

Abstract
Psychosocial stress is associated with altered immune function and development of psychological disorders including anxiety and depression. Here we show that repeated social defeat in mice increased c-Fos staining in brain regions associated with fear and threat appraisal and promoted anxiety-like behavior in a β-adrenergic receptor-dependent manner. Repeated social defeat also significantly increased the number of CD11b+/CD45high/Ly6Chigh macrophages that trafficked to the brain. In addition, several inflammatory markers were increased on the surface of microglia (CD14, CD86, and TLR4) and macrophages (CD14 and CD86) after social defeat. Repeated social defeat also increased the presence of deramified microglia in the medial amygdala, prefrontal cortex, and hippocampus. Moreover, mRNA analysis of microglia indicated that repeated social defeat increased levels of interleukin (IL)-1β and reduced levels of glucocorticoid responsive genes [glucocorticoid-induced leucine zipper (GILZ) and FK506 binding protein-51 (FKBP51)]. The stress-dependent changes in microglia and macrophages were prevented by propranolol, a β-adrenergic receptor antagonist. Microglia isolated from socially defeated mice and cultured ex vivo produced markedly higher levels of IL-6, tumor necrosis factor-α, and monocyte chemoattractant protein-1 after stimulation with lipopolysaccharide compared with microglia from control mice. Last, repeated social defeat increased c-Fos activation in IL-1 receptor type-1-deficient mice, but did not promote anxiety-like behavior or microglia activation in the absence of functional IL-1 receptor type-1. These findings indicate that repeated social defeat-induced anxiety-like behavior and enhanced reactivity of microglia was dependent on activation of β-adrenergic and IL-1 receptors.
Introduction
Psychosocial stressors activate neuroendocrine pathways to release catecholamines, glucocorticoids, and cytokines. These stressor-induced pathways have a profound influence on behavior, immunity, and physiology in both humans and rodents (Blanchard et al., 2001; Kiecolt-Glaser and Glaser, 2002; Kinsey et al., 2007; Cole et al., 2010). For example, repeated social defeat in mice promotes anxiety-like behavior (Kinsey et al., 2007; Krishnan et al., 2007) and alters immune responses to viral and bacterial challenges (Bailey et al., 2009; Mays et al., 2010). These immune changes are related to a decreased sensitivity of peripheral myeloid cells (CD11b+) to glucocorticoid (GC) anti-inflammatory feedback (Stark et al., 2001). Moreover, social defeat promotes trafficking of these GC-resistant CD11b+ cells from the bone marrow to the spleen and lung (Engler et al., 2004; Curry et al., 2010). The GC-resistant CD11b+ cells from socially defeated mice are more inflammatory with increased production of the inflammatory cytokines, interleukin (IL)-6, tumor necrosis factor (TNF)-α, and IL-1β following antigen stimulation (Stark et al., 2001; Avitsur et al., 2003; Bailey et al., 2009). The increased inflammatory profile and GC-insensitivity of these myeloid cells may be driven by IL-1β. In support of this premise, CD11b+ cells from mice lacking interleukin-1 receptor type-1 (IL-1r1−/−) do not develop GC-resistance following repeated social defeat (Engler et al., 2008). Although these findings indicate that repeated social defeat increases the proinflammatory profile of peripheral CD11b+ cells, the degree to which repeated social defeat affects the inflammatory profile of resident CD11b+ cells of the CNS is unknown.
This is relevant because CNS macrophages and microglia are CD11b+ cells that play a pivotal role in interpreting and propagating inflammatory signals in the brain (Nguyen et al., 2002; Davalos et al., 2005; Nimmerjahn et al., 2005). For example, several stressors including inescapable shock (Frank et al., 2007) and restraint stress (Nair and Bonneau, 2006; Tynan et al., 2010) enhance the inflammatory profile of microglia. The pathway by which microglia and macrophages are activated by stress is unclear. It is plausible, however, that activation of β-adrenergic receptors during stress enhances inflammation within the CNS by increasing the expression of IL-1β (Johnson et al., 2008; McNamee et al., 2010). Moreover, repeated social defeat promotes a similar pattern of inflammation and β-adrenergic receptor-mediated-gene expression (e.g., GATA1) in the cortex of mice that is consistent with the increased peripheral inflammatory profile detected in humans after prolonged social stress (Cole et al., 2010).
Therefore, the purpose of this study was to understand how the inflammatory signals that are initiated in response to social defeat are interpreted and propagated in the brain. Our data indicate that repeated social defeat increased c-Fos activation in brain regions associated with threat and fear appraisal and promoted anxiety-like behavior. Social defeat also enhanced the inflammatory profile of CD11b+ microglia and macrophages in the brain. The social defeat-induced c-Fos activation, anxiety-like behavior, and neuroinflammation were blocked by β-adrenergic receptor antagonism. Last, the absence of a functional IL-1 receptor type-1 ablated the social defeat-induced microglial activation and anxiety-like behavior.
Materials and Methods
Animals
Male C57BL/6 (6–8 weeks old) and CD-1 (12 months, retired breeders) mice were obtained from Charles River Breeding Laboratories and allowed to acclimate to their surroundings for 7–10 d before experimentation. IL-1r1−/− mice on a C57BL/6 background were a gift from Dr. Ning Quan (Ohio State University, Columbus, OH). C57BL/6 mice were housed in cohorts of three and CD-1 mice were singly housed in 11.5 × 7.5 × 6 inch polypropylene cages. Rooms were maintained at 21°C under a 12:12 h light:dark cycle with ad libitum access to water and rodent chow. Mice were randomly selected for inclusion in experimental treatment groups. In addition, propranolol and vehicle injections were administered throughout cages to avoid any intracage confounds. All procedures were in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals and were approved by the Ohio State University Institutional Laboratory Animal Care and Use Committee.
Social disruption stress
Social disruption stress (SDR) was performed as previously described (Avitsur et al., 2001). In brief, a male intruder CD-1 mouse (12 months retired breeder) was introduced into cages of established male cohorts (three per cage) of C57BL/6 mice for 2 h between 17:00 and 19:00 for 6 consecutive nights. During each cycle, submissive behavior including upright posture, fleeing, and crouching (Avitsur et al., 2001; Stark et al., 2001) was observed. If the intruder did not initiate an attack within 5–10 min or was attacked by any of the resident mice, then a new intruder was introduced. At the end of the 2 h period, the intruder was removed and the residents were left undisturbed until the following day when the paradigm was repeated. Different intruders were used on consecutive nights. Home-cage control (HCC) mice were also housed in cohorts of three, but were left undisturbed in a separate room. The health status of the mice was determined throughout the experiment. Mice that were injured or moribund were removed from the study. Consistent with previous studies using repeated social defeat (Avitsur et al., 2001; Stark et al., 2001), <5% of mice (15 of 300) met the early removal criteria. Following administration of propranolol, there were no noticeable differences in interactions between the intruder mouse and the subordinate mice compared with vehicle controls.
Experimental protocols
In the first study, adult male C57BL/6 mice were subjected to one or three cycles of social defeat. Mice were deeply anesthetized and transcardially perfused with PBS followed by 4% formaldehyde. Brains were collected, sectioned, and used for c-Fos analysis (n = 3).
In the second study, adult male C57BL/6 mice received a subcutaneous injection of vehicle (0.2% ethanol in sterile saline) or 10 mg/kg propranolol (propranolol hydrochloride; Sigma-Aldrich) before each of the six cycles of social defeat. Propranolol is a lipophilic nonselective β-adrenergic receptor antagonist that crosses the blood brain barrier and is active for at least 12 h following peripheral administration (Johnsson and Regàrdh, 1976; Salako et al., 1979). Immediately following the cessation of the sixth cycle of social defeat, mice were deeply anesthetized and transcardially perfused with PBS followed by 4% formaldehyde. Brains were collected, sectioned, and used for c-Fos analysis (n = 4–10).
In the third study, adult male C57BL/6 mice were injected subcutaneously with vehicle or propranolol (10 mg/kg) 1 h before each of the six cycles of social defeat. Light/dark preference was determined 14 h after the last cycle of social defeat (n = 10–16). Mice were killed by CO2 asphyxiation after the completion of behavioral testing. Spleen weights were determined (n = 7–18). Blood was collected by cardiac puncture with EDTA lined 1 ml syringes. Whole blood was centrifuged, plasma was collected, and IL-6 levels were determined (n = 8–10).
In the fourth set of studies, adult male (6–8 week old) C57BL/6 mice were injected subcutaneously with vehicle or propranolol (10 mg/kg) 1 h before each of the six cycles of social defeat. Fourteen hours after the sixth cycle of social defeat, mice were killed by CO2 asphyxiation and microglia/macrophages were isolated by discontinuous Percoll density gradient. Cells were used for flow cytometric analyses of CD11b, CD45, TLR4, CD14, MHC-II, CD86, and Ly6C expression or for total RNA isolation/quantitative PCR (IL-1β, GR, GILZ, and FKBP51) (n = 6–8). Subsets of mice were transcardially perfused before cell isolation and flow analysis. In a related set of studies, C57BL/6 mice were subjected to six cycles of social defeat and 14 h later were killed. Cells were isolated by discontinuous Percoll density gradient and cultured ex vivo.
In the fifth set of studies, adult male (6–8 week old) C57BL/6 mice were injected subcutaneously with vehicle or propranolol (10 mg/kg) 1 h before each of the six cycles of social defeat. Fourteen hours after the sixth cycle of social defeat, mice were deeply anesthetized and transcardially perfused with PBS followed by 4% formaldehyde. Brains were collected, sectioned, and used for ionized calcium-binding adapter molecule-1 (Iba-1) analysis (n = 3–4).
In the last set of studies, adult male (6–8 week old) C57BL/6 wild-type or IL-1r1−/− mice were subjected to repeated social defeat. Fourteen hours after the sixth cycle, light/dark preference was determined. Following behavioral testing, mice were deeply anesthetized, transcardially perfused with PBS followed by 4% formaldehyde, and brains were removed for Iba-1 and c-Fos analyses (n = 4–5).
c-Fos staining.
c-Fos staining was performed as previously described (Trainor et al., 2008). In brief, brains were collected and fixed in 5% acrolein for 24 h at 4°C, washed, and incubated in 30% sucrose for an additional 24 h. Preserved brains were frozen and then sectioned (40 μm) using a Microm HM550 cryostat (Mikron Instruments). The brain regions were identified anatomically by reference markers in accordance with the stereotaxic mouse brain atlas (Paxinos and Franklin, 2004). Antigens were unmasked with 1% sodium borohydride and sections were blocked with 20% normal goat serum and 0.3% hydrogen peroxide. Next, sections were incubated with a rabbit anti-mouse c-Fos antibody (Calbiochem) with 1% NGS at 4°C overnight. Sections were washed and then incubated with a biotinylated goat anti-rabbit secondary antibody for 2 h. c-Fos staining was developed using the 3,3′-diaminobenzidine (DAB) protocol. Sections were placed in porcine gelatin (10%), transferred to slides, and coverslipped. Images were captured using a Nikon Optishot microscope with a Sanyo CCD video camera and the number of c-Fos-positive cells was determined using NIH ImageJ software.
Anxiety-like behavior.
Anxiety-like behavior was determined using the light/dark preference test as previously described (Kinsey et al., 2007). In brief, the test apparatus consisted of a 40 × 40 × 25 cm Plexiglas box that was divided into equal zones (i.e., light and dark zones) with a doorway connecting the two sides. The dark zone was enclosed and had significantly less light (<3 lux). To initiate testing, mice were placed in the light side and locomotor activity was recorded for 5 min using an automated system (AccuScan Instruments) and analyzed using VersaMap software.
IL-6.
IL-6 was determined from plasma using the BD OptEIA Mouse IL-6 ELISA according to the manufacturer's instructions (BD Biosciences). In brief, 96-well enzyme immunoassay plates were coated with anti-mouse IL-6 capture antibody and incubated overnight at 4°C. Samples and IL-6 standards (0–1000 pg/ml) were added and incubated for 2 h at room temperature (RT). Plates were washed and incubated with biotinylated anti-mouse IL-6 antibody. Plates were washed and incubated with streptavidin-horseradish peroxidase conjugate. After 1 h incubation at RT, plates were washed and incubated with tetramethylbenzidine liquid substrate for 15 min. Reactions were terminated and absorbance was read at 450 nm using a Spectramax Plus384 plate reader (Molecular Devices). The assay was sensitive to 10 ng/ml IL-6 and the interassay and intra-assay coefficients of variation were <10%.
Isolation of microglia.
Microglia were isolated from whole-brain homogenates as described previously (Henry et al., 2008, 2009; Wynne et al., 2010). In brief, brains were homogenized in HBSS, pH 7.4, by passing through a 70 μm nylon cell strainer. Resulting homogenates were centrifuged at 600 × g for 6 min. Supernatants were removed and cell pellets were resuspended in 70% isotonic Percoll (GE Healthcare) at room temperature. A discontinuous Percoll density gradient was layered as follows: 70%, 50%, 35%, and 0% isotonic Percoll. The gradient was centrifuged for 20 min at 2000 × g and microglia were collected from the interphase between the 70% and 50% Percoll layers (Frank et al., 2006a; Nair et al., 2007). Cells were washed and then resuspended in sterile HBSS. The number of viable cells was determined using a hemacytometer and 0.1% trypan blue staining. Each brain extraction yielded ∼3 × 105 viable cells. These cells will be referred to as enriched microglia based on previous studies demonstrating that viable cells isolated from brain homogenates through Percoll density gradient yields >90% microglia (Henry et al., 2009; Wynne et al., 2010).
Microglial staining and flow cytometry.
Staining of microglia surface antigens was performed as previously described (Henry et al., 2008, 2009). In brief, Fc receptors were blocked with anti-CD16/CD32 antibody (eBioscience). Cells were washed and then incubated with the appropriate conjugated antibodies CD45, CD11b, CD14, TLR4, or CD45, CD11b, CD86, MHC-II or CD45, CD11b (eBioscience), and Ly6C (BD Biosciences) for 45 min. Cells were washed and then resuspended in FACS buffer (2% FBS in HBSS with 1 mg/ml sodium azide) for analysis. Nonspecific binding was assessed by using nonspecific, isotype-matched antibodies. Antigen expression was determined using a Becton-Dickinson FACSCaliber four-color cytometer. Ten thousand events were recorded for each sample and isotype-matched conjugate. Data were analyzed using FlowJo software (Tree Star) and gating for each antibody was determined based on nonspecific binding of appropriate negative isotype stained controls.
Morphological analysis of microglia.
To stain for Iba-1, mice were deeply anesthetized and transcardially perfused with sterile PBS (pH 7.4 with EDTA) and 4% formaldehyde. Brains were postfixed in 4% formaldehyde for 24 h and incubated in 20% sucrose for an additional 24 h. Fixed brains were frozen using isopentane (−78°C) and sectioned (20 μm) using a Microm HM550 cryostat (Mikron Instruments). Brain regions were identified by reference markers in accordance with the stereotaxic mouse brain atlas (Paxinos and Franklin, 2004). Sections were mounted on slides and blocked with 5% normal goat serum. Next, sections were washed in PBS with 1% BSA and incubated with a rabbit anti-mouse Iba-1 antibody (Wako Chemicals). For fluorescent staining, sections were washed in PBS and then incubated with a fluorochrome-conjugated secondary antibody (Alexa Fluor 594). Sections were counterstained with a DNA stain, 4′,6-diamidino-2-phenylindole. Sections were coverslipped in a 25% glycerol solution and stored at −20°C. For nonfluorescent Iba-1 staining, brains were prepared and processed as above. Sections were incubated in a biotinylated goat anti-rabbit secondary antibody and developed using the DAB protocol. These sections were coverslipped with Permount (Fischer-Scientific). Fluorescent and DAB stained sections were visualized using an epi-fluorescent Leica DM5000B microscope. Images were captured using a Leica DFC300 FX camera and imaging software. To quantify the phenotypic changes of microglia, digital image analysis (DIA) (Donnelly et al., 2009) of Iba-1 staining was performed. Six representative images were taken from each brain region at 20× magnification. A threshold for positive staining was determined for each image and was processed by densitometric scanning of the threshold targets using ImageJ software. Proportional area was reported as the average percentage area in the positive threshold for all representative pictures.
RNA isolation and real-time PCR.
RNA was isolated from Percoll-isolated cells using the RNeasy plus mini-kit (Qiagen), RNA concentration was determined by spectrophotometry (Eppendorf), and RNA was reverse transcribed to cDNA using an RT-RETROscript kit (Ambion). Quantitative PCR was performed using the Applied Biosystems Assay-on-Demand Gene Expression protocol as previously described (Godbout et al., 2005). In brief, cDNA was amplified by real-time PCR where a target cDNA (e.g., IL-1β, GR, GILZ, and FKBP51) and a reference cDNA (glyceraldehyde-3-phosphate dehydrogenase) were amplified simultaneously using an oligonucleotide probe with a 5′ fluorescent reporter dye (6-FAM) and a 3′ quencher dye (NFQ or TAMRA). Fluorescence was determined on an ABI PRISM 7300-sequence detection system (Applied Biosystems). Data were analyzed using the comparative threshold cycle method and results were expressed as fold difference.
Primary microglia cultures and cytokine measurement by cytometric bead array.
Microglia collected by Percoll gradient separation were counted and plated at 100,000 cells on poly-l-lysine-coated 96-well plates. Cells were placed in complete RPMI (containing 10% heat-inactivated fetal bovine serum, 0.075% sodium bicarbonate, 10 mm HEPES buffer, 100U/ml penicillin G, 100 μg/ml streptomycin sulfate, 1.5 nm l-glutamine, and 0.0035% 2-mercaptoethanol) and were stimulated with saline or 0.40 μg/ml lipopolysaccharide (LPS; Sigma), vehicle (0.2% ethanol), and 0.10 or 5 μm corticosterone (Sigma) for 18 h at 37°C and 5% CO2. Supernatants were harvested and the concentration of secreted cytokines (TNF-α, IL-12p70, IL-10, IFN-γ, MCP-1, and IL-6) were detected using a mouse inflammatory cytokine cytometric bead array (CBA) kit according to the manufacturer's instructions (BD Biosciences). For comparison, data were normalized and presented as the percentage of noncorticosterone-treated cultures from the same treatment group.
Statistical analysis.
To ensure a normal distribution, data were subjected to Shapiro-Wilk test using Statistical Analysis Systems (SAS) statistical software. Observations >3 interquartile ranges from the first and third quartile were considered outliers and were excluded in the subsequent analysis. To determine significant main effects and interactions between main factors, data were analyzed using one- (stress, propranolol), two- (stress × propranolol), or three-way (stress × propranolol × region) ANOVA using the general linear model procedures of SAS. When appropriate, differences between treatment means were evaluated by an F-protected t test using the least-significant difference procedure of SAS. All data are expressed as treatment means ± SEM.
Results
Repeated social defeat-induced c-Fos activation in the CNS was β-adrenergic receptor-dependent
Repeated social defeat promotes anxiety-like behavior in random-bred CD-1 and inbred C57BL/6 mice (Kinsey et al., 2007). To begin to understand how social defeat is interpreted within the CNS, c-Fos immunoreactivity was determined in several brain regions associated with fear and threat appraisal, including the prefrontal cortex (PFC), lateral septum (LS), bed nucleus of stria terminalis (BNST), paraventricular nucleus of the hypothalamus (PVN), medial amygdala (MeA), and hippocampus (HPC) (Kollack-Walker et al., 1997; Martinez et al., 2002; Kovács, 2008). In the first experiment, C57BL/6 mice were subjected to one or three cycles of social defeat and c-Fos staining was determined. The number of social defeat cycles is significant because it takes at least three repeated exposures to cause glucocorticoid resistance in peripheral CD11b+ cells (Avitsur et al., 2002). Representative images of c-Fos staining from the PFC are shown in Figure 1A. Figure 1B shows that one and three cycles of social defeat increased the number of c-Fos-positive cells in the brain (main effect of stress; F(2,69) = 61.58, p < 0.0001) in a brain region-dependent manner (stress × region interaction; F(10,69) = 3.95, p < 0.0005). For instance, one and three cycles of social defeat significantly increased c-Fos staining in the PFC (p < 0.0001), LS (p < 0.002), BNST (p < 0.02), PVN (p < 0.005), and MeA (p < 0.0001).
Figure 1.

Repeated social defeat-induced c-Fos activation in the CNS was β-adrenergic-dependent. Male C57BL/6 mice were subjected to one or three cycles of SDR, brains were collected immediately following the final cycle, and c-Fos staining was determined in the PFC, LS, BNST, PVN, MeA, and HPC. A, Representative pictures of c-Fos staining from the prefrontal cortex (20×) are shown. B, Average number of c-Fos-positive cells. C, D, In a related study, male C57BL/6 mice were injected subcutaneously with vehicle or propranolol (10 mg/kg) before each of the six cycles of social defeat. Brains were collected immediately after the sixth cycle of social disruption and c-Fos staining was determined as above. C, Representative pictures of c-Fos staining from the prefrontal cortex (20×) are shown. D, Average number of c-Fos-positive cells. Error bars represent the mean ± SEM (n = 6–8). Means with different letters (a, b, or c) are significantly different (p < 0.05) from each other.
Because these brain regions are integrated with catecholaminergic pathways (Ulrich-Lai and Herman, 2009), we next examined the degree to which the nonselective β-adrenergic receptor antagonist, propranolol, prevented repeated social defeat-induced c-Fos expression. Although Figure 1, A and B, shows that c-Fos expression was enhanced after one and three cycles of social defeat, six cycles of social defeat was selected in these propranolol studies because six cycles promotes GC-resistance in peripheral CD11b+ cells and induces prolonged anxiety-like behavior (Avitsur et al., 2002; Kinsey et al., 2007). In these studies, C57BL/6 mice were administered vehicle or propranolol before each of the six cycles of social defeat and brains were collected immediately following the sixth cycle of social defeat. Representative images of c-Fos staining from the PFC are shown in Figure 1C. In Figure 1D, the average number of c-Fos-positive cells in each brain region is shown. ANOVA revealed that repeated social defeat enhanced c-Fos staining in the brain (main effect of stress, F(1,154) = 22.38, p < 0.0001) and this increase was dependent on brain region (F(5,154) = 1.98, p = 0.09). Post hoc analysis indicated that repeated social defeat increased c-Fos staining in the PFC (p < 0.002), LS (p < 0.002), PVN (p < 0.0001), and MeA (p < 0.0001). In the regions examined, c-Fos levels were highest in the PVN and MeA after repeated social defeat, but were unchanged in the HPC and BNST. Moreover, pretreatment with propranolol prevented the stress-induced induction of c-Fos (stress × propranolol interaction, F(1,154) = 22.8, p < 0.0001) that was detected in the PFC (p=0.07), LS (p=0.08), PVN (p < 0.0001), and MeA (p < 0.0001). Collectively, these data indicate that social defeat promoted a unique pattern of c-Fos activation in the brain that was blocked by pretreatment with a β-adrenergic receptor antagonist.
Repeated social defeat-induced anxiety-like behavior was prevented by β-adrenergic receptor antagonism
Because β-adrenergic receptor blockade with propranolol reduced c-Fos activation, the affect of propranolol on repeated social defeat-induced anxiety-like behavior was examined. C57BL/6 mice were administered propranolol before each cycle of social defeat and light/dark preference was determined 14 h after the sixth cycle of social defeat. This time point was selected because repeated social defeat-induced anxiety-like behavior was detected in CD-1 and C57BL/6 mice at this time (Kinsey et al., 2007, 2008). Figure 2A shows representative motion plots of the mice in the light/dark preference paradigm. Mice subjected to repeated social defeat entered the dark zone of the testing apparatus faster (17.2 ± 4.9 s) than HCC mice (35.2 ± 6.4 s) (main effect of stress, F(1,45) = 4.37, p < 0.05) (Fig. 2B). Figure 2C shows that repeated social defeat caused a marked increase in the time spent in the dark zone (main effect of stress, F(1,45) = 11.55, p < 0.002). Moreover, pretreatment of mice with propranolol reduced anxiety-like behavior with increased time to enter the dark zone (main effect of propranolol, F(1,45) = 5.39, p < 0.03) (Fig. 2B) and decreased the total time spent in the dark zone (main effect of propranolol, F(1,45) = 7.26, p < 0.002) (Fig. 2C). It is important to note that propranolol treatment alone tended to reduce baseline anxiety in HCC mice (p = 0.10).
Figure 2.

Repeated propranolol pretreatment blocked repeated social defeat-induced anxiety-like behavior. Male C57BL/6 mice were injected subcutaneously with vehicle or propranolol (10 mg/kg) before each of the six cycles of SDR. Light/dark preference was determined 14 h after the sixth cycle of social defeat. A, Representative motion paths of the mice used in the light/dark testing paradigm. B, Average time to enter the dark zone. C, Average time spent in the dark zone. Bars represent the mean ± SEM (n = 10–16). Mice were treated as above. D, E, Spleen weight (D) and plasma IL-6 levels (E) were determined. Bars represent the mean ± SEM (n = 9). Means with different letters (a or b) are significantly different (p < 0.05) from each other.
Along with promoting anxiety-like behavior, repeated social defeat increases spleen weight (i.e., splenomegaly) (Avitsur et al., 2003), plasma levels of IL-6 (Stark et al., 2001, 2002), and corticosterone levels (Avitsur et al., 2001). These changes were used to gauge the robustness of the response to repeated social defeat. Corticosterone was significantly increased after each cycle of social defeat; however, levels of corticosterone returned to baseline 14 h after the sixth cycle of social defeat (data not shown). Therefore, spleen weight and plasma IL-6 levels were determined 14 h after the sixth cycle of social defeat. As expected, social defeat increased spleen weight (main effect of stress, F(1,56) = 9.74, p < 0.003) (Fig. 2D), and this increase was blocked by propranolol pretreatment (stress × propranolol interaction, F(1,56) = 7.24, p < 0.01). Figure 2E shows a similar interaction between stress and propranolol on IL-6 levels. Plasma IL-6 was increased after repeated social defeat (main effect of stress, F(1,35) = 7.72, p < 0.01) and was prevented by propranolol pretreatment (stress × propranolol interaction, F(1,35) = 9.53 p < 0.005). Together, these data indicate that propranolol pretreatment blocked the repeated social defeat-induced splenomegaly, reduced the increased plasma levels of IL-6, and returned anxiety-like behavior back to baseline in C57BL/6 mice.
Repeated social defeat increased the percentage of CD11b+/CD45high/Ly6Chigh macrophages in the CNS
Previous studies demonstrate that repeated social defeat markedly altered the phenotype of myeloid-derived CD11b+ cells (e.g., macrophages, dendritic cells, and neutrophils) in the periphery (Bailey et al., 2009; Powell et al., 2009; Curry et al., 2010) and increased their propensity to traffic from the bone marrow to the spleen and lungs (Engler et al., 2004; Kinsey et al., 2008; Curry et al., 2010). Therefore, we next sought to determine the degree to which CD11b+ cells of the CNS were altered by repeated social defeat. In these experiments, mice received vehicle or propranolol before each cycle of social defeat. Fourteen hours after the final cycle of social defeat, microglia and macrophages were collected by Percoll density gradient. Figure 3A shows representative bivariate dot plots of CD11b and CD45 staining for each experimental condition; microglia (CD11b+/CD45low) and macrophages (CD11b+/CD45high) were differentiated based on CD45 expression (Nair et al., 2007). Figure 3B shows that the percentage of macrophages was increased after repeated social defeat (main effect of stress, F(1,80) = 31.10, p < 0.0001). For example, the approximate percentage of macrophages was increased from 2.44% in HCC–vehicle mice to 7.36% in social disruption stress (SDR)–vehicle mice (Fig. 3A,B). Moreover, the increase in macrophages after repeated social defeat was ablated by propranolol pretreatment (stress × propranolol interaction, F(1,80) = 7.37, p < 0.008).
Figure 3.

Repeated social defeat increased the percentage of CD11b+/ CD45high/Ly6Chigh macrophages in the CNS. Male C57BL/6 mice were injected subcutaneously with vehicle or propranolol (10 mg/kg) before each of the six cycles of SDR. Brains were collected 14 h after the sixth cycle of social defeat, CD11b+ cells were enriched by Percoll gradient separation, and CD11b, CD45, and Ly6C expression levels were determined through flow cytometry. A, Representative bivariate dot plots of CD11b/CD45 staining. B, Average number of CD11b+/CD45high macrophages (n = 10). C, Representative bivariate dot plots of Ly6C/CD45 staining. D, Average number of CD11b+/CD45high/Ly6Chigh macrophages detected (n = 5). Bars represent the mean ± SEM. Means with different letters (a or b) are significantly different (p < 0.05) from each other. E, In a related study, mice were subjected to repeated social defeat and half the mice were perfused before collecting brains for flow cytometric analysis (n = 6). Bars represent the mean ± SEM. Means with different letters (a or b) are significantly different (p < 0.05) from each other. APC, Allophycocyanin; PerCP, peridinin chlorophyll protein.
Because trafficking of macrophages to the CNS is associated with increased expression of Ly6C (Mildner et al., 2007), Ly6C levels were determined after repeated social defeat. Figure 3C shows representative bivariate dot plots of Ly6C and CD45 staining. The percentage of Ly6Chigh macrophages was markedly increased in SDR–vehicle mice compared with all other treatment groups (p < 0.02) (Fig. 3D). In addition, propranolol pretreatment blocked the increased percentage of Ly6Chigh macrophages in the CNS after repeated social defeat (main effect of propranolol, F(1,26) = 10.21, p < 0.004) (Fig. 3D). It is important to note that repeated social defeat did not increase Ly6C expression on microglia (Fig. 3C).
To ensure that the increased macrophage population of the CNS was not confounded by the presence of CD11b+ monocytes circulating in the vasculature of the CNS, a subset of mice were subjected to repeated social defeat and transcardially perfused with sterile PBS 14 h after the sixth cycle of social defeat. Microglia and macrophages were collected for flow cytometric analysis. Figure 3E shows that repeated social defeat increased the percentage of macrophages in the CNS and this increase was unaffected by perfusion (main effect of stress, F(1,15) = 19.01, p < 0.0006; effect of perfusion, F(1,18) = 1.05, p < 0.33). Together, these data indicate that repeated social defeat increased the percentage of Ly6Chigh macrophages that traffic to the CNS.
Increased protein expression of inflammatory markers on the surface of microglia and CNS macrophages after repeated social defeat
To further characterize the profile of microglia (CD45low) and CNS macrophages (CD45high) after repeated social defeat, these cells were analyzed for several inflammatory-related surface markers. Surface expression of two markers associated with innate immunity, CD14 and toll-like receptor 4 (TLR4), and two markers associated with antigen presentation, major histocompatibility complex (MHC)-II and CD86, were determined 14 h after the sixth cycle of social defeat. Figure 4A shows representative bivariate dot plots of CD11b and CD14 staining for microglia and macrophages. Figure 4, B–D, shows that repeated social defeat increased the percentage of CD14+ (F(1,17) = 29.56, p < 0.0001) (Fig. 4B), TLR4+ (F(1,17) = 33.83, p < 0.001) (Fig. 4C), and CD86+ (F(1,16) = 26.32, p < 0.0001) (Fig. 4D) microglia. Moreover, repeated social defeat also enhanced the percentage of CD14+ (F(1,16) = 22.98, p < 0.0002) (Fig. 4B) and CD86+ (F(1,16) = 15.77, p < 0.002) (Fig. 4D) macrophages. TLR4 expression, however, was not significantly enhanced on macrophages after repeated social defeat (Fig. 4C). Furthermore, repeated social defeat did not affect MHC-II expression on either microglia or macrophages (Fig. 4E). These data indicate that repeated social defeat increases the inflammatory profile of microglia and CNS macrophages.
Figure 4.

Increased surface expression of inflammatory markers on microglia and CNS macrophages after repeated social defeat. Male C57BL/6 mice were subjected to six cycles of SDR. Brains were collected 14 h after the sixth cycle of social defeat, CD11b+ cells were enriched by Percoll gradient separation, and CD11b, CD45, CD14, TLR4, and CD86 expression levels were determined. Cells were gated on microglia (CD45low) or macrophages (CD45high). A, Representative bivariate dot plots are shown for CD11b/CD14. B–E, Average percentages of positive cells for CD14 (B), TLR4 (C), CD86 (D), and MHC-II (E) are shown. Bars represent the mean ± SEM (n = 9). Asterisks indicate HCC and SDR within cell types are significantly different (p < 0.05) from each other. In a separate but related study, male C57BL/6 mice were injected subcutaneously with vehicle or propranolol (10 mg/kg) before each of the six cycles of social defeat. Brains were collected 14 h after the sixth cycle of social defeat and CD11b, CD45, and CD14 levels were determine as above. F, G, Representative bivariate dot plots and the average percentage of positive cells for CD14. Bars represent the mean ± SEM (n = 10–12). Means with different letters (a, b, or c) are significantly different (p < 0.05) from each other. APC, Allophycocyanin; FITC, fluorescein isothiocyanate.
In a related experiment, mice were pretreated with vehicle or propranolol and then subjected to six cycles of social defeat. Cell-surface expression of CD14 was determined on microglia. Representative bivariate dot plots of CD11b/CD14 staining are shown in Figure 4F. Consistent with the above experiment (Fig. 3B), repeated social defeat increased the percentage of CD14+ microglia (main effect of stress, F(1,44) = 5.20, p < 0.03). This stress-induced increase in CD14 surface expression was blocked by pretreatment with propranolol (stress × propranolol interaction, F(1,44) = 4.29, p < 0.05) (Fig. 4G). These data indicate that repeated social defeat-induced changes in inflammatory protein expression are mediated, in part, by β-adrenergic pathways.
Activated microglial morphology induced by repeated social defeat was prevented by propranolol
Because several markers of inflammation were detected on the surface of microglia after repeated social defeat (Fig. 4), alterations in microglia morphology using anti-Iba-1 antibody was determined in the PFC, MeA, PVN, and HPC. Iba-1 is used to differentiate morphological changes associated with microglial activation (Imai and Kohsaka, 2002). Figure 5A, i and ii, shows representative fluorescent staining for Iba-1 in the MeA of HCC and SDR mice, respectively. These images illustrate that microglia from HCC mice have long and thin processes (i.e., ramified), but microglia from socially defeated mice exhibit hypertrophy with shorter and thicker processes (i.e., deramified). To quantify these differences, DIA for Iba-1 staining was determined (Donnelly et al., 2009). The results from the DIA (Fig. 5B–E) confirmed that microglia from socially defeated mice had an increased proportional area compared with microglia from control mice (main effect of stress, F(1,52) = 15.52, p < 0.0003). Post hoc analysis revealed that repeated social defeat significantly increased the proportional area of microglia in all brain areas examined including the MeA (p < 0.01) (Fig. 5B), PFC (p < 0.04) (Fig. 5C), HPC (p < 0.0002) (Fig. 5D), and PVN (p < 0.0001) (Fig. 5E). Moreover, representative images indicate that the increase in activated microglia after repeated social defeat was blocked by propranolol pretreatment in the MeA (Fig. 5Aiii,iv). DIA confirmed that propranolol pretreatment prevented the stress-induced increase in proportional area of microglia (stress × propranolol interaction, F(1,52) = 11.83, p < 0.002) in the PFC, MeA, and HPC. In the PVN, however, the stress-induced increase in proportional area of Iba-1 staining was unaffected by propranolol pretreatment (Fig. 5E). Together, these data indicate that repeated social defeat caused morphological changes in activated microglia within the MeA, PFC, and HPC, and this was blocked by propranolol.
Figure 5.
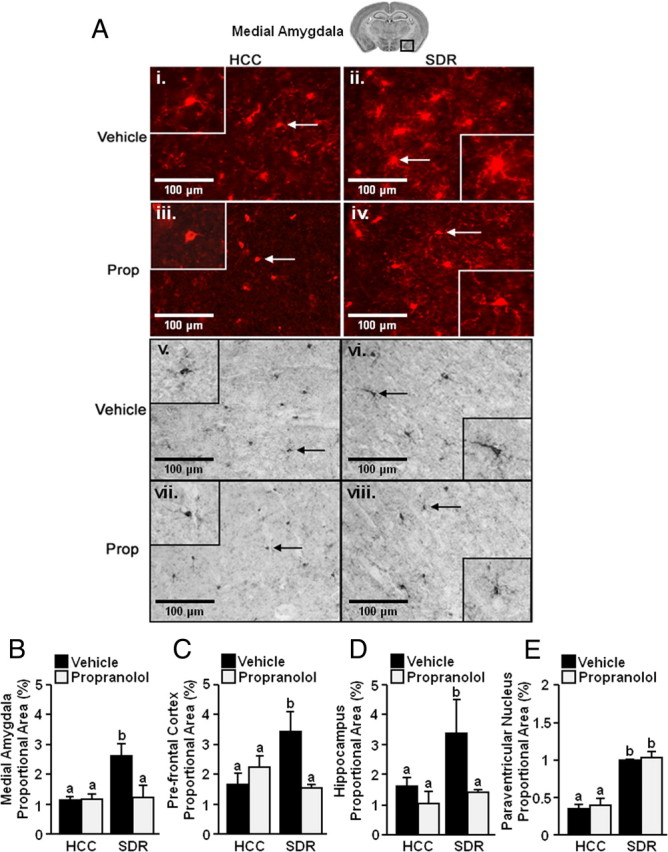
Increased activated morphology of microglia after repeated social defeat. Male C57BL/6 mice were injected subcutaneously with vehicle or propranolol (Prop; 10 mg/kg) before each of the six cycles of SDR. Mice were perfused and brains were collected 14 h after the sixth cycle of social defeat for Iba-1 staining. A, Representative pictures of Iba-1 staining [fluorescent (i–iv) and DAB (v–viii) from the medial amygdala (40×) are shown). Inset includes enlarged image of Iba-1+ cell indicated by arrow. B–E, Proportional area for Iba-1 staining in the medial amygdala (B, n = 3), prefrontal cortex (C, n = 3), hippocampus (D, n = 3), and paraventricular nucleus (E, n = 3). Bars represent the mean ± SEM. Means with different letters (a or b) are significantly different (p < 0.05) from each other.
Repeated social defeat increased IL-1β and decreased GILZ and FKBP51 mRNA levels in enriched microglia cultures
Because CD11b+ cells in the periphery are GC-resistant and produce higher levels of inflammatory cytokines after repeated social defeat (Stark et al., 2001; Bailey et al., 2009; Powell et al., 2009), the next experiment examined gene expression of IL-1β, glucocorticoid receptor (GR), and two genes containing glucocorticoid responsive elements (GILZ and FKBP51) in enriched microglia (Fig. 3A). Table 1 shows that repeated social defeat increased IL-1β mRNA levels in enriched microglia (main effect of stress, F(1,23) = 4.50, p < 0.05). Although GR mRNA expression was unaffected following repeated social defeat, the mRNA levels of GILZ (main effect of stress, F(1,28) = 19.83, p < 0.0001) and FKBP51 (main effect of stress, F(1,26) = 8.84, p < 0.007) were significantly decreased by repeated social defeat. Moreover, propranolol pretreatment blocked the repeated social defeat-induced increase in IL-1β (p < 0.04) and reduction in GILZ (p < 0.006) and FKBP51 (p < 0.0007). Furthermore, β-adrenergic receptor antagonism enhanced mRNA levels of FKBP51 in mice subjected to repeated social defeat. These data indicate that repeated social defeat increased IL-1β mRNA and decreased mRNA for two GC-responsive genes in enriched microglia, whereas blockade of β-adrenergic receptor stimulation restored IL-1β levels to baseline and enhanced FKBP51 levels.
Table 1.
Repeated social defeat increased IL-1β and decreased glucocorticoid responsive element mRNA in enriched microglia
| Gene | HCC (fold change) | SDR vehicle (fold change) | SDR propranolol (fold change) |
|---|---|---|---|
| IL-1β | 1.10 ± 0.26a | 2.83 ± 0.63b | 1.00 ± 0.10a |
| GR | 1.10 ± 0.16a | 1.26 ± 0.10a | 1.34 ± 0.48a |
| GILZ | 1.13 ± 0.18a | 0.62 ± 0.05b | 1.47 ± 0.53a |
| FKBP51 | 0.8 ± 0.12a | 0.43 ± 0.06b | 2.46 ± 0.85c |
Table represents average ± SEM.
a,b,cMeans are significantly different (p < 0.05) from each other.
Repeated social defeat increased IL-6, TNF-α, and MCP-1 protein levels in supernatants of enriched microglia cultures
Based on the protein, morphological, and mRNA data, the objective of the next study was to determine the functional consequence of the increased inflammatory profile of microglia after repeated social defeat. To address this issue, ex vivo cultures of enriched microglia were established from HCC and SDR mice. Cells were plated at 1 × 105 cells per well in a 96-well culture plate in growth media and were treated with 0.40 μg/ml LPS in the presence and absence of corticosterone. Supernatants were collected and levels of IL-6, TNF-α, IL-12p70, IL-10, IFN-γ, and MCP-1 protein were determined by CBA. IFN-γ and IL-12 were not increased by LPS in HCC- or SDR-derived cultures of enriched microglia (data not shown). Figure 6 shows that treatment with LPS induced higher production of IL-6, TNF-α, and MCP-1 in cultures from socially defeated mice compared with cultures established from HCC mice (main effects of stress: IL-6, F(1,7) = 25.35 p < 0.005; TNF-α, F(1,7) = 2.83 p = 0.14; MCP-1, F(1,7) = 21.88 p < 0.005). A similar response to LPS was detected for IL-10 (data not shown). Moreover, glucocorticoids lowered the cytokine production in a dose-dependent manner (IL-6, F(1,23) = 14.22 p < 0.001; TNF-α, F(1,23) = 12.80 p < 0.001; MCP-1, F(1,23) = 58.09 p < 0.001), but this reduction was independent of repeated social defeat. Collectively, these data indicate enriched microglia isolated from socially defeated mice produced significantly higher levels of IL-6, TNF-α, and MCP-1 after LPS compared with enriched microglia obtained from HCC mice.
Figure 6.

Repeated social defeat increased the reactivity of microglia. Male C57BL/6 mice were subjected to six cycles of SDR. Brains were collected 14 h after the sixth cycle of social defeat. Enriched microglia were collected by Percoll gradient separation and cultured ex vivo. A–C, Cells were incubated with LPS and corticosterone (Cort) (0, 0.1, or 5 μm) and IL-6 (A), TNF-α (B), MCP-1 (C) protein levels were determined in supernatants collected 18 h later. Bars represent the percentage increase over controls ± SEM. Means with different letters (a, b, or c) are significantly different (p < 0.05) from each other.
Repeated social defeat enhanced c-Fos activation in IL-1r1−/− mice but did not promote anxiety-like behavior or microglial activation
Social defeat increased the inflammatory profile of microglia and increased mRNA levels of IL-1β. Thus, the extent to which social defeat induced c-Fos activation, anxiety-like behavior, and microglia activation was determined in IL-1r1−/− mice. Representative images of c-Fos staining from the MeA of IL-1r1−/− mice are shown in Figure 7A. Similar to the results obtained in wild type (WT) mice (Fig. 1C,D), repeated social defeat enhanced c-Fos staining in the MeA of IL-1r1−/− mice compared with IL-1r1−/−–HCC mice (main effect of stress, F(1,10) = 14.52, p < 0.004). A similar pattern of c-Fos activation was detected in other brain regions (data not shown). In the light/dark preference test, WT mice subjected to repeated social defeat decreased time to enter the dark zone (p < 0.005) (Fig. 7C) and increased time spent in the dark zone (p < 0.01) (Fig. 7D). The effect of repeated social defeat, however, was dependent on expression of functional IL-1 receptor type-1 (genotype × stress interaction, F(1,24) = 10.39, p < 0.005). IL-1r1−/− mice subjected to repeated social defeat did not display anxiety-like behavior. It is important to note that IL-1r1−/−–HCC mice had higher baseline anxiety-like behavior than WT–HCC mice. These baseline behavioral differences in IL-1r1−/− mice, however, were unaffected by repeated social defeat.
Figure 7.

Repeated social defeat enhanced c-Fos activation, but did not affect behavior or microglial activation in IL-1r1−/− mice. Male IL-1r1−/− C57BL/6 mice were subjected to six cycles of SDR. A, Representative pictures of c-Fos staining from MeA (20×). B, Average number of c-Fos-positive cells in the MeA (n = 5). C–F, In a separate set of studies, WT or IL-1r1−/− C57BL/6 mice were subjected to repeated social defeat and light/dark preference was determined 14 h after the sixth cycle of social defeat. C, Average time to enter the dark zone. D, Average time spent in the dark zone (n = 6–8). Following behavioral testing, mice were perfused and brains were collected for Iba-1 immunohistology. E, Representative images of Iba-1 staining from MeA (40×). Insets include enlarged image of Iba-1+ cell indicated by arrow. F, Proportional area for Iba-1 staining in the MeA (n = 3). Bars represent the mean ± SEM. Asterisks indicate significant differences within the genotype (p < 0.05). n.s., Not significant.
To determine the level of microglia activation, Iba-1 staining was performed in the MeA. Figure 7E shows representative staining for Iba-1 in the MeA of WT–HCC, WT–SDR, and IL-1r1−/−–SDR mice. Results from the DIA indicate that microglia from WT–SDR mice showed increased proportional area in the MeA compared with microglia from control mice (stress × genotype interaction, F(1,11) = 46.77, p < 0.0001). These changes in proportional area of microglia were not detected in socially defeated IL-1r1−/− mice. Together, these data indicate that, although repeated social defeat increased c-Fos activation in the brain, it did not induce anxiety-like behavior or microglial activation in IL-1r1−/− mice.
Discussion
Repeated social defeat significantly alters peripheral immune responses and induces prolonged anxiety-like behavior (Stark et al., 2001; Engler et al., 2004; Berton et al., 2006; Kinsey et al., 2007; Krishnan et al., 2007; Bailey et al., 2009; Powell et al., 2009; Curry et al., 2010; Mays et al., 2010). We show that anxiety-like behavior induced by social defeat coincides with a unique pattern of c-Fos activation in brain regions associated with fear and threat appraisal. Moreover, stress-induced anxiety-like behavior and c-Fos activation were prevented by propranolol pretreatment (Figs. 1, 2). Social defeat also increased the number of Ly6Chigh macrophages in the CNS (Fig. 3) and increased the inflammatory profile of microglia/macrophages (Figs. 4, 5; Table 1). The stress-induced inflammatory changes in microglia/macrophages were blocked by propranolol. One functional consequence of the increased inflammatory profile of enriched microglia was exaggerated cytokine production following ex vivo stimulation with LPS (Fig. 6B). Last, repeated social defeat increased c-Fos activation in the medial amygdala of IL-1r1−/− mice, but did not promote anxiety-like behavior or microglial activation in mice lacking functional IL-1 receptor type-1.
The finding that repeated social defeat increased anxiety-like behavior (Fig. 2) is consistent with previous studies of social defeat (Berton et al., 2006; Kinsey et al., 2007; Krishnan et al., 2007). Our findings indicate that c-Fos expression was increased in the brain after one exposure to social defeat and activation was either maintained or enhanced with repeated cycles (Fig. 1). The regions that were activated after social defeat are integrated in catecholaminergic pathways and implicated in fear and threat appraisal (Samuels and Szabadi, 2008; Roozendaal et al., 2009; Ulrich-Lai and Herman, 2009). Other studies of social defeat in rodents show c-Fos activation in similar brain regions (Martinez et al., 2002). Although exposure to one cycle of social defeat was sufficient to induce c-Fos activation, it is likely that repeated cycles of defeat are required to promote prolonged anxiety-like behavior that is detectable 8 d after the cessation of the stressor (Kinsey et al., 2007). For example, three social defeat cycles are required to establish longer-lasting immunological changes associated with the stressor (Avitsur et al., 2002). Our current studies extend these previous findings and indicate that pretreatment with propranolol blocks the induction of c-Fos expression and anxiety-like behavior caused by repeated social defeat (Figs. 1, 2). Although the induction of c-Fos was dependent on activation of β-adrenergic receptors, it did not depend on the presence of functional IL-1 receptor type-1 (Fig. 7A). It is important to mention that an acute injection of propranolol after six cycles of social defeat did not attenuate any of the physiological or behavioral responses to the stressor (data not shown). Therefore, these collective data indicate that activation of β-adrenergic receptors play an active role in the initial interpretation of threat and fear signals and the induction of anxiety-like behavior following repeated social defeat.
Social defeat increases the egress of myeloid cells from the bone marrow and trafficking of these cells to the spleen and lung (Engler et al., 2004; Curry et al., 2010). Here we provide novel data that repeated social defeat significantly increased the percentage of Ly6Chigh macrophages that trafficked to the CNS (Fig. 3). The presence of these macrophages after social defeat was unaffected by transcardial perfusion (Fig. 3E), which suggests that these macrophages are present in either the perivascular space or parenchyma. Because Ly6Chigh macrophages have a propensity to traffic to inflamed tissue (Mildner et al., 2007; King et al., 2009), the presence of these cells in the brain supports our conclusion that repeated social defeat enhances neuroinflammation.
Repeated social defeat also enhanced the inflammatory profile of microglia and CNS macrophages. For example, CD14 and TLR4, innate immune markers responsible for recognizing LPS, were enhanced on the surface of microglia (Fig. 4A,B). CD14 was also increased on CNS macrophages (Fig. 4A). Moreover, CD86, a costimulatory molecule necessary for antigen presentation, was enhanced on microglia and CNS macrophages after social defeat (Fig. 4D). In addition, there was an increased presence of deramified microglia in all brain regions examined (Fig. 5) and microglial IL-1β mRNA was increased after repeated social defeat (Fig. 6). The stress-induced changes in inflammatory markers, morphology, and mRNA indicate that repeated social defeat primes microglia and CNS macrophages.
Primed or reactive microglia are increased with aging (Godbout et al., 2005), early life infection (Bilbo et al., 2005, 2008), neurodegenerative disease (Cunningham et al., 2005), and autoimmunity (Raivich and Banati, 2004). Increased microglial priming is associated with an exaggerated inflammatory response in the brain after an immune stimulus. For instance, primed microglia from aged or prion-infected mice produce exaggerated levels of inflammatory cytokines, including IL-1β, after LPS stimulation (Cunningham et al., 2005; Frank et al., 2006b; Henry et al., 2009). Moreover, primed microglia from socially defeated mice stimulated ex vivo with LPS produced amplified levels of IL-6, TNF-α, and MCP-1 compared with controls (Fig. 6B–D). Similar findings were shown with inescapable foot shock, where LPS increased IL-1β mRNA expression in hippocampal microglia ex vivo (Frank et al., 2007). The increased surface expression of TLR4 and CD14, the primary receptor and coreceptor for LPS, is likely related to increased sensitivity to LPS following repeated social defeat (Fig. 4). These data provide direct evidence that repeated social defeat increased the sensitivity of microglia to subsequent inflammatory challenges.
Previous reports have shown that other stressors, including inescapable footshock, increased IL-1β mRNA in the brain and that increased IL-1β was prevented by β-adrenergic receptor antagonism (Johnson et al., 2005, 2008). In addition, activation of microglia was implicated because minocycline, a purported microglial inhibitor, also reduced the stress-associated increase of IL-1β mRNA in the CNS (Blandino et al., 2006). In other studies, administration of β-adrenergic receptor agonists increased IL-1β mRNA expression in the brain (McNamee et al., 2010). Furthermore, a recent microarray and transcription element listening system analysis of the cortex from socially defeated mice found evidence of inflammatory gene induction (GATA-1) related to β-adrenergic receptor stimulation (Cole et al., 2010). In addition, higher levels of IL-1β in the CNS are linked to behavioral changes, including social withdrawal, anxiety, and depressive-like behaviors (Anisman et al., 2005; Berton et al., 2006; Koo and Duman, 2009). Because repeated social defeat-induced anxiety was blocked by propranolol (Fig. 1), it is plausible that β-adrenergic receptor activation promotes microglia/macrophage activation that contributes to the promotion of anxiety-like behavior. In support of this notion, our data show that functional IL-1 receptor type-1 is required for the social defeat-induced anxiety and microglial activation (Fig. 7). Moreover, recent evidence indicates that NFκB signaling, which is a pathway activated by inflammatory cytokines including IL-1β (Nadjar et al., 2003), is essential to social avoidance and increased dendritic spine density caused by chronic social defeat (Christoffel et al., 2011).
Increased MHC-II expression is associated with microglia priming in several model systems, but it was not increased 14 h after repeated social defeat (Fig. 4E). This differs from studies using inescapable shock (Frank et al., 2007), where MHC-II mRNA levels were increased in microglia. Nonetheless, our data are consistent with a study showing that MHC-II was not increased on Iba-1+ cells in the CNS after restraint stress (Tynan et al., 2010). Studies using restraint stress also indicate there is proliferation (Nair and Bonneau, 2006) or migration of microglia (Tynan et al., 2010). Whereas repeated social defeat promoted an increase in macrophage trafficking to the CNS, analyses of microglia did not show any evidence of proliferation or migration (data not shown). These differences may lie in the nature of the stressor (Sheridan et al., 2004).
Although the present data point to β-adrenergic receptor-dependent changes in the CNS after stress, it is important to consider the influence of glucocorticoids on microglia and CNS macrophages. A recent study demonstrated that pretreatment of primary macrophages with glucocorticoids enhanced inflammatory cytokine secretion following LPS stimulation (Frank et al., 2010). In addition, peripheral CD11b+ cells from socially defeated mice produce more inflammatory cytokines and are GC-insensitive (Avitsur et al., 2001, 2002; Stark et al., 2001). Our data indicate that ex vivo LPS stimulation of enriched microglia from socially defeated mice caused exaggerated cytokine production compared with controls. There was no difference in the glucocorticoid-mediated reduction of inflammatory proteins induced by LPS or glucocorticoid-induced apoptosis of microglia (data not shown). Nonetheless, the mRNA profile of microglia from socially defeated mice point to a reduction of in vivo glucocorticoid responsiveness. For instance, repeated social defeat decreased mRNA levels in microglia of two GC-responsive genes, GILZ and FKBP51, and propranolol pretreatment prevented the downregulation of these genes in socially defeated mice (Table 1). Thus, these mRNA data indicate that social defeat may cause microglia to become less sensitive to the anti-inflammatory effects of glucocorticoids.
In conclusion, this work demonstrates that repeated social defeat caused anxiety-like behavior and enhanced the inflammatory state on the CNS in a β-adrenergic- and IL-1 receptor-dependent manner. Moreover, repeated social defeat primed microglia and CNS macrophages and increased their reactivity to subsequent inflammatory stimuli. These findings are relevant because they begin to establish a mechanism by which social stress increases the proinflammatory state of the CNS and promotes behavioral-related complications.
Footnotes
This work was supported by National Institutes of Mental Health Grant R01-MH046801-18 to J.F.S. and National Institute on Aging Grant R01-AG-033028 to J.P.G. M.L.H. and E.S.W. are supported by an National Institute of Dental and Craniofacial Research Training Grant T32-DE-01-4320 to J.F.S. We thank Dustin Donnelly and Dr. Phillip G. Popovich for their help with the digital image analysis.
References
- Anisman H, Merali Z, Poulter MO, Hayley S. Cytokines as a precipitant of depressive illness: animal and human studies. Curr Pharm Des. 2005;11:963–972. doi: 10.2174/1381612053381701. [DOI] [PubMed] [Google Scholar]
- Avitsur R, Stark JL, Sheridan JF. Social stress induces glucocorticoid resistance in subordinate animals. Horm Behav. 2001;39:247–257. doi: 10.1006/hbeh.2001.1653. [DOI] [PubMed] [Google Scholar]
- Avitsur R, Stark JL, Dhabhar FS, Padgett DA, Sheridan JF. Social disruption-induced glucocorticoid resistance: kinetics and site specificity. J Neuroimmunol. 2002;124:54–61. doi: 10.1016/s0165-5728(02)00010-3. [DOI] [PubMed] [Google Scholar]
- Avitsur R, Padgett DA, Dhabhar FS, Stark JL, Kramer KA, Engler H, Sheridan JF. Expression of glucocorticoid resistance following social stress requires a second signal. J Leukoc Biol. 2003;74:507–513. doi: 10.1189/jlb.0303090. [DOI] [PubMed] [Google Scholar]
- Bailey MT, Kinsey SG, Padgett DA, Sheridan JF, Leblebicioglu B. Social stress enhances IL-1beta and TNF-alpha production by Porphyromonas gingivalis lipopolysaccharide-stimulated CD11b+ cells. Physiol Behav. 2009;98:351–358. doi: 10.1016/j.physbeh.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Levkoff LH, Mahoney JH, Watkins LR, Rudy JW, Maier SF. Neonatal infection induces memory impairments following an immune challenge in adulthood. Behav Neurosci. 2005;119:293–301. doi: 10.1037/0735-7044.119.1.293. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Barrientos RM, Eads AS, Northcutt A, Watkins LR, Rudy JW, Maier SF. Early-life infection leads to altered BDNF and IL-1beta mRNA expression in rat hippocampus following learning in adulthood. Brain Behav Immun. 2008;22:451–455. doi: 10.1016/j.bbi.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Blanchard RJ, McKittrick CR, Blanchard DC. Animal models of social stress: effects on behavior and brain neurochemical systems. Physiol Behav. 2001;73:261–271. doi: 10.1016/s0031-9384(01)00449-8. [DOI] [PubMed] [Google Scholar]
- Blandino P, Jr, Barnum CJ, Deak T. The involvement of norepinephrine and microglia in hypothalamic and splenic IL-1beta responses to stress. J Neuroimmunol. 2006;173:87–95. doi: 10.1016/j.jneuroim.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Christoffel DJ, Golden SA, Dumitriu D, Robison AJ, Janssen WG, Ahn HF, Krishnan V, Reyes CM, Han MH, Ables JL, Eisch AJ, Dietz DM, Ferguson D, Neve RL, Greengard P, Kim Y, Morrison JH, Russo SJ. IkappaB kinase regulates social defeat stress-induced synaptic and behavioral plasticity. J Neurosci. 2011;31:314–321. doi: 10.1523/JNEUROSCI.4763-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SW, Arevalo JM, Takahashi R, Sloan EK, Lutgendorf SK, Sood AK, Sheridan JF, Seeman TE. Computational identification of gene-social environment interaction at the human IL6 locus. Proc Natl Acad Sci U S A. 2010;107:5681–5686. doi: 10.1073/pnas.0911515107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C, Wilcockson DC, Boche D, Perry VH. Comparison of inflammatory and acute-phase responses in the brain and peripheral organs of the ME7 model of prion disease. J Virol. 2005;79:5174–5184. doi: 10.1128/JVI.79.8.5174-5184.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry JM, Hanke ML, Piper MG, Bailey MT, Bringardner BD, Sheridan JF, Marsh CB. Social disruption induces lung inflammation. Brain Behav Immun. 2010;24:394–402. doi: 10.1016/j.bbi.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Donnelly DJ, Gensel JC, Ankeny DP, van Rooijen N, Popovich PG. An efficient and reproducible method for quantifying macrophages in different experimental models of central nervous system pathology. J Neurosci Methods. 2009;181:36–44. doi: 10.1016/j.jneumeth.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler H, Bailey MT, Engler A, Sheridan JF. Effects of repeated social stress on leukocyte distribution in bone marrow, peripheral blood and spleen. J Neuroimmunol. 2004;148:106–115. doi: 10.1016/j.jneuroim.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Engler H, Bailey MT, Engler A, Stiner-Jones LM, Quan N, Sheridan JF. Interleukin-1 receptor type 1-deficient mice fail to develop social stress-associated glucocorticoid resistance in the spleen. Psychoneuroendocrinology. 2008;33:108–117. doi: 10.1016/j.psyneuen.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Wieseler-Frank JL, Watkins LR, Maier SF. Rapid isolation of highly enriched and quiescent microglia from adult rat hippocampus: immunophenotypic and functional characteristics. J Neurosci Methods. 2006a;151:121–130. doi: 10.1016/j.jneumeth.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Frank MG, Barrientos RM, Biedenkapp JC, Rudy JW, Watkins LR, Maier SF. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol Aging. 2006b;27:717–722. doi: 10.1016/j.neurobiolaging.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun. 2007;21:47–59. doi: 10.1016/j.bbi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Frank MG, Miguel ZD, Watkins LR, Maier SF. Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain Behav Immun. 2010;24:19–30. doi: 10.1016/j.bbi.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, Johnson RW. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 2005;19:1329–1331. doi: 10.1096/fj.05-3776fje. [DOI] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, Sheridan JF, Godbout JP. Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J Neuroinflammation. 2008;5:15. doi: 10.1186/1742-2094-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23:309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Kohsaka S. Intracellular signaling in M-CSF-induced microglia activation: role of Iba1. Glia. 2002;40:164–174. doi: 10.1002/glia.10149. [DOI] [PubMed] [Google Scholar]
- Johnson JD, Campisi J, Sharkey CM, Kennedy SL, Nickerson M, Greenwood BN, Fleshner M. Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience. 2005;135:1295–1307. doi: 10.1016/j.neuroscience.2005.06.090. [DOI] [PubMed] [Google Scholar]
- Johnson JD, Cortez V, Kennedy SL, Foley TE, Hanson H, 3rd, Fleshner M. Role of central beta-adrenergic receptors in regulating proinflammatory cytokine responses to a peripheral bacterial challenge. Brain Behav Immun. 2008;22:1078–1086. doi: 10.1016/j.bbi.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsson G, Regàrdh CG. Clinical pharmacokinetics of beta-adrenoreceptor blocking drugs. Clin Pharmacokinet. 1976;1:233–263. doi: 10.2165/00003088-197601040-00001. [DOI] [PubMed] [Google Scholar]
- Kiecolt-Glaser JK, Glaser R. Depression and immune function: central pathways to morbidity and mortality. J Psychosom Res. 2002;53:873–876. doi: 10.1016/s0022-3999(02)00309-4. [DOI] [PubMed] [Google Scholar]
- King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood. 2009;113:3190–3197. doi: 10.1182/blood-2008-07-168575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Bailey MT, Sheridan JF, Padgett DA, Avitsur R. Repeated social defeat causes increased anxiety-like behavior and alters splenocyte function in C57BL/6 and CD-1 mice. Brain Behav Immun. 2007;21:458–466. doi: 10.1016/j.bbi.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Bailey MT, Sheridan JF, Padgett DA. The inflammatory response to social defeat is increased in older mice. Physiol Behav. 2008;93:628–636. doi: 10.1016/j.physbeh.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollack-Walker S, Watson SJ, Akil H. Social stress in hamsters: defeat activates specific neurocircuits within the brain. J Neurosci. 1997;17:8842–8855. doi: 10.1523/JNEUROSCI.17-22-08842.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo JW, Duman RS. Interleukin-1 receptor null mutant mice show decreased anxiety-like behavior and enhanced fear memory. Neurosci Lett. 2009;456:39–43. doi: 10.1016/j.neulet.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács KJ. Measurement of immediate-early gene activation: c-Fos and beyond. J Neuroendocrinol. 2008;20:665–672. doi: 10.1111/j.1365-2826.2008.01734.x. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- Martinez M, Calvo-Torrent A, Herbert J. Mapping brain response to social stress in rodents with c-Fos expression: a review. Stress. 2002;5:3–13. doi: 10.1080/102538902900012369. [DOI] [PubMed] [Google Scholar]
- Mays JW, Bailey MT, Hunzeker JT, Powell ND, Papenfuss T, Karlsson EA, Padgett DA, Sheridan JF. Influenza virus-specific immunological memory is enhanced by repeated social defeat. J Immunol. 2010;184:2014–2025. doi: 10.4049/jimmunol.0900183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamee EN, Griffin EW, Ryan KM, Ryan KJ, Heffernan S, Harkin A, Connor TJ. Noradrenaline acting at beta-adrenoceptors induces expression of IL-1beta and its negative regulators IL-1ra and IL-1RII, and drives an overall anti-inflammatory phenotype in rat cortex. Neuropharmacology. 2010;59:37–48. doi: 10.1016/j.neuropharm.2010.03.014. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Brück W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Nadjar A, Combe C, Layé S, Tridon V, Dantzer R, Amédée T, Parnet P. Nuclear factor kappaB nuclear translocation as a crucial marker of brain response to interleukin-1: a study in rat and interleukin-1 type I deficient mouse. J Neurochem. 2003;87:1024–1036. doi: 10.1046/j.1471-4159.2003.02097.x. [DOI] [PubMed] [Google Scholar]
- Nair A, Bonneau RH. Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation. J Neuroimmunol. 2006;171:72–85. doi: 10.1016/j.jneuroim.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Nair A, Hunzeker J, Bonneau RH. Modulation of microglia and CD8(+) T cell activation during the development of stress-induced herpes simplex virus type-1 encephalitis. Brain Behav Immun. 2007;21:791–806. doi: 10.1016/j.bbi.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Julien JP, Rivest S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci. 2002;3:216–227. doi: 10.1038/nrn752. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The mouse brain in stereotaxic coordinates. 2nd edition. San Diego: Academic; 2004. [Google Scholar]
- Powell ND, Bailey MT, Mays JW, Stiner-Jones LM, Hanke ML, Padgett DA, Sheridan JF. Repeated social defeat activates dendritic cells and enhances Toll-like receptor dependent cytokine secretion. Brain Behav Immun. 2009;23:225–231. doi: 10.1016/j.bbi.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich G, Banati R. Brain microglia and blood-derived macrophages: molecular profiles and functional roles in multiple sclerosis and animal models of autoimmune demyelinating disease. Brain Res Rev. 2004;46:261–281. doi: 10.1016/j.brainresrev.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, McEwen BS, Chattarji S. Stress, memory and the amygdala. Nat Rev Neurosci. 2009;10:423–433. doi: 10.1038/nrn2651. [DOI] [PubMed] [Google Scholar]
- Salako LA, Falase AO, Aderounmu AF. Comparative beta-adrenoreceptor-blocking effects and pharmacokinetics or propranolol and pindolol in hypertensive Africans. Clin Sci (Lond) 1979;57(Suppl 5):393s–396s. doi: 10.1042/cs057393s. [DOI] [PubMed] [Google Scholar]
- Samuels ER, Szabadi E. Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part I: principles of functional organisation. Curr Neuropharmacol. 2008;6:235–253. doi: 10.2174/157015908785777229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan JF, Padgett DA, Avitsur R, Marucha PT. Experimental models of stress and wound healing. World J Surg. 2004;28:327–330. doi: 10.1007/s00268-003-7404-y. [DOI] [PubMed] [Google Scholar]
- Stark JL, Avitsur R, Padgett DA, Campbell KA, Beck FM, Sheridan JF. Social stress induces glucocorticoid resistance in macrophages. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1799–R1805. doi: 10.1152/ajpregu.2001.280.6.R1799. [DOI] [PubMed] [Google Scholar]
- Stark JL, Avitsur R, Hunzeker J, Padgett DA, Sheridan JF. Interleukin-6 and the development of social disruption-induced glucocorticoid resistance. J Neuroimmunol. 2002;124:9–15. doi: 10.1016/s0165-5728(02)00004-8. [DOI] [PubMed] [Google Scholar]
- Trainor BC, Finy MS, Nelson RJ. Paternal aggression in a biparental mouse: parallels with maternal aggression. Horm Behav. 2008;53:200–207. doi: 10.1016/j.yhbeh.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tynan RJ, Naicker S, Hinwood M, Nalivaiko E, Buller KM, Pow DV, Day TA, Walker FR. Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav Immun. 2010;24:1058–1068. doi: 10.1016/j.bbi.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. 2009;10:397–409. doi: 10.1038/nrn2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynne AM, Henry CJ, Huang Y, Cleland A, Godbout JP. Protracted downregulation of CX(3)CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behav Immun. 2010;24:1190–1201. doi: 10.1016/j.bbi.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]