malinen M, Rieck M, Vaisanen S, Huotari A, Herzig KH, Muller R, Carlberg C. Three Members of the Human Pyruvate Dehydrogenase Kinase Gene Family Are Direct Targets of the Peroxisome Proliferator-activated Receptor beta/delta. J Mol Biol. 2007;372:341–55. doi: 10.1016/j.jmb.2007.06.091. [DOI] [PubMed] [Google Scholar]
- Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–88. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- Duplus E, Forest C. Is there a single mechanism for fatty acid regulation of gene transcription? Biochem Pharmacol. 2002;64:893–901. doi: 10.1016/s0006-2952(02)01157-7. [DOI] [PubMed] [Google Scholar]
- Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. Journal of Clinical Investigation. 2006;116:615–22. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation. 2007;115:2540–8. doi: 10.1161/CIRCULATIONAHA.107.670588. [DOI] [PubMed] [Google Scholar]
- Gobin S, Bonnefont JP, Prip-Buus C, Mugnier C, Ferrec M, Demaugre F, Saudubray JM, Rostane H, Djouadi F, Wilcox W, Cederbaum S, Haas R, Nyhan WL, Green A, Gray G, Girard J, Thuillier L. Organization of the human liver carnitine palmitoyltransferase 1 gene (CPT1A) and identification of novel mutations in hypoketotic hypoglycaemia. Hum Genet. 2002;111:179–89. doi: 10.1007/s00439-002-0752-0. [DOI] [PubMed] [Google Scholar]
- Harris RA, Bowker-Kinley MM, Huang B, Wu P. Regulation of the activity of the pyruvate dehydrogenase complex. Adv Enzyme Regul. 2002;42:249–59. doi: 10.1016/s0065-2571(01)00061-9. [DOI] [PubMed] [Google Scholar]
- Holness MJ, Bulmer K, Smith ND, Sugden MC. Investigation of potential mechanisms regulating protein expression of hepatic pyruvate dehydrogenase kinase isoforms 2 and 4 by fatty acids and thyroid hormone. Biochem J. 2003;369:687–95. doi: 10.1042/BJ20021509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell G, 3rd, Deng X, Yellaturu C, Park EA, Wilcox HG, Raghow R, Elam MB. N-3 polyunsaturated fatty acids suppress insulin-induced SREBP-1c transcription via reduced trans-activating capacity of LXRalpha. Biochim Biophys Acta. 2009;1791:1190–6. doi: 10.1016/j.bbalip.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Wu P, Bowker-Kinley MM, Harris RA. Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptor-alpha ligands, glucocorticoids, and insulin. Diabetes. 2002;51:276–83. doi: 10.2337/diabetes.51.2.276. [DOI] [PubMed] [Google Scholar]
- Jackson-Hayes L, Song S, Lavrentyev EN, Jansen MS, Hillgartner FB, Tian L, Wood PA, Cook GA, Park EA. A thyroid hormone response unit formed between the promoter and first intron of the carnitine palmitoyltransferase-Ialpha gene mediates the liver-specific induction by thyroid hormone. J Biol Chem. 2003;278:7964–72. doi: 10.1074/jbc.M211062200. [DOI] [PubMed] [Google Scholar]
- Jansen MS, Cook GA, Song S, Park EA. Thyroid hormone regulates carnitine palmitoyltransferase Ialpha gene expression through elements in the promoter and first intron. J Biol Chem. 2000;275:34989–97. doi: 10.1074/jbc.M001752200. [DOI] [PubMed] [Google Scholar]
- Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest. 1999;103:1489–98. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo SH, Satoh H, Herzig S, Lee CH, Hedrick S, Kulkarni R, Evans RM, Olefsky J, Montminy M. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat Med. 2004;10:530–4. doi: 10.1038/nm1044. [DOI] [PubMed] [Google Scholar]
- Kwon HS, Huang B, Unterman TG, Harris RA. Protein kinase B-alpha inhibits human pyruvate dehydrogenase kinase-4 gene induction by dexamethasone through inactivation of FOXO transcription factors. Diabetes. 2004;53:899–910. doi: 10.2337/diabetes.53.4.899. [DOI] [PubMed] [Google Scholar]
- Lagor WR, Heller R, de Groh ED, Ness GC. Functional analysis of the hepatic HMG-CoA reductase promoter by in vivo electroporation. Exp Biol Med (Maywood) 2007;232:353–61. [PubMed] [Google Scholar]
- Le May C, Cauzac M, Diradourian C, Perdereau D, Girard J, Burnol AF, Pegorier JP. Fatty acids induce L-CPT I gene expression through a PPARalpha-independent mechanism in rat hepatoma cells. J Nutr. 2005;135:2313–9. doi: 10.1093/jn/135.10.2313. [DOI] [PubMed] [Google Scholar]
- Lehman JJ, Kelly DP. Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin Exp Pharmacol Physiol. 2002;29:339–45. doi: 10.1046/j.1440-1681.2002.03655.x. [DOI] [PubMed] [Google Scholar]
- Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci U S A. 1999;96:7473–8. doi: 10.1073/pnas.96.13.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–70. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–35. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Louet JF, Chatelain F, Decaux JF, Park EA, Kohl C, Pineau T, Girard J, Pegorier JP. Long-chain fatty acids regulate liver carnitine palmitoyltransferase I gene (L-CPT I) expression through a peroxisome-proliferator-activated receptor alpha (PPARalpha)-independent pathway. Biochem J. 2001;354:189–97. doi: 10.1042/0264-6021:3540189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louet JF, Hayhurst G, Gonzalez FJ, Girard J, Decaux JF. The coactivator PGC-1 is involved in the regulation of the liver carnitine palmitoyltransferase I gene expression by cAMP in combination with HNF4 alpha and cAMP-response element-binding protein (CREB) J Biol Chem. 2002;277:37991–8000. doi: 10.1074/jbc.M205087200. [DOI] [PubMed] [Google Scholar]
- Ma K, Zhang Y, Elam MB, Cook GA, Park EA. Cloning of the Rat Pyruvate Dehydrogenase Kinase 4 Gene Promoter: Activation of pyruvate dehydrogenase kinase by the peroxisome proliferator-activated receptor {gamma} coactivator. J Biol Chem. 2005;280:29525–32. doi: 10.1074/jbc.M502236200. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–50. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- McGarry JD, Brown NF. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur J Biochem. 1997;244:1–14. doi: 10.1111/j.1432-1033.1997.00001.x. [DOI] [PubMed] [Google Scholar]
- Mynatt RL, Park EA, Thorngate FE, Das HK, Cook GA. Changes in carnitine palmitoyltransferase-I mRNA abundance produced by hyperthyroidism and hypothyroidism parallel changes in activity. Biochem Biophys Res Commun. 1994;201:932–7. doi: 10.1006/bbrc.1994.1791. [DOI] [PubMed] [Google Scholar]
- Napal L, Marrero PF, Haro D. An intronic peroxisome proliferator-activated receptor-binding sequence mediates fatty acid induction of the human carnitine palmitoyltransferase 1A. J Mol Biol. 2005;354:751–9. doi: 10.1016/j.jmb.2005.09.097. [DOI] [PubMed] [Google Scholar]
- Park EA, Cook GA. Differential regulation in the heart of mitochondrial carnitine palmitoyltransferase-I muscle and liver isoforms. Mol Cell Biochem. 1998;180:27–32. [PubMed] [Google Scholar]
- Park EA, Mynatt RL, Cook GA, Kashfi K. Insulin regulates enzyme activity, malonyl-CoA sensitivity and mRNA abundance of hepatic carnitine palmitoyltransferase-I. Biochem J. 1995;310(Pt 3):853–8. doi: 10.1042/bj3100853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EA, Steffen ML, Song S, Park VM, Cook GA. Cloning and characterization of the promoter for the liver isoform of the rat carnitine palmitoyltransferase I (L-CPT I) gene. Biochem J. 1998;330(Pt 1):217–24. doi: 10.1042/bj3300217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegorier JP, Le May C, Girard J. Control of gene expression by fatty acids. J Nutr. 2004;134:2444S–2449S. doi: 10.1093/jn/134.9.2444S. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–39. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Rhee J, Inoue Y, Yoon JC, Puigserver P, Fan M, Gonzalez FJ, Spiegelman BM. Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc Natl Acad Sci U S A. 2003;100:4012–7. doi: 10.1073/pnas.0730870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld MG, Glass CK. Coregulator codes of transcriptional regulation by nuclear receptors. J Biol Chem. 2001;276:36865–8. doi: 10.1074/jbc.R100041200. [DOI] [PubMed] [Google Scholar]
- Song S, Zhang Y, Ma K, Jackson-Hayes L, Lavrentyev EN, Cook GA, Elam MB, Park EA. Peroxisomal proliferator activated receptor gamma coactivator (PGC-1alpha) stimulates carnitine palmitoyltransferase I (CPT-Ialpha) through the first intron. Biochim Biophys Acta. 2004;1679:164–73. doi: 10.1016/j.bbaexp.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Steffen ML, Harrison WR, Elder FF, Cook GA, Park EA. Expression of the rat liver carnitine palmitoyltransferase I (CPT-Ialpha) gene is regulated by Sp1 and nuclear factor Y: chromosomal localization and promoter characterization. Biochem J. 1999;340(Pt 2):425–32. [PMC free article] [PubMed] [Google Scholar]
- Sugden MC, Bulmer K, Gibbons GF, Knight BL, Holness MJ. Peroxisome-proliferator-activated receptor-alpha (PPARalpha) deficiency leads to dysregulation of hepatic lipid and carbohydrate metabolism by fatty acids and insulin. Biochem J. 2002;364:361–8. doi: 10.1042/BJ20011699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am J Physiol Endocrinol Metab. 2003;284:E855–62. doi: 10.1152/ajpendo.00526.2002. [DOI] [PubMed] [Google Scholar]
- Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–76. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wende AR, Huss JM, Schaeffer PJ, Giguere V, Kelly DP. PGC-1alpha coactivates PDK4 gene expression via the orphan nuclear receptor ERRalpha: a mechanism for transcriptional control of muscle glucose metabolism. Mol Cell Biol. 2005;25:10684–94. doi: 10.1128/MCB.25.24.10684-10694.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Peters JM, Harris RA. Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor alpha. Biochem Biophys Res Commun. 2001;287:391–6. doi: 10.1006/bbrc.2001.5608. [DOI] [PubMed] [Google Scholar]
- Wu Y, Delerive P, Chin WW, Burris TP. Requirement of helix 1 and the AF-2 domain of the thyroid hormone receptor for coactivation by PGC-1. J Biol Chem. 2002;277:8898–905. doi: 10.1074/jbc.M110761200. [DOI] [PubMed] [Google Scholar]
- Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, Spiegelman BM. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–8. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ma K, Sadana P, Chowdhury F, Gaillard S, Wang F, McDonnell DP, Unterman TG, Elam MB, Park EA. Estrogen-related receptors stimulate pyruvate dehydrogenase kinase isoform 4 gene expression. J Biol Chem. 2006;281:39897–906. doi: 10.1074/jbc.M608657200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ma K, Song S, Elam MB, Cook GA, Park EA. Peroxisomal proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1 alpha) enhances the thyroid hormone induction of carnitine palmitoyltransferase I (CPT-I alpha) J Biol Chem. 2004;279:53963–71. doi: 10.1074/jbc.M406028200. [DOI] [PubMed] [Google Scholar]