

Abstract
According to the theory of neuronal health, neurons exist in a spectrum of states ranging from highly resilient to vulnerable. An unhealthy neuron may be rendered dysfunctional or non-viable by an insult that would ordinarily be non-toxic to a healthy neuron. Over the years it has become clear that a neuron’s health is subject to dynamic regulation by electrical or synaptic activity. This review highlights recently identified activity dependent signalling events which boost neuronal health through the transcriptional control of pro- and anti-apoptotic genes, the enhancement of antioxidant defences, and the regulation of mitochondrial and neurotrophic factor availability. Furthermore, activity dependent signals have recently been shown to influence a variety of events specific to individual neurodegenerative diseases, which will also be highlighted.
Introduction
Neuronal health is more complex than that offered by the binary classification of either dead or alive. It is better considered a dynamic spectrum of physiological states ranging from protected and fully functional to vulnerable and dysfunctional [1]. A neuron’s position within this spectrum can be influenced by both detrimental and beneficial external cues (Figure 1). The concept that electrical activity promotes neuronal health, originated from studies in which activity blockade (either pharmacological or through deafferentation) caused death in the disconnected target neurons [2].
Figure 1.
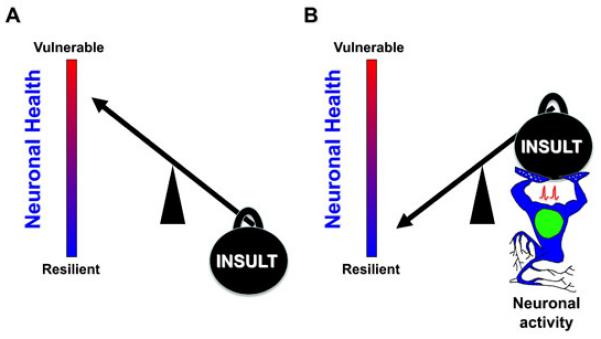
Neurons exist within a dynamic spectrum of health ranging from vulnerable to resilient as shown by the red to blue gradients. A neuron’s position within this spectrum is constantly influenced by a multitude of both beneficial and detrimental cues. In (A), we see that the weight of an “INSULT”, which represents a variety of detrimental events [e.g. oxidative stress, excitotoxicity, ischemia, pathology-associated burdens (amyloid beta protein, mutant huntingtin etc.)], burdens the neuron such that health is compromised and it is rendered vulnerable. In (B) however, neuronal activity, as represented by an active and muscular neuron, is able to boost neuronal health, enabling the neuron to withstand the same “INSULT” burden, whilst remain resilient, emphasizing the important neuroprotective influence of activity on neuronal health.
In vivo studies on the chick and later mammalian developing retinotectal/retino-superior collicular pathway revealed that neuronal survival required firing activity, as similarly seen in other systems such as the spiral ganglion [3,4]. Activity-dependent neuroprotection also appears relevant to mature neurons, despite this being difficult to assess in many brain regions given the complexity of incoming afferents. Deafferentation of adult cerebellar granule cells promotes apoptosis, and olfactory bulb ablation triggers deafferentation dependent apoptosis in piriform cortical neurons[2]. Activity-dependent survival has been recapitulated in multiple cultured neuronal types including those from the spinal cord, cerebellum (granule cells and Purkinje cells) hippocampus, neocortex, hypothalamus and several sensory ganglia [2]. Artificial manipulation of neuronal electrical activity both in vivo (principally spiral ganglion and retinal ganglion cells) and in culture also promotes neuronal survival in a variety of experimental systems and trauma models, including models of apoptosis, oxidative stress and excitotoxicity/ischemia [2,3,5-7].
Ca2+ influx is a key mediator of activity-dependent health-promoting pathways, and the synaptic N-methyl-D-aspartate receptor (NMDAR) is an important route for such influx. Impaired synaptic NMDAR activity promotes neuronal death in vitro and in vivo in development [8,9], in adults NMDAR blockade exacerbates neuronal loss during ongoing neurodegeneration or post traumatic brain injury [10] and impairs survival of new-born neurons in the dentate gyrus [11]. The protective effects of physiological patterns of synaptic NMDAR activity are in marked contrast to the destructive effects of excessive NMDAR activity, particularly that mediated by extrasynaptic NMDARs [12,13].
Other sources of activity-dependent Ca2+ elevation are also relevant to neuroprotective signaling, including release from internal stores [14], as well as firing activity promoting influx through voltage-gated Ca2+ channels [15], indeed, different sources may cooperate in the creation of the transient. As will be discussed, neuronal activity triggers neuroprotection through the complimentary and coincident regulation of numerous health-promoting pathways that alter vulnerability to apoptotic, excitotoxic and oxidative insults, as will be outlined below. We will also discuss the emerging role of synaptic activity in influencing a number of processes specific to individual neurodegenerative diseases.
Synaptic activity induces expression of survival genes and suppresses pro-death genes
Neuronal activity and resultant Ca2+ influx is an important mechanism of dialogue between the synapse and nucleus, enabling both activation of second messengers and gene transcription. Activity-dependent induction of gene expression mediated by the transcription factor CREB is a classic example of an activity dependent health promoting process which confers resistance to both apoptotic and excitotoxic insults [16-18]. More recently, a group of genes termed the activity-regulated inhibitors of death (AID), with visible neuroprotective effects both in vitro and in vivo, were identified as being activity and nuclear Ca2+ regulated [19]. Some of these genes appear to be CREB targets, possibly promoting protection through an enhancement of mitochondrial resistance to stress or toxicity [19]. The Ca2+ responsive transcription factor NFAT (nuclear factor of activated T cells), was also recently implicated in NMDAR-dependent neuroprotection [20]. In addition to nuclear Ca2+ signalling, other neuroprotective signalling cascades are also triggered at the synaptic NMDAR, including the PI3K-Akt and Erk1/2 pathways [9], and the more recently identified nitric oxide synthase/Erk dependent activation of neuroprotective transcription factor NFI-A (nuclear factor I subtype A [21]). NFI-A joins CREB as a transcription factor implicated in mediating both NMDAR-dependent neuroprotection, as well as conferring resistance to NMDAR-dependent excitotoxicity.
Neuronal activity is also intimately involved in regulating the expression, processing, transport and release of neurotrophic factors, many of which have well-characterized neuroprotective effects. In vivo electrical stimulation upregulates fibroblast growth factor 2 (Fgf2) [22] and delays photoreceptor death, preserving retinal function [23], and also promotes IGF-1 secretion by Mueller glia. Elevated expression and release of BDNF is associated with elevated synaptic activity which contributes to neuroprotection [12,24,25], and BDNF is up-regulated by environmental enrichment in vivo [26]. The release and maturation of pro-NGF has also been identified as being activity-dependent [27]. Moreover, activity also promotes growth factor trafficking from the periphery in the case of IGF-1, enabling a coordinated delivery of trophic support to active areas [28].
In addition to pro-survival gene induction, synaptic activity results in the transcriptional suppression of core components of the intrinsic apoptotic cascade including Puma, Apaf1, Casp9, Casp3, and Trp53, leading to enhanced resistance to apoptotic insults [29,30]. While Puma suppression is central to apoptosis prevention, by preventing insult-induced cytochrome c release, additional downstream mechanisms such as the downregulation of Apaf-1 and caspase-9 also exist [29,30]. The transcriptional suppression of these apoptotic genes may be linked to the activity-dependent inactivation of the transcription factors that control their expression, including p53 and the forkhead box protein O (FOXO) class of transcription factors [29,31,32]
Given that apoptotic features are found in a number of neurodegenerative conditions including Alzheimer’s, Parkinson’s and Huntington’s diseases (AD, PD, HD respectively), the above findings are of clinical relevance, highlighting the possibility that less active neurons are more likely to undergo apoptosis in these conditions. Activity-dependent caspase regulation may be particularly relevant to AD as non-apoptotic, caspase-dependent events have been observed in several aspects of the pathology. Caspases have been shown to cleave the presenilin-1 subunit of γ-secretase, creating a complex which preferentially releases a higher yield of the more toxic 42-form of Aβ [33]. Moreover, caspases are activated following Death Receptor 6 binding by an APP cleavage product, leading to caspase-dependent axonal and cell body degeneration [34]. Caspase activation has also been linked to both tangle formation [35] and synaptic dysfunction [36] in animal models of AD. Given that neurodegeneration is a characteristic feature of AD, disrupted neuronal activity might be a contributing factor to resultant pathology through any of the above described mechanisms or through enhanced vulnerability to apoptosis itself. It should however be noted that caspases can mediate important non-apoptotic physiological events, including forms of long term depression and AMPA receptor internalization which rely on caspase-3 activation [37]. As such, the fact that the Casp3 gene is itself subject to activity-dependent inactivation [30] may be relevant to physiological as well as pathological processes.
As well as restricting the apoptotic potential of neurons, synaptic activity also boosts neuronal health through an enhancement of intrinsic antioxidant defences. Previously active neurons withstand oxidative insults better than less active neurons, a mechanism which at least partially involves changes within the thioredoxin/peroxiredoxin antioxidant system [6]. Synaptic NMDAR activity reduces the level of oxidant-induced peroxiredoxin hyperoxidation and enhances expression of sulfiredoxin 1 (Srxn1) and sestrin 2 (Sesn2), the genes whose products are believed to mediate this. Interestingly, in addition to boosting expression of these genes, synaptic NMDAR activity also suppresses the expression of the thioredoxin inhibitor Txnip, a FOXO target gene [6]. Also relevant to the control of antioxidant defences is the influence of synaptic activity on gene expression promoted by the co-activator peroxisome proliferator-activated receptor-γ co-activator 1α (PGC-1α), a master regulator of neuronal antioxidant defences [38,39]. Synaptic activity post-translationally enhances the transactivating potential of PGC-1α, induces transcription of the PGC-1α gene, and triggers the nuclear export of the corepressor SMRT [40] which, when nuclear, can block the antioxidant effects of PGC-1α [39].
Activity dependent regulation of mitochondrial availability and mitochondria related genes
Neuronal health is influenced by energy demands and Ca2+ homeostasis, two events which are regulated by mitochondria. By regulating mitochondrial fission/fusion and intracellular trafficking, neuronal activity triggers events which balance energy demands with localized availability [41]. For example, synaptic activity enhances mitochondrial fission, reduces mitochondrial mobility and localizes mitochondria to dendritic spines [42-44] and presynaptic sites [45] where energy demands are high, as is in line with the finding that mitochondrial mobility is accelerated in areas rich in ATP and slowed in areas high in ADP [46].
One mechanism through which mitochondria detect changes in intracellular Ca2+ and subsequently alter movement, involves the GTPase Miro, which has two EF-hand Ca2+ binding domains. Following Ca2+ influx, Miro unhinges mitochondria from transport machinery, enabling stoppage in specific areas [45,47,48]. Mitochondrial movement into dendritic spines and filopodia is also activity-dependent and is mediated by Wiskott-Aldrich syndrome protein (WASP)-family verprolin homologous protein 1 (WAVE1) whose inhibitory phosphorylation by Cdk5 is in turn suppressed by NMDAR activity [49]. In addition to motility, neuronal activity also promotes gene transcription which enhances mitochondrial health, such as the previously mentioned activation of AID genes [19] or PGC1α [39] which is capable of controlling mitochondrial biogenesis [50].
Neuronal activity can influence aspects of disease pathology
We have hitherto focused on general neuroprotective events triggered by neuronal activity, which may be relevant to the survival of neurons in response to a variety of insults (Figure 2). However, recent studies have illustrated how activity can inhibit molecular events specific to individual neurodegenerative disease processes (Figure 3). As such, neuronal hypo-activity could be an exacerbating factor in certain diseases and since this could also be a consequence of disease pathology, could form a feed-forward cycle of disease progression.
Figure 2.
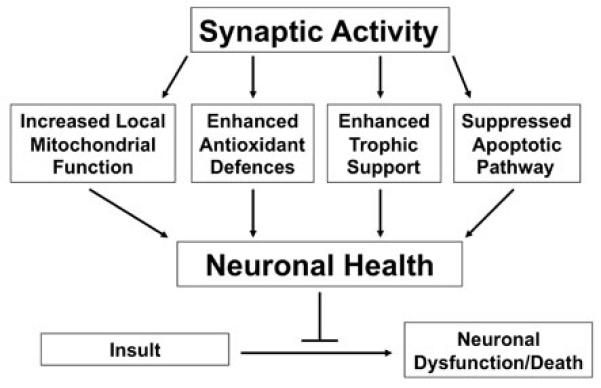
Schematic demonstrating the mechanisms through which synaptic activity boosts neuronal health, enabling the cell to better resist dysfunction or death. For full description see text.
Figure 3.
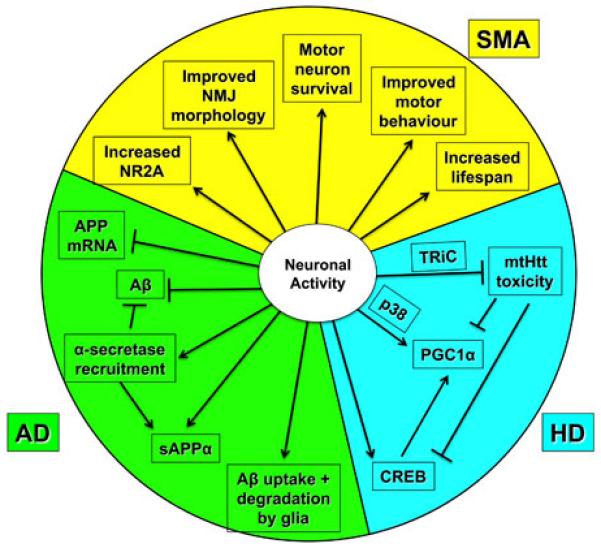
Neuronal activity has been shown to influence specific disease processes in a number of experimental models of neurodegenerative disease. Summarized in graphic format are disease-specific pathways reported as being influenced by neuronal activity, in experimental models of Alzheimer’s disease (AD), Huntington’s disease (HD) and Spinal Muscular Atrophy (SMA), as reviewed in greater detail in the text. Aβ = amyloid beta protein, APP = amyloid precursor protein, mtHtt = mutant Huntingtin, NMJ = neuromuscular junction, PGC1α = peroxisome proliferator-activated receptor-γ co-activator 1α, sAPPα = soluble Amyloid Precursor Protein alpha, TriC = T complex-1 (TCP-1) ring complex.
Alzheimer’s Disease
Neuronal activity, specifically that involving synaptic NMDAR activation, appears to have a suppressive effect on multiple aspects of AD-related amyloid processing. Synaptic NMDAR activation promotes protective non-amyloidogenic processing, over amyloidogenic processing, as seen by the activity-dependent recruitment of putative α-secretase ADAM-10 [51], increased non-amyloidogenic pathway components c83 and soluble APPα [52], reduced Aβ production and release [51,52], reduced APP695 mRNA expression [53] and reduced intraneuronal Aβ [54]. Interestingly, synaptic activity blockade in vivo in a transgenic mouse model of AD increased intraneuronal Aβ, reduced synaptophysin-immunoreactivity, increased synapse loss and worsened behaviour [55], highlighting the importance of neuronal activity in ameliorating pathology. Adrenergic neuronal activity may also be relevant, as β2 adrenergic receptor activation enhances glial Aβ uptake and degradation [56]. One can envisage that any stochastic or disease-related reduction in neuronal activity could worsen neuronal health and accelerate pathogenesis through the removal of the above-mentioned brakes on Aβ-related pathology.
Spinal Muscular Atrophy and Amyotrophic Lateral Sclerosis
Both Spinal Muscular Atrophy (SMA) and Amyotrophic Lateral Sclerosis (ALS) are characterized by degeneration of motor neurons, an event which is influenced by modest levels of NMDAR activity. In a transgenic mouse model of SMA, exercise was found to delay motor neuron death, and upregulate NMDAR subunit NR2A, an effect which was ablated by NMDAR inhibition [57]. Daily in vivo NMDA administration to transgenic SMA-like mice, accelerated postnatal skeletal muscle maturation, reduced motor neuron death, improved neuromuscular junction morphology and motor behaviour and prolonged lifespan [58]. In an ALS-like transgenic mouse model, significant decreases in both NR2A and αCaMKII autophosphorylation were identified with coincident alterations in synaptic plasticity and reduced dendritic outgrowth, prior to symptom onset [59], alterations which led the authors to suggest that reduced glutamatergic signaling may contribute to disease pathogenesis.
Hungtington’s Disease
Neuronal activity has also been shown to profoundly impact HD pathology, as largely demonstrated through the use of the YAC 128 HD mouse model. Synaptic activity increased formation of non-toxic mutant huntingtin (mtHtt) inclusions [60] via the NMDA-dependent transcriptional upregulation of chaperonin TRiC (T complex-1 (TCP-1) ring complex), which mediates non-toxic inclusion formation in neurons, rendering neurons more resistant to mtHtt-mediated death [60]. Synaptic activity also enhances regulation of CREB target gene PGC1α, a co-activator which, as previously mentioned, regulates mitochondrial density and antioxidant defences [38,39,50], and which is lowered in the striatum of HD patients [61], an area particularly vulnerable to PGC1α deficiency [62]. Interestingly, mtHtt directly interferes with CREB-dependent PGC1α expression, leading to mitochondrial dysfunction and metabolic defects [61,63]. Blocking synaptic activity decreases both CREB activity and PGC1α protein levels, while activation has the opposite effect, suggesting a mechanism whereby synaptic NMDAR activity enhances neuronal health and resilience to insult via the protective CREB-PGC1α pathway and promotion of mtHtt inclusion formation [60]. Extrasynaptic NMDA receptor activation has been known for some time to dominate over protective CREB dependent signalling originating from synaptic NMDAR [12,13]. This is particularly relevant given that the YAC 128 mouse model displays an increase in extrasynaptic NMDAR expression and currents, along with an associated decrease in CREB activity [64]. Thus, the beneficial effects of synaptic NMDAR activity are negated by increased, dominating pro-death signalling from extrasynaptic NMDAR. Interestingly, in vivo low-dose memantine administration, which preferentially blocks extrasynaptic NMDAR while preserving synaptic NMDAR signalling [65], improved neuropathological and behavioural outcomes [60,64]. In contrast, higher doses of memantine, which also block synaptic NMDARs, exacerbated disease pathology and symptoms [60] highlighting the importance of maintaining synaptic activity to neuronal health and viability.
Conclusions
The myriad routes by which neuronal activity influences neuronal health are becoming clearer. Not only are general antioxidant, mitochondrial and apoptotic pathways subject to control, they are also central to a number of neurodegenerative diseases. Knowledge of such endogenous neuroprotective pathways points to ways in which they may be mimicked or boosted for therapeutic effect. Furthermore, they underline the fact that activity-dependent signals are important contributors to neuronal robustness and that successful therapies should leave these beneficial effects intact.
Acknowledgements
The authors’ work is funded by the a Medical Research Council Senior Research Fellowship (GEH), the Wellcome Trust, the Biotechnology and Biological Sciences Research Council, and the EMBO Young Investigator Programme
References
* of special interest
** of outstanding interest
- 1.Isacson O. On neuronal health. Trends Neurosci. 1993;16:306–308. doi: 10.1016/0166-2236(93)90104-t. [DOI] [PubMed] [Google Scholar]
- 2.Mennerick S, Zorumski CF. Neural activity and survival in the developing nervous system. Mol Neurobiol. 2000;22:41–54. doi: 10.1385/MN:22:1-3:041. [DOI] [PubMed] [Google Scholar]
- 3.Corredor RG, Goldberg JL. Electrical activity enhances neuronal survival and regeneration. J Neural Eng. 2009;6:055001. doi: 10.1088/1741-2560/6/5/055001. [DOI] [PubMed] [Google Scholar]
- 4.Bessero AC, Clarke PG. Neuroprotection for optic nerve disorders. Curr Opin Neurol. 2010;23:10–15. doi: 10.1097/WCO.0b013e3283344461. [DOI] [PubMed] [Google Scholar]
- 5.Tauskela JS, Fang H, Hewitt M, Brunette E, Ahuja T, Thivierge JP, Comas T, Mealing GA. Elevated synaptic activity preconditions neurons against an in vitro model of ischemia. J Biol Chem. 2008;283:34667–34676. doi: 10.1074/jbc.M805624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Papadia S, Soriano FX, Leveille F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat.Neurosci. 2008;11:476–487. doi: 10.1038/nn2071. * This paper demonstrated that synaptic NMDA receptor activation triggers a coordinated upregulation of intrinsic antioxidant defences within the thioredoxin-peroxiredoxin system, enabling enhanced resilience to oxidative insult in active neurons specifically.
- 7.Fishbein I, Segal M. Active cortical innervation protects striatal neurons from slow degeneration in culture. J Neural Transm. 2010 doi: 10.1007/s00702-010-0505-5. [DOI] [PubMed] [Google Scholar]
- 8.Olney JW, Wozniak DF, Jevtovic-Todorovic V, Barber N, Bittigau P, Ikonomidou C. Drug-induced apoptotic neurodegeneration in the developing brain. Brain Pathology. 2002;12:488–498. doi: 10.1111/j.1750-3639.2002.tb00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hardingham GE. Pro-survival signalling from the NMDA receptor. Biochem Soc Trans. 2006;34:936–938. doi: 10.1042/BST0340936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ikonomidou C, Stefovska V, Turski L. Neuronal death enhanced by N-methyl-D-aspartate antagonists. Proc.Natl.Acad.Sci.U.S.A. 2000;97:12885–12890. doi: 10.1073/pnas.220412197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tashiro A, Sandler VM, Toni N, Zhao C, Gage FH. NMDA-receptor-mediated, cell-specific integration of new neurons in adult dentate gyrus. Nature. 2006;442:929–933. doi: 10.1038/nature05028. [DOI] [PubMed] [Google Scholar]
- 12.Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat.Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- 13.Hardingham GE. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem Soc Trans. 2009;37:1147–1160. doi: 10.1042/BST0371147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardingham GE, Arnold FJ, Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci. 2001;4:261–267. doi: 10.1038/85109. [DOI] [PubMed] [Google Scholar]
- 15.Saha RN, Dudek SM. Action potentials: to the nucleus and beyond. Exp Biol Med (Maywood) 2008;233:385–393. doi: 10.3181/0709-MR-241. [DOI] [PubMed] [Google Scholar]
- 16.Lee B, Butcher GQ, Hoyt KR, Impey S, Obrietan K. Activity-Dependent Neuroprotection and cAMP Response Element-Binding Protein (CREB): Kinase Coupling, Stimulus Intensity, and Temporal Regulation of CREB Phosphorylation at Serine 133. J Neurosci. 2005;25:1137–1148. doi: 10.1523/JNEUROSCI.4288-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papadia S, Stevenson P, Hardingham NR, Bading H, Hardingham GE. Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. J.Neurosci. 2005;25:4279–4287. doi: 10.1523/JNEUROSCI.5019-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soriano FX, Hardingham GE. Compartmentalized NMDA receptor signalling to survival and death. J Physiol. 2007;584:381–387. doi: 10.1113/jphysiol.2007.138875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang SJ, Zou M, Lu L, Lau D, Ditzel DA, Delucinge-Vivier C, Aso Y, Descombes P, Bading H. Nuclear calcium signaling controls expression of a large gene pool: identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet. 2009;5:e1000604. doi: 10.1371/journal.pgen.1000604. ** This study identified a novel group of nuclear calcium regulated genes termed the activity-regulated inhibitors of death. Activation of these genes was shown to afford significant neuroprotection both in vitro and in vivo in animal models of neurodegeneration.
- 20.Vashishta A, Habas A, Pruunsild P, Zheng JJ, Timmusk T, Hetman M. Nuclear factor of activated T-cells isoform c4 (NFATc4/NFAT3) as a mediator of antiapoptotic transcription in NMDA receptor-stimulated cortical neurons. J Neurosci. 2009;29:15331–15340. doi: 10.1523/JNEUROSCI.4873-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng S, Eacker SM, Hong SJ, Gronostajski RM, Dawson TM, Dawson VL. NMDA-induced neuronal survival is mediated through nuclear factor I-A in mice. J Clin Invest. 2010;120:2446–2456. doi: 10.1172/JCI33144. * This study identified the novel activation of neuroprotective transcription factor NFI-A following sublethal NMDAR activation. NFI-A knockdown with siRNA reduced the neuroprotection induced by sublethal doses of NMDA and Nfia −/− neurons were highly susceptible to NMDA-induced excitotoxicity. Moreover, Nfia +/− mice were more susceptible to NMDA-induced intrastriatal lesions than were wildtype mice.
- 22.Ciavatta VT, Kim M, Wong P, Nickerson JM, Shuler RK, Jr., McLean GY, Pardue MT. Retinal expression of Fgf2 in RCS rats with subretinal microphotodiode array. Invest Ophthalmol Vis Sci. 2009;50:4523–4530. doi: 10.1167/iovs.08-2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morimoto T, Fujikado T, Choi JS, Kanda H, Miyoshi T, Fukuda Y, Tano Y. Transcorneal electrical stimulation promotes the survival of photoreceptors and preserves retinal function in royal college of surgeons rats. Invest Ophthalmol Vis Sci. 2007;48:4725–4732. doi: 10.1167/iovs.06-1404. [DOI] [PubMed] [Google Scholar]
- 24.Soriano FX, Papadia S, Hofmann F, Hardingham NR, Bading H, Hardingham GE. Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J.Neurosci. 2006;26:4509–4518. doi: 10.1523/JNEUROSCI.0455-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lipsky RH, Marini AM. Brain-derived neurotrophic factor in neuronal survival and behavior-related plasticity. Ann N Y Acad Sci. 2007;1122:130–143. doi: 10.1196/annals.1403.009. [DOI] [PubMed] [Google Scholar]
- 26.Angelucci F, De Bartolo P, Gelfo F, Foti F, Cutuli D, Bossu P, Caltagirone C, Petrosini L. Increased concentrations of nerve growth factor and brain-derived neurotrophic factor in the rat cerebellum after exposure to environmental enrichment. Cerebellum. 2009;8:499–506. doi: 10.1007/s12311-009-0129-1. [DOI] [PubMed] [Google Scholar]
- 27.Bruno MA, Cuello AC. Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc Natl Acad Sci U S A. 2006;103:6735–6740. doi: 10.1073/pnas.0510645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishijima T, Piriz J, Duflot S, Fernandez AM, Gaitan G, Gomez-Pinedo U, Verdugo JM, Leroy F, Soya H, Nunez A, et al. Neuronal activity drives localized blood-brain-barrier transport of serum insulin-like growth factor-I into the CNS. Neuron. 2010;67:834–846. doi: 10.1016/j.neuron.2010.08.007. Nishijima, T. Neuron, 2010. ** This paper demonstrates that neuronal activity regulates the transport of peripherally produced neurotrophic factor IGF-1 through the blood brain barrier (BBB) into the CNS. The authors identify that glutamatergic synaptic activity triggers vasodilation and localized recruitment of circulating IGF-1 to the BBB interface where it is cleaved and transported into the CNS by activity regulated machinery, to specific areas of need.
- 29.Lau D, Bading H. Synaptic activity-mediated suppression of p53 and induction of nuclear calcium-regulated neuroprotective genes promote survival through inhibition of mitochondrial permeability transition. J Neurosci. 2009;29:4420–4429. doi: 10.1523/JNEUROSCI.0802-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leveille F, Papadia S, Fricker M, Bell KF, Soriano FX, Martel MA, Puddifoot C, Habel M, Wyllie DJ, Ikonomidou C, et al. Suppression of the intrinsic apoptosis pathway by synaptic activity. J Neurosci. 2010;30:2623–2635. doi: 10.1523/JNEUROSCI.5115-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Al-Mubarak B, Soriano FX, Hardingham GE. Synaptic NMDAR activity suppresses FOXO1 expression via a cis-acting FOXO binding site: FOXO1 is a FOXO target gene. Channels (Austin) 2009;3:233–238. doi: 10.4161/chan.3.4.9381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dick O, Bading H. Synaptic activity and nuclear calcium signaling protect hippocampal neurons from death signal-associated nuclear translocation of FoxO3a induced by extrasynaptic N-methyl-D-aspartate receptors. J Biol Chem. 2010;285:19354–19361. doi: 10.1074/jbc.M110.127654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hedskog L, Petersen CA, Svensson AI, Welander H, Tjernberg LO, Karlstrom H, Ankarcrona M. Gamma-secretase Complexes Containing Caspase-cleaved Presenilin-1 Increase Intracellular Abeta42/Abeta40 Ratio. J Cell Mol Med. doi: 10.1111/j.1582-4934.2010.01208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT. Caspase activation precedes and leads to tangles. Nature. 2010;464:1201–1204. doi: 10.1038/nature08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, Diamantini A, De Zio D, Carrara P, Battistini L, et al. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat Neurosci. 2010;14:69–76. doi: 10.1038/nn.2709. [DOI] [PubMed] [Google Scholar]
- 37.Li Z, Jo J, Jia JM, Lo SC, Whitcomb DJ, Jiao S, Cho K, Sheng M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell. 141:859–871. doi: 10.1016/j.cell.2010.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 39.Soriano FX, Leveille F, Papadia S, Bell KF, Puddifoot C, Hardingham GE. Neuronal activity controls the antagonistic balance between PGC-1alpha and SMRT in regulating antioxidant defences. Antioxid Redox Signal. 2010 doi: 10.1089/ars.2010.3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mckenzie GJ, Stephenson P, Ward G, Papadia S, Bading H, Chawla S, Privalsky M, Hardingham GE. Nuclear Ca2+ and CaM kinase IV specify hormonal- and Notch-responsiveness. J. Neurochem. 2005;93:171–185. doi: 10.1111/j.1471-4159.2005.03010.x. [DOI] [PubMed] [Google Scholar]
- 41.MacAskill AF, Kittler JT. Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 2010;20:102–112. doi: 10.1016/j.tcb.2009.11.002. * This study demonstrated that the protein Miro 1, which links mitochondria to microtubule transport machinery, is a key regulator of mitochondrial transport. Miro 1 is capable of detecting and binding calcium through its EF hand domains, which causes a subsequent dissociation from transport machinery. This activity dependent event enables coordinated mitochondrial delivery to active areas within the neuron where calcium levels and energy requirements are high.
- 42.Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 43.Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci. 2003;23:7881–7888. doi: 10.1523/JNEUROSCI.23-21-07881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang DT, Honick AS, Reynolds IJ. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J Neurosci. 2006;26:7035–7045. doi: 10.1523/JNEUROSCI.1012-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, Schwarz TL. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mironov SL. ADP regulates movements of mitochondria in neurons. Biophys J. 2007;92:2944–2952. doi: 10.1529/biophysj.106.092981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnoczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A. 2008;105:20728–20733. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61:541–555. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sung JY, Engmann O, Teylan MA, Nairn AC, Greengard P, Kim Y. WAVE1 controls neuronal activity-induced mitochondrial distribution in dendritic spines. Proc Natl Acad Sci U S A. 2008;105:3112–3116. doi: 10.1073/pnas.0712180105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wareski P, Vaarmann A, Choubey V, Safiulina D, Liiv J, Kuum M, Kaasik A. PGC-1{alpha} and PGC-1{beta} regulate mitochondrial density in neurons. J Biol Chem. 2009;284:21379–21385. doi: 10.1074/jbc.M109.018911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marcello E, Gardoni F, Mauceri D, Romorini S, Jeromin A, Epis R, Borroni B, Cattabeni F, Sala C, Padovani A, et al. Synapse-associated protein-97 mediates alpha-secretase ADAM10 trafficking and promotes its activity. J Neurosci. 2007;27:1682–1691. doi: 10.1523/JNEUROSCI.3439-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoey SE, Williams RJ, Perkinton MS. Synaptic NMDA receptor activation stimulates alpha-secretase amyloid precursor protein processing and inhibits amyloid-beta production. J Neurosci. 2009;29:4442–4460. doi: 10.1523/JNEUROSCI.6017-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bordji K, Becerril-Ortega J, Nicole O, Buisson A. Activation of Extrasynaptic, But Not Synaptic, NMDA Receptors Modifies Amyloid Precursor Protein Expression Pattern and Increases Amyloid-{beta} Production. J Neurosci. 30:15927–15942. doi: 10.1523/JNEUROSCI.3021-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tampellini D, Rahman N, Gallo EF, Huang Z, Dumont M, Capetillo-Zarate E, Ma T, Zheng R, Lu B, Nanus DM, et al. Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J Neurosci. 2009;29:9704–9713. doi: 10.1523/JNEUROSCI.2292-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tampellini D, Capetillo-Zarate E, Dumont M, Huang Z, Yu F, Lin MT, Gouras GK. Effects of synaptic modulation on beta-amyloid, synaptophysin, and memory performance in Alzheimer’s disease transgenic mice. J Neurosci. 30:14299–14304. doi: 10.1523/JNEUROSCI.3383-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kong Y, Ruan L, Qian L, Liu X, Le Y. Norepinephrine promotes microglia to uptake and degrade amyloid beta peptide through upregulation of mouse formyl peptide receptor 2 and induction of insulin-degrading enzyme. J Neurosci. 30:11848–11857. doi: 10.1523/JNEUROSCI.2985-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Biondi O, Grondard C, Lecolle S, Deforges S, Pariset C, Lopes P, Cifuentes-Diaz C, Li H, della Gaspera B, Chanoine C, et al. Exercise-induced activation of NMDA receptor promotes motor unit development and survival in a type 2 spinal muscular atrophy model mouse. J Neurosci. 2008;28:953–962. doi: 10.1523/JNEUROSCI.3237-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Biondi O, Branchu J, Sanchez G, Lancelin C, Deforges S, Lopes P, Pariset C, Lecolle S, Cote J, Chanoine C, et al. In vivo NMDA receptor activation accelerates motor unit maturation, protects spinal motor neurons, and enhances SMN2 gene expression in severe spinal muscular atrophy mice. J Neurosci. 30:11288–11299. doi: 10.1523/JNEUROSCI.1764-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spalloni A, Origlia N, Sgobio C, Trabalza A, Nutini M, Berretta N, Bernardi G, Domenici L, Ammassari-Teule M, Longone P. Postsynaptic Alteration of NR2A Subunit and Defective Autophosphorylation of alphaCaMKII at Threonine-286 Contribute to Abnormal Plasticity and Morphology of Upper Motor Neurons in Presymptomatic SOD1G93A Mice, a Murine Model for Amyotrophic Lateral Sclerosis. Cereb Cortex. doi: 10.1093/cercor/bhq152. [DOI] [PubMed] [Google Scholar]
- 60.Okamoto S, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE, Zaidi R, Clemente A, Kaul M, Graham RK, et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat.Med. 2009;15:1407–1413. doi: 10.1038/nm.2056. ** This paper identified the capacity of synaptic activity to beneficially influence pathology in a mouse model of HD, as shown through the activity dependent enhancement of mtHtt inclusion bodies, which renders neurons more resistant to mtHtt-mediated cell death. Through the use of low versus high dose memantine, whichc specifically targets extrasynaptic NMDARs, the study also demonstrated the therapeutic benefit of sparing synaptic signalling in vivo.
- 61.Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 62.Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 63.Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, Gilbert ML, Morton GJ, Bammler TK, Strand AD, et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 64.Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, Ko RW, Vasuta OC, Graham RK, Hayden MR, et al. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron. 65:178–190. doi: 10.1016/j.neuron.2010.01.008. * This paper identified the important finding that extrasynaptic, but not synaptic NMDAR currents are specifically elevated in an animal model of HD and that these enhanced currents contribute to phenotype onset. The study also demonstrated the beneficial effect of memantine treatment in ameliorating pathology associated signaling and motor learning deficits, highlighting the importance of negating extrasynaptic signaling while maintaining protective synaptic-based signalling events.
- 65.Xia P, Chen HS, Zhang D, Lipton SA. Memantine Preferentially Blocks Extrasynaptic Over Synaptic NMDA Receptor Currents in Hippocampal Autapses. J Neurosci. 2010;30:11246–11250. doi: 10.1523/JNEUROSCI.2488-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]