

Abstract
Apolipoprotein E (apoE) is a satiation factor. While central apoE administration reduces food intake, the specific intracellular signaling mechanisms activated by apoE remain largely unknown. Using primary cultured hypothalamic neurons, we demonstrated that apoE treatment (50 nM) elicited rapid activation of the phosphatidylinositol-3-kinase (PI3K)/Akt signaling cascade. Specifically, apoE induced the phosphorylation of Akt, peaking at 30 min, and the increased phosphorylation of Akt was significantly attenuated after pretreatment with LY294002 (50 μM), an inhibitor of the PI3K signaling pathway. To determine whether the activation of PI3K by apoE is required for the ability of apoE to reduce food intake, LY294002 (1 nmol) was infused into the 3rd-cerebral ventricle before injection of an anorectic dose of apoE. Consistent with our previous report, apoE (4 μg) exerted significant reduction of food intake in the 4-h fasted rats, compared with saline. Pretreatment with LY294002 significantly attenuated the potency of exogenous apoE to induce satiation, while the same dose of PI3K inhibitor by itself caused only a slight non-significant decrease of food intake. These results indicate that the activation of the PI3K/Akt pathway is necessary for the acute effects of apoE on food intake.
Keywords: apolipoprotein, food intake, body weight, signaling pathway
1. Introduction
Apolipoprotein E (apoE) is essential for the normal catabolism of triglyceride-rich lipoprotein constituents. Although apoE is produced in most organs, its major production sites are the liver and brain [1]. ApoE was initially recognized for its importance in lipoprotein metabolism and cardiovascular disease. More recently, it has been studied for its role in the control of energy intake [2–4]. We have reported that intracerebroventricular (icv) apoE significantly decreases food intake without causing malaise, whereas icv infusion of apoE antiserum stimulates feeding, implying that endogenous apoE tonically inhibits food intake [2]. Consistent with these observations, apoE is present in the hypothalamus, a brain site intimately involved in the integration of signals for energy homeostasis. Fasted rats have significantly decreased apoE gene expression in the hypothalamus, and refeeding evokes a significant increase of hypothalamic apoE mRNA levels. Genetically obese (ob/ob) mice, and rats with high-fat diet-induced obesity, have significantly reduced hypothalamic apoE mRNA levels compared with their lean control counterparts, suggesting that decreased apoE may contribute to hyperphagia in these obese animals [2].
Phosphatidylinositol 3-kinases (PI3Ks) are heterodimeric complexes composed of regulatory and catalytic subunits that recruit lipids as second messengers and control a broad spectrum of cellular functions including survival, growth and metabolism [5]. The PI3K signaling pathway has been found to be important in the regulation of food intake and energy balance by hypothalamic neurons [6–10]. As occurs in many peripheral cells, neurons also respond to hormonal or neuropeptide signals that activate intracellular pathways to regulate membrane potential, firing rate, the synthesis and release of neurotransmitters, and gene expression. Many of these functions relate to the ability of PI3K to activate protein kinase B (Akt) as occurs in the PI3K/Akt pathway. In the present experiments we sought to determine 1) whether the PI3K/Akt signaling pathway in hypothalamic neurons is modulated by apoE, and 2) whether activation of PI3K/Akt mediates the anorectic effect of apoE in the hypothalamus using a rat model. We found that acute icv administration of apoE in rats activates the hypothalamic PI3K and reduces food intake, and that this is attenuated by pre-infusion of an inhibitor of PI3K.
2. Material and methods
2.1 Animals
Adult male Long-Evans rats (Harlan Industries, Indianapolis, IN) weighing 275–320 g were housed individually in a temperature-controlled vivarium (22 ± 2° C) on a 12/12-h light/dark (lights on at 0600 h) cycle. Purina 5001 laboratory chow (Purina, St. Louis, MO) and water were available ad libitum except where noted. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Cincinnati and conformed to the guidelines of the National Institutes of Health.
2.2. Materials
The PI3K inhibitor LY294002, mouse anti-phosphorylated Akt (anti-pAkt) monoclonal and rabbit anti-Akt polyclonal antibodies were purchased from Cell Signaling Technologies (Beverly, MA). With Western blot analysis, all antibodies gave clear signals at the predicted molecular sizes of the investigated proteins.
2.3. Intracerebroventricular cannulation
At least one week after arrival in the laboratory, rats were anesthetized with ketamine (80 mg/kg)/xylazine (1.6 mg/kg) and implanted with 22-gauge stainless steel guide cannulas (Plastics One) aimed at the 3rd-cerebral ventricle. Coordinates were 2.2 mm posterior to bregma and 7.4 mm ventral to dura as described previously [11;12]. Rats were given 7 days to recover, after which the placement of cannulas was confirmed by administration of 10 ng of angiotensin II in saline through the cannula while the animals were water replete. Animals that did not drink at least 5 ml of water within 30 min were considered to have failed cannula placement and were not used in the experiments.
2.4. Primary hypothalamic neuron cultures and apoE treatment
Primary neuronal cells were prepared from Long-Evans rat embryos on the 18th day of gestation as described previously [13]. Briefly, hypothalamic tissues dissected from the fetal brains were triturated by repeated pipetting, plated at a density of 1 × 105 cells/well in 12-well plates, and cultured in a serum-free neurobasal medium supplemented with B-27 at 37° C. After a 3-day incubation, half of the medium was replaced with fresh medium containing cytosine arabinofuranoside (Ara-C, 10 μM; Sigma) but without L-glutamic acid, and additional incubation was carried out. On Day 7, the cells were treated with either apoE (50 nM) or saline. Then, at different time points, the cells were washed with ice-cold PBS, and lysed in buffer containing protease- and phosphatase-inhibitor cocktails on ice. Lysates were cleared at 12,000 × g for 20 min at 4° C, and protein were stored at −80° C until ready for Western blot analysis.
2.5. Pre-treatment with PI3K inhibitor
Primary hypothalamic cells cultured for 7 days as described above were pre-treated with the PI3K inhibitor LY294002 (50 μM) or vehicle (50% dimethylsulfoxide/saline). Sixty min later, the cells were treated with apoE (50 nM) or saline for an additional 30 min; then the cells were harvested for Western blot analysis.
2.6. In vivo experimental protocols
To investigate the ability of the PI3K inhibitor LY294002 to reverse apoE-induced reduction of food intake, 24 male adult Long-Evans rats with 3rd-ventricular cannulas were divided into 4 treatment groups: vehicle-saline, vehicle-apoE, LY294002-vehicle, and LY294002-apoE (n = 6–7/group). On the test day, these rats were fasted for 4 h before lights off, and received icv LY294002 (1 nmol) [14] or vehicle (50% dimethylsulfoxide/saline) 60 min prior to lights off. We selected this dose of LY294002 because it had no effect on food intake when administered alone, but significantly attenuated the decrease of food intake induced by leptin [6;15] and the reduction of blood glucose induced by insulin [14]. Sixty min later, the rats were administered icv apoE (4 μg), an effective dose at reducing food intake [2], or vehicle (saline). All infusions were given over a period of 2 min in a volume of 3 μl. The injections were separated by 60 min to guarantee sufficient diffusion of LY294002 before the relatively fast action of icv apoE. Food was provided at the onset of dark, and intake was measured after 2 and 4 h.
After recovery (7 days) from the feeding test, the same rats were re-divided into 4 groups as described above. The 4-h fasted rats received icv LY294002 (1 nmol/3μl) or vehicle 60 min prior to dark. Sixty min later, the rats were treated icv with apoE (4 μg) or saline. No food was provided this time. The rats were sacrificed at 30 min after apoE or saline injection. The brains were rapidly removed, hypothalami were dissected and immediately snap frozen in liquid nitrogen. The tissues were homogenized, and proteins were extracted in buffer containing protease- and phosphatase-inhibitor cocktails, and stored at −80° C.
2.7. SDS-PAGE and Western blot analysis
Protein concentration was determined by the BCA protein assay kit (Thermo Fisher Scientific, Rockford, IL). An aliquot of the sample from cultured cells or hypothalamus was diluted with an equal volume of 2 × electrophoresis sample buffer and boiled for 10 min. Twenty micrograms of proteins were separated by size on a 10% SDS-polyacrylamide gel using the Laemmli buffer system and transferred to Immobilon-P polyvinylidene difluoride (PVDF) membranes in Towbin-transfer buffer. After transfer, blots were washed with PBS containing 0.05%Tween 20 (PBST). The membranes were blocked in PBST containing 5% nonfat dry milk and 1% bovine serum albumin for 1 h at room temperature with agitation, and then incubated with primary anti-phospho-Akt antibody (1:1000 dilution) overnight at 4° C. After washing, the membranes were incubated with horseradish peroxidase-labeled secondary rabbit anti-mouse antiserum (1:5000 dilution) for 1 h at room temperature. Immunoreactive bands were visualized with enhanced chemiluminescence (ECL kit; GE Healthcare) after exposure to Hyperfilm ECL (Amersham Biosciences, Piscataway, NJ). Membranes were then stripped by incubation in stripping buffer (Thermo Fisher Scientific, Rockford, IL), for 30 min at 50° C with gentle agitation. Membranes were blocked and reprobed with total rabbit polyclonal antibody to Akt (1:1000 dilution) to verify equal loading as described above. Film autoradiograms were analyzed and quantified by computer-assisted densitometry.
2.8. Statistics
Data were analyzed using one-way or repeated-measures two-way ANOVA by parametric statistics (SigmaStat version 3.5), and statistical significance was determined using Tukey’s multiple comparison. P values less than 0.05 were considered statistically significant.
3. Results
3.1. ApoE rapidly phosphorylated Akt in cultured primary hypothalamic neurons
To test the hypothesis that apoE signaling in the hypothalamus involves action of the PI3K cascade, we assayed apoE-associated PI3K activity in cultured primary hypothalamic neurons. As depicted in Fig. 1, the phosphorylation of Akt in the cultured neuronal cells was significantly induced after apoE treatment with a maximal effect at 30 min. ApoE-induced Akt activation significantly dropped off by 60 min after being normalized to total Akt protein.
Fig. 1.

Time course of apoE (50 nM)-induced activation of Akt in cultured hypothalamic cells. Proteins were analyzed using Western blot with phospho-specific anti-pAkt antibody or antibody that recognizes the total Akt protein as loading control. A, representative immunoblots of pAkt and total Akt; B, quantitative analysis. Data represent mean ± SEM; n = 3. Bars that do not share the same letter are significantly different from each other.
3.2. PI3K inhibitor reduced apoE-induced phosphorylation of Akt in cultured primary hypothalamic neurons
If the phosphorylation of Akt is mediated by the PI3K signaling pathway, apoE-induced action of Akt should be attenuated by pharmacological inhibition of the PI3K signaling pathway. Consistent with this hypothesis, LY294002, a PI3K signaling pathway inhibitor, significantly reduced the pAkt levels induced by apoE (Fig. 2).
Fig. 2.
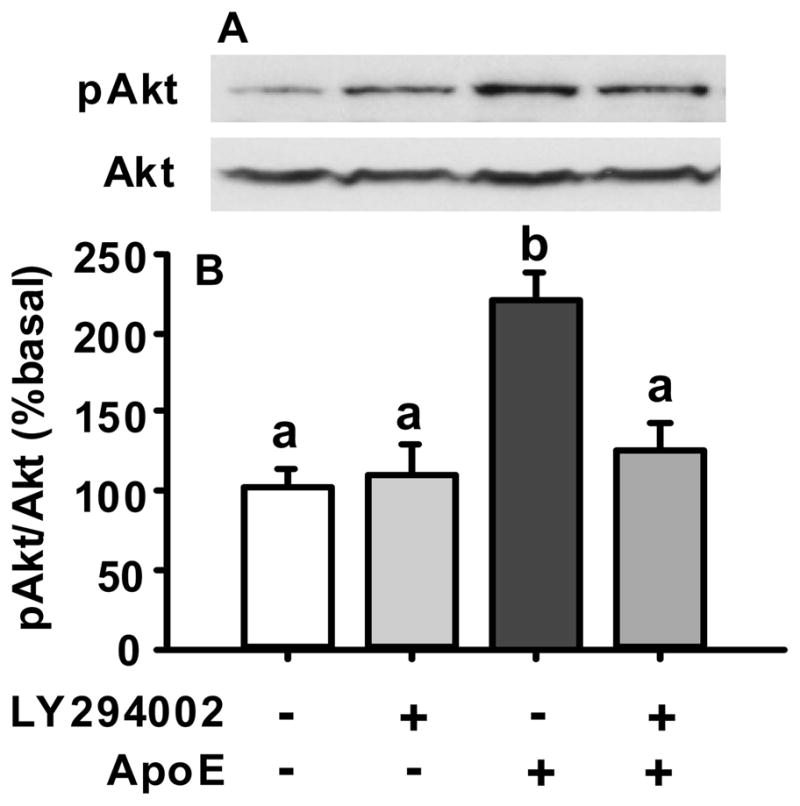
Inhibition of apoE-induced activation of pAkt by LY294002 (50 μM), an inhibitor of the PI3K signaling pathway, in cultured hypothalamic cells. A, representative immunoblots of pAkt and total Akt; B, quantitative analysis. Data represent mean ± SEM; n = 3. Bars that do not share the same letter are significantly different from each other.
3.3. Inhibition of the PI3K signaling cascade decreased the potency of apoE to reduce food intake in rats
To determine whether the activation of PI3K by apoE is required for the ability of apoE to reduce food intake, we infused the inhibitor of PI3K into the 3rd-cerebral ventricle before injection of an anorectic dose of apoE (4 μg). Consistent with our previous report [2], apoE exerted the expected significant reduction of food intake in the 4-h fasted rats, compared with saline (Fig. 3). Pretreatment with LY294002 significantly attenuated the potency of exogenous apoE to induce satiation. The same dose of PI3K inhibitor by itself caused only a slight non-significant decrease of food intake (Fig. 3), indicating that the LY294002 did not independently influence food intake. Because PI3K is the only enzyme known to be inhibited by LY294002 [16], these data suggest that apoE’s ability to reduce food intake is dependent on the activation of PI3K.
Fig. 3.

ApoE-induced anorexigenic effect requires hypothalamic PI3K/Akt signaling. Third-ventricular administration of apoE (4 μg) before onset of the dark cycle significantly suppressed food intake of rats during the first 2 and 4 h of the dark cycle compared to vehicle (saline). Pretreatment with LY294002 (LY, 1 nmol) 60 min before apoE administration significantly attenuated apoE-induction anorexia. Data represent mean ± SEM; n = 6–7, *P <0.05, **P < 0.01.
3.4. Exogenous apoE activated the PI3K signaling cascade in the hypothalamus
To verify the influence of PI3K inbibitor on apoE-induced activation of the PI3K cascade in vivo, 4-h fasted rats received icv LY294002 (1 nmol) or vehicle prior to dark. Sixty min later, the rats were treated icv with apoE (4 μg) or saline. The rats were sacrificed at 30 min after apoE or saline injection. As depicted in Fig. 4, apoE significantly increased Akt phosphorylation (P < 0.05). Pretreatment with LY294002 significantly (P < 0.05) attenuated apoE-induced Akt phosphorylation by about 45%, whereas LY294002 administration alone did not change it.
Fig. 4.

ApoE induced the activation of PI3K/Akt signaling pathway in rat hypothalamus. 4-h fasted rats received icv LY294002 (1 nmol) or vehicle prior to dark. Sixty min later, the rats were treated icv with apoE (4 μg) or saline. After 30 min, hypothalami were microdissected and the protein extracted. Proteins were analyzed using Western blot with phospho-specific anti-pAkt antibody or antibody that recognizes the total Akt protein as loading control. Pretreatment with LY294002 (LY) attenuated apoE-induced Akt phosphorylation in the hypothalamus. A, representative immunoblots of pAkt and total Akt; B, quantitative analysis. Data represent mean ± SEM; n = 6–7, *P <0.05.
4. Discussion
The brain is a major site of apoE expression in humans and rodents with only the liver making more of this apolipoprotein [1;17]. In our previous studies, we have reported that centrally administered apoE suppresses food intake dose-dependently and without causing malaise. Since the fact that comparable doses of apoE administered systemically have no effect on food intake, the anorectic action seems likely to occur within the brain [2]. The hypothalamus is a specialized brain area that evolved for energy homeostasis [18;19]. Given that centrally administered apoE reduces food intake acutely, it is unlikely that meal termination is controlled by transcriptional events because they typically require more time. We therefore considered that a receptor-activated signaling cascade is likely responsible for the reduced intake.
Phosphatidylinositol 3-kinases (PI3Ks) are a family of enzymes involved in cellular functions such as cell growth, proliferation, differentiation, motility, and intracellular trafficking [20]. In numerous cell types, PI3K acts in a heterodimeric form consisting of one 85-kDa regulatory (p85) and one 110-kDa catalytic (p110) subunit [21]. Activated PI3K rapidly mediates the phosphorylation of PI 4,5-biphosphate to PI 3,4,5-trisphoshpate (PIP3), a key signaling intermediate that recruits and activates downstream molecules, including serine-threonine kinases, tyrosine kinases, GTPases, and others [16]. One key downstream target of PI3K is protein kinase B (PKB)/Akt, which is activated via serine and threonine phosphorylation [22]. Recent studies suggest that hypothalamic PI3K signaling is important in the regulation of both energy balance and glucose metabolism [10;23].
We sought to determine whether this signaling pathway also mediates actions of apoE in the hypothalamus. We first determined PI3K activity after apoE treatment using cultured primary hypothalamic neurons, and found that apoE, at a concentration of 50 nM, significantly induced the phosphorylation of Akt. This phosphorylation was maximal at 30 min, and significantly dropped by 60 min after apoE treatment (Fig. 1). If the phosphorylation of Akt is mediated by the PI3K signaling pathway, the increased pAkt levels induced by apoE should be attenuated by pharmacological inhibition of the PI3K signaling pathway. Consistent with this hypothesis, LY294002, an inhibitor of PI3K, was found to significantly reduce the phosphorylation of Akt induced by apoE (Fig. 2). These data suggest that intracellular PI3K signaling mediates the downstream actions of receptors for apoE in hypothalamic neurons.
To investigate the functional significance of this activation, we further determined whether apoE-induced anorexia can be affected by icv pretreatment with the PI3K inhibitor. When LY294002 (1 nmol) was infused into the 3rd ventricle 60 min prior to central apoE (4 μg) administration, the inhibitory effect on food intake induced by apoE was significantly attenuated (Fig. 3). To verify that the PI3K inbibitor did, in fact, block apoE-induced activation of the PI3K cascade in vivo, we repeated the experiment [icv LY294002 (1 nmol) or vehicle, 60 min later, icv apoE (4 μg) or saline], and the rats were sacrificed 30 min after apoE or saline injection. ApoE-induced Akt activation was significantly attenuated by LY294002, whereas LY294002 alone did not significantly influence Akt phosphorylation (Fig. 4). While the mechanism of apoE-induced activation of PI3K signaling pathway remains to be investigated, these results support our hypothesis that apoE engages the PI3K signaling pathway and its downstream effector mechanisms to activate hypothalamic neuronal cells and exert its anorectic effect.
A difference in the response to apoE treatment between in vitro and in vivo studies was observed, i.e. the activation of Akt phosphorylation in tested animals was less than that in cultured neuronal cells after apoE administration. Several factors may cause such a difference. First, in the in vitro study, the apoE was directly added into the medium, so the cultured cells were theoretically evenly-exposed to the apolipoprotein. However, apoE, after icv infusion, may only reach a subset of potentially responsive neurons in rat hypothalamus due to limited diffusion in some brain areas. Second, the doses were different. The final concentration of apoE in the in vitro medium was 50 nM. Since apoE is also produced in the CNS, the degree to which the icv-infused apoE (4 μg) might change local apoE level in the interstitial fluid is unknown. We presume that whereas apoE levels in brain interstitial fluid is increased following 3rd-ventricular administration, its levels are nonetheless lower than the concentration in the cultured medium, leading to less activation of pAkt in rat brain. Additionally, other unknown endogenous factors in the in vivo experiment could act in a compensatory manner to preclude further elevations of activated pAkt.
Our data extend the integrative role of PI3K/Akt in the regulation of food intake in the hypothalamus. For example, leptin is a key hormonal regulator of energy balance that acts upon hypothalamic neurons to reduce food intake. Niswender et al. demonstrated that systemic administration of leptin in rats activates PI3K in the hypothalamus and that icv infusion of LY294002 prevents leptin-induced anorexia [6;8]. Insulin also acts in the brain to regulate food intake, and its central administration in rats causes a significant reduction in food intake and body weight [7]. This effect was also completely blocked by the administration of the PI3K inhibitor LY294002 given one hour before insulin treatment [7;8]. Thus, the PI3K/Akt signaling in the hypothalamus may be a common pathway that integrates metabolic cues to provide a coordinated control of energy homeostasis.
In conclusion, our results identify an essential cellular mechanism of apoE’s action within the brain, particularly within the hypothalamus. We demonstrated that apoE triggers the activation of the PI3K cascade within hypothalamic neurons, and that the anorectic action of apoE within the hypothalamus is mediated by a local apoE-induced activation of the PI3K cascade. Thus, manipulations of the brain apoE system, by apoE agonism or via the specific activation of the PI3K cascade within selected brain regions, may provide novel therapeutic strategies for the presentment and treatment of obesity and its related disorders.
Acknowledgments
This work was supported in part by research grants DK70992 and DK63907 to ML and DK 17844 to SCW.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest. 1985;76:1501–1513. doi: 10.1172/JCI112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shen L, Tso P, Woods SC, Clegg DJ, Barber KL, Carey K, Liu M. Brain apolipoprotein E: an important regulator of food intake in rats. Diabetes. 2008;57:2092–2098. doi: 10.2337/db08-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen L, Tso P, Wang DQ, Woods SC, Davidson WS, Sakai R, Liu M. Up-regulation of apolipoprotein E by leptin in the hypothalamus of mice and rats. Physiol Behav. 2009;98:223–228. doi: 10.1016/j.physbeh.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen L, Carey K, Wang DQ, Woods SC, Liu M. Food-entrained rhythmic expression of apolipoprotein E expression in the hypothalamus of rats. Brain Res. 2009;1273:66–71. doi: 10.1016/j.brainres.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donato J, Jr, Frazao R, Elias CF. The PI3K signaling pathway mediates the biological effects of leptin. Arq Bras Endocrinol Metabol. 2010;54:591–602. doi: 10.1590/s0004-27302010000700002. [DOI] [PubMed] [Google Scholar]
- 6.Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG, Jr, Schwartz MW. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature. 2001;413:794–795. doi: 10.1038/35101657. [DOI] [PubMed] [Google Scholar]
- 7.Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers MG, Jr, Seeley RJ, Schwartz MW. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes. 2003;52:227–231. doi: 10.2337/diabetes.52.2.227. [DOI] [PubMed] [Google Scholar]
- 8.Mirshamsi S, Laidlaw HA, Ning K, Anderson E, Burgess LA, Gray A, Sutherland C, Ashford ML. Leptin and insulin stimulation of signalling pathways in arcuate nucleus neurones: PI3K dependent actin reorganization and KATP channel activation. BMC Neurosci. 2004;5:54. doi: 10.1186/1471-2202-5-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao AZ, Huan JN, Gupta S, Pal R, Sahu A. A phosphatidylinositol 3-kinase phosphodiesterase 3B-cyclic AMP pathway in hypothalamic action of leptin on feeding. Nat Neurosci. 2002;5:727–728. doi: 10.1038/nn885. [DOI] [PubMed] [Google Scholar]
- 10.Morton GJ, Gelling RW, Niswender KD, Morrison CD, Rhodes CJ, Schwartz MW. Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. 2005;2:411–420. doi: 10.1016/j.cmet.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 11.Liu M, Maiorano N, Shen L, Pearson K, Tajima D, Zhang DM, Woods SC, Seeley RJ, Davidson WS, Tso P. Expression of biologically active rat apolipoprotein AIV in Escherichia coli. Physiol Behav. 2003;78:149–155. doi: 10.1016/s0031-9384(02)00959-9. [DOI] [PubMed] [Google Scholar]
- 12.Chavez M, Seeley RJ, Woods SC. A comparison between effects of intraventricular insulin and intraperitoneal lithium chloride on three measures sensitive to emetic agents. Behav Neurosci. 1995;109:547–550. [PubMed] [Google Scholar]
- 13.Shen L, Tso P, Woods SC, Sakai RR, Davidson WS, Liu M. Hypothalamic Apolipoprotein A-IV Is Regulated by Leptin. Endocrinology. 2007;148:2681–2689. doi: 10.1210/en.2006-1596. [DOI] [PubMed] [Google Scholar]
- 14.Gelling RW, Morton GJ, Morrison CD, Niswender KD, Myers MG, Jr, Rhodes CJ, Schwartz MW. Insulin action in the brain contributes to glucose lowering during insulin treatment of diabetes. Cell Metab. 2006;3:67–73. doi: 10.1016/j.cmet.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 15.Rahmouni K, Haynes WG, Morgan DA, Mark AL. Intracellular mechanisms involved in leptin regulation of sympathetic outflow. Hypertension. 2003;41:763–767. doi: 10.1161/01.HYP.0000048342.54392.40. [DOI] [PubMed] [Google Scholar]
- 16.Niswender KD, Gallis B, Blevins JE, Corson MA, Schwartz MW, Baskin DG. Immunocytochemical detection of phosphatidylinositol 3-kinase activation by insulin and leptin. J Histochem Cytochem. 2003;51:275–283. doi: 10.1177/002215540305100302. [DOI] [PubMed] [Google Scholar]
- 17.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 18.Woods SC, Seeley RJ, Porte D, Jr, Schwartz MW. Signals that regulate food intake and energy homeostasis. Science. 1998;280:1378–1383. doi: 10.1126/science.280.5368.1378. [DOI] [PubMed] [Google Scholar]
- 19.Kamohara S, Burcelin R, Halaas JL, Friedman JM, Charron MJ. Acute stimulation of glucose metabolism in mice by leptin treatment. Nature. 1997;389:374–377. doi: 10.1038/38717. [DOI] [PubMed] [Google Scholar]
- 20.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo J, Cantley LC. The negative regulation of phosphoinositide 3-kinase signaling by p85 and it’s implication in cancer. Cell Cycle. 2005;4:1309–1312. doi: 10.4161/cc.4.10.2062. [DOI] [PubMed] [Google Scholar]
- 22.Saltiel AR, Pessin JE. Insulin signaling pathways in time and space. Trends Cell Biol. 2002;12:65–71. doi: 10.1016/s0962-8924(01)02207-3. [DOI] [PubMed] [Google Scholar]
- 23.Choudhury AI, Heffron H, Smith MA, Al Qassab H, Xu AW, Selman C, Simmgen M, Clements M, Claret M, Maccoll G, Bedford DC, Hisadome K, Diakonov I, Moosajee V, Bell JD, Speakman JR, Batterham RL, Barsh GS, Ashford ML, Withers DJ. The role of insulin receptor substrate 2 in hypothalamic and beta cell function. J Clin Invest. 2005;115:940–950. doi: 10.1172/JCI24445. [DOI] [PMC free article] [PubMed] [Google Scholar]