

Abstract
AIM: To identify methylation profile and novel tumor marker of extrahepatic cholangiocarcinoma (CCA) with high throughout microarray.
METHODS: Differential methylation profile was compared between normal bile duct epithelial cell lines and CCA cell lines by methyl-DNA immunoprecipitation (MeDIP) microarray. Bisulfite-polymerase chain reaction (BSP) was performed to identify the methylated allels of target genes. Expression of target genes was investigated before and after the treatment with DNA demethylating agent. Expression of candidate genes was also evaluated by immunofluorescence in 30 specimens of CCA tissues and 9 normal bile duct tissues.
RESULTS: Methylation profile of CCA was identified with MeDIP microarray in the respects of different gene functions and signaling pathways. Interestingly, 97 genes with hypermethylated CpG islands in the promoter region were homeobox genes. The top 5 hypermethylated homeobox genes validated by BSP were HOXA2 (94.29%), HOXA5 (95.38%), HOXA11 (91.67%), HOXB4 (90.56%) and HOXD13 (94.38%). Expression of these genes was reactivated with 5’-aza-2’-deoxycytidine. Significant expression differences were found between normal bile duct and extrahepatic CCA tissues (66.67%-100% vs 3.33%-10%).
CONCLUSION: HOXA2, HOXA5, HOXA11, HOXB4 and HOXD13 may work as differential epigenetic biomarkers between malignant and benign biliary tissues.
Keywords: DNA methylation, Epigenetic, Promoter microarray, Cholangiocarcinoma
INTRODUCTION
Cholangiocarcinoma (CCA), including intra-hepatic cholangiocarcinoma (ICC) and extra-hepatic cholangiocarcinoma (ECC), is a cancer arising from the neoplastic transformation of cholangiocytes. Several epidemiological studies show that the incidence and mortality rates of CCA have been increasing worldwide in the past decades. In Europe, approximately 50 000 new cases of primary liver cancer are diagnosed each year. Data from the Cancer Incidence in Five Continents indicates that approximately 20% of those cases are attributed to CCA[1]. Although surgical excision is considered as the most effective therapeutic approach, the 5-year survival rate is still lower than 5%. CCA is characterized by late diagnosis, poor prognosis and lack of response to both chemotherapy and radiation therapy. In addition, the only effective surgical excision is not frequently applicable because of delayed diagnosis[2]. For these reasons, specific and sensitive biomarkers for CCA will facilitate early detection and surgical treatment of CCA.
In recent years, much has been learned about epigenetic changes which were confirmed to be an important mechanism in multiple tumors. Epigenetic is defined as heritable modifications of the genome that are not accompanied by changes in DNA sequence[3]. Aberrant promoter methylation is initiated at about 1% of all CpG islands, and as much as 10% CpG islands become methylated CpG islands during multistep processes of tumorigenesis[4]. Methylation is restricted to CpG dinucleotides which are largely depleted from the genomes excepted at short genomic regions called CpG islands, which commonly represent promoters[5]. Promoter CpG islands hypermethylation can result in gene silencing, which is an alternative mechanism of gene inactivation and contributes to tumor formation, including CCA. One of the most promising molecular markers for CCA is aberrant DNA methylation marker[6].
For decades, scientists have been engaged in methylation profile of CCA. Lots of epigenetically silenced genes have already been identified in CCA, such as p14ARF, RASSFIA, TMS1/ASC, APC, E-cadherin, DAPK, and RUNX3. These silenced genes are involved in important molecular pathways, such as cell cycle regulation, apoptosis, DNA repair and cell adhesion[7-9]. Although the list of epigenetically silenced genes is increasing, it is still the iceberg of the whole methylation profile of CCA, and only a few genes show promise as tumor biomarkers for early diagnosis and prognosis. One reason for this status is that most of the studies focused on the limited tumor suppressor genes that have historically been shown to be inactivated by classical mutations/deletions[10]. Thus, investigation of aberrant DNA methylation of CCA with a high throughput assay will contribute to present the whole and complete methylation pattern of CCA.
In this study, we compared the differential methylation pattern between normal epithelial cell line of bile duct and ECC cell line by genome-wide CpG methylation profiling to discover candidate genes. We also investigated and evaluated the promising candidate genes as differential epigenetic biomarkers between malignant and benign biliary tissues.
MATERIALS AND METHODS
Cell lines and tissue samples
ECC cell line TFK-1 was purchased from the official cell bank (DSMZ, Germany). Normal bile duct epithelial cell line bile duct epithelial cells (BEC) was generously provided by Hiromi Ishibashi (Japan).
TFK-1 cells were cultured in RPMI-1640 medium (GIBCO, USA) with the presence of 10% FBS (GIBCO, USA) at 37°C in a humidified atmosphere containing 5% CO2. According to the previous description[11], BEC cells were resuspended and cultured in BEC medium (1:1 mixture of Ham’s F12 and DMEM, supplemented with 5% fetal calf serum, epithelial growth factor 10 ng/mL, cholera toxin 10 ng/mL, hydrocortisone 0.4 μg/mL, tri-iodo-thyronine 1.3 μg/L, transferring 5 μg/mL, insulin 5 μg/mL, adenine 24.3 μg/mL, and hepatocyte growth factor 10 ng/mL) (all from GIBCO, USA).
Normal and neoplastic specimens were obtained from resected ECC samples at the Tongji Hospital. Nine normal extrahepatic bile duct samples were obtained from discarded tissues from non-invasive CCA patients who underwent bile duct resections. Thirty extrahepatic bile duct samples were obtained from ECC at different pathologic staging. Of these patients, 3 patients were at stage I/II and 27 patients were at stage III/IV. And 11 were younger than 60 years and 28 were ≥ 60 years old (range, 49-74 years, mean age at diagnosis, 63.7 years).
DNA extraction and bisulfate treatment
DNA was isolated as described before. Briefly, total DNA was extracted and purified by DNeasy Blood and Tissue Kit (Qiagen, Germany) according to the manufacturer’s instructions. Concentration of DNA was assessed by spectrophotometry, confirmed by gel electrophoresis and stored at -20°C. In order to convert all unmethylated cytosine to uracil, genomic DNA (2 μg) from cells was subjected to sodium bisulfate using the EpiTect Bisulfite Kit (Qiagen, Germany) according the manufacturer’s instructions.
Methyl-DNA immunoprecipitation and microarray hybridization
Methyl-DNA immunoprecipitation (MeDIP) was performed as described previously[12-14]. Genomic DNA was sonicated to produce random fragments in size of 200-600 bp. Four micrograms of fragmented DNA was used for a standard MeDIP assay as described. After denaturation at 95°C for 10 min, immunoprecipitation was performed using 10 μg monoclonal antibody against 5-methylcytidine in a final volume of 500 μL IP buffer (10 mmol/L sodium phosphate, pH 7.0), 140 mmol/L NaCl, 0.05% Triton X-100) at 4°C for 2 h. Immunoprecipitated complexes were collected with Dynabeads Protein A and M-280 sheep anti-mouse IgG (Roche, Germany) at 4°C for 12 h, washed with 1 × IP buffer for 4 times, treated with Proteinase K at 50°C for 4 h, and purified by phenol-chloroform extraction and isopropanol precipitation.
Immunoprecipitated methylated DNA was labeled with Cy5 fluorophere and the input genomic DNA was labeled with Cy3 fluorophere. Labeled DNA from the enriched and the input pools was combined (1-2 μg) and hybridized to NimbleGen HG18CpG promoter (Roche, Germany), which contained 28 226 all known CpG islands annotated by UCSC (University of California Santa Cruz) and all well-characterized RefSeq promoter regions [from -800 bp to +200 bp transcription start sites (TSSs)] totally covered by 385 000 probes. Arrays were then washed and scanned with NimbleScanTM2.2 (NimbleGen) microarray scanner. After normalization, raw data was input into SignalMap software (v1.9, Roche-NimbleGen) to observe and evaluate the differential methylation peaks between cell lines.
A customized peak-finding algorithm provided by NimbleGen was applied to analyze methylation data from MeDIP-microarray (NimbleScan v2.5; Roche-NimbleGen) as previously described. The algorithm was used to perform the modified Kolmogorov-Smirnov test on several adjacent probes using sliding windows to predict enriched regions across the array. We filtered the differential methylation peaks according to the principles suggested by Pälmke et al[15] : (1) At least one coverage of probes is located inside the peak; and (2) The mean log-ratio (> 2) across the peak has to be positive for at least one of the two samples.
Bisulfite-polymerase chain reaction
The bisulfate modified DNA was amplified with forward and reverse primers for target genes. Table 1 shows the sequences of primers and annealing temperature used in the polymerase chain reaction (PCR) reaction. One microliter PCR products were cloned into pCR-TOPO using the TOPO TA cloning kit (Invitrogen, USA) according to the manufacturer’s instructions. One microliter reaction products were transformed into One Shot TOP 10 chemically competent cells and cultured for 1 h in 250 μL Super Optimal Broth with Catabolite Repression medium at 225 r/min in the incubator shaker. After overnight growth on Luria-Bertani agar plates containing 50 μg/mL kanamycin, plasmid DNA of 10 positive clones of each gene was extracted by EaZy nucleic acid TM Plasmid Mini-Kit II (OMEGA, USA) and sequenced using T7 or M13 forward and reverse primers.
Table 1.
Primer sequences, fragments and annealing temperature used for bisulfite-polymerase chain reaction and reverse transcription-polymerase chain reaction
| Genes | Primer 5'→3' | Annealing temperature (°C) | Size (bp) |
| BSP | |||
| HOXA2 | L: TGTTTTAATAGAATTTATGTGGTTGG | 56 | 190 |
| R: ATAACTACCCTCTACCTCCCCC | |||
| HOXA5 | L: GTTTTGGAGAAATATTATATAAAAGTTATT | 50 | 145 |
| R: CAATTAAAAAAATAAATCCTACCC | |||
| HOXA11 | L: TGAGTATAAGTATGTTGTATGGGGG | 60 | 148 |
| R: TTATAACCACCTCAAAAAAAACAAC | |||
| HOXB4 | L: TAGAGGTGAGGTAGAATAGGAGGGT | 60 | 204 |
| R: ACCCAACACCAAAATTTACATAAAA | |||
| HOXD13 | L: GTGGGTTTAGTTAGGTTTGGGT | 60 | 239 |
| R: TCTAACCCTCTCTCCCTCTATAAAC | |||
| RT-PCR | |||
| HOXA2 | F: GTCACTCTTTGAGCAAGCCC | 59 | 345 |
| R: TAGGCCAGCTCCACAGTTCT | |||
| HOXA5 | F: ACTCCGGCAGGTACGGCTACG | 62 | 259 |
| R: -CCGCTGGAGTTGCTTAGGGAG | |||
| HOXA11 | F: TGCCAAGTTGTACTTACTACGTC | 61 | 181 |
| R: GTTGGAGGAGTAGGAGTATGTA | |||
| HOXB4 | F: GTGCAAAGAGCCCGTCGTCTA CC | 65 | 161 |
| R: CGTGTCAGGTAGCGGTTGTAGTG | |||
| HOXD13 | F: TGCTCCTCTTCTGCCGTTGT | 55 | 467 |
| R: CCTGTGGCTGGTCCTTGGT |
BSP: Bisulfite-polymerase chain reaction; RT-PCR: Reverse transcription-polymerase chain reaction.
Treatment with 5-Aza-2’-deoxycytidine
1 × 105 TFK-1 cells were seeded into each well of a 6-well plate and cultured for 24 h. In the following 3 d, cells were treated continuously with 1 μmol/L (refreshed daily) 5-Aza-2’-deoxycytidine (Sigma, USA). Cells without drug treatment were used as control.
RNA purification and reverse transcription-PCR
Total RNA from cells was extracted using the TRIZOL reagent (Invitrogen, USA). Reverse transcription reaction was performed using MMLV-RT (TOYOBA, Japan). cDNA was amplified by PCR according to the manufacturer’s instructions. The primer sequences for target genes used in the reaction are shown in Table 1.
Immunofluorescence
Forty micrometers-thick sections of tissue samples were cut and mounted on slides. The sections were subsequently rinsed with phosphate-buffered saline (PBS) for 10 min, incubated with 0.5% Triton X-100 for 60 min, and with block solution (5% cattle serum + 0.5% Triton X-100) at room temperature for 1.5 h. After aspiration of block solution, sections were incubated with diluted rabbit polyclonal antibody (Santa Cruz, USA) in block solution overnight at 4°C. The reaction was stopped by three washes with PBS. Then sections were incubated with diluted secondary antibodies (goat anti-rabbit 1:400) conjugated with CY3 NHS Ester for 2 h at 37°C. Slides were washed in PBS and counterstained with 1 μg/mL 4’,6-diamidiono-2-phenylindole for 10 min. The slides were finally mounted with aqueous mounting medium containing anti-fade. Images were obtained by fluorescent microscopy (OLYMPUS IX70).
Statistical analysis
Values were presented as mean ± SD. The mean values of the two subgroups were compared by Student’s t test. For each HOX gene, the median expression was noted as a percentage of immunoreactive samples. Fisher exact test was performed to assess the differences in the median expression values between tissue specimens. All P values were two-sided and P < 0.05 was considered statistically significant. All experiments were performed at least 3 times independently.
RESULTS
MeDIP
We identified 2013 differential hypermethylated CpG islands including 1672 known CpG islands and 531 unknown CpG islands, and 1385 hypomethylated CpG islands including 858 known CpG islands and 527 unknown CpG islands (Figure 1A). Although they spread on different chromosomes, the cluster tendency was obvious that hypermethylated regions preferred to cluster on specific chromosomes, including Chr2, Chr17 and Chr7. The result was different from the former reports that 3p21.3 was methylation hot spot of ECC, but stood in line with other reports that Chr17 and Chr2 were methylation hot spots of myeloid leukemia, bladder cancer and lung cancer[16-18]. Lots of genes have been identified related to tumorigenesis of CCA, such as BCL2, COX2, IGF2, NEUROG1, RAR DAPK1, CDH1[19].
Figure 1.

Identification of differentially hypo- or hypermethylated genes by Methylated DNA Immunoprecipitation and functional analysis of differentially hypermethylated genes by Molecule Annotation System. A: Differentially hypermethylated and hypomethylated genes between TFK-1 and bile duct epithelial cells (BEC) cell lines; B: Functional categories of differentially hypermethylated genes identified by the Molecule Annotation System.
It is well known that aberrant promoter hypermethylation of TSS is an alternative mechanism of gene inactivation and contributes to the carcinogenesis of human neoplasmas, so we investigated the methylation pattern of CpG islands in TSS. We further filtered the experimental data by screening differential hypermethylation peaks overlapping the promoter region of the relevant transcript (-800 to +200 bp). We identified 970 genes with hypermethylated CpG islands overlapping promoter region, which included 317 unknown genes and 653 known genes. We then checked the function of these genes annotated in UCSC and RefSeq gene database, and found that these genes were involved in different cellular process, such as cell-cell adhesion, cell migration, signal transduction and cell repair (Figure 1B).
Interestingly, we also observed a phenomenon that 97 of 970 genes with hypermethylated CpG islands in the promoter region belonged to homeobox gene clusters. These homeobox genes were mainly distributed on HOX, ANTP, PRD, LIM and SINE subclasses. However, aberrant epigenetic changes of HOX genes would disrupt the HOX gene expression and affect various pathways that promote tumorigenesis and metastasis in different cancers, such as breast cancer, lung cancer, ovarian cancer and prostate cancer[20-22], and research of HOX genes was limited in choangiocarcinoma. To further evaluate the influence of DNA methylation changes on the expression of HOX genes, we picked 11 homeobox genes showing hypermethylated status for further analysis (Table 2).
Table 2.
Methylated HOX genes identified by methyl-DNA immunoprecipitation microarray
| No. | Genes | Description | CpG location | Strand | Position of gene |
| 1 | HOXA2 | HomeoboxA2 | chr7:27109706-27110004 | - | Promoter |
| 2 | HOXA5 | HomeoboxA5 | chr7:27149138-27152087 | - | Promoter |
| 3 | HOXA11 | HomeoboxA11 | chr7:27191575-27192154 | - | Promoter |
| 4 | HOXB2 | HomeoboxB2 | chr17:43982786-43983443 | - | Promoter |
| 5 | HOXB4 | HomeoboxB4 | chr17:44010214-44010603 | - | Promoter |
| 6 | HOXB5 | HomeoboxB5 | chr17:44025521-44026457 | - | Promoter |
| 7 | HOXC10 | HomeoboxC10 | chr12:52664963-52666369 | + | Promoter |
| 8 | HOXD1 | HomeoboxD1 | chr2:176761203-176762596 | + | Promoter |
| 9 | HOXD9 | HomeoboxD9 | chr2:176694670-176696537 | + | Promoter |
| 10 | HOXD12 | HomeoboxD12 | chr2:176672308-176673755 | + | Promoter |
| 11 | HOXD13 | HomeoboxD13 | chr2:176665300-176666525 | + | Promoter |
Bisulfite-PCR
To further verify the hypermethylation of HOX genes, we performed bisulfite-PCR (BSP) to validate the results of MeDIP. We observed that HOXA2, HOXA5, HOXA11, HOAB4 and HOXD13 were the top 5 methylated genes in TFK-1 cells. As shown in Figure 2B, the frequency of promoter hypermethylation was 94.29% for HOXA2, 95.38% for HOXA5, 91.67% for HOXA11, 90.56% for HOXB4 and 94.38% for HOXD13 in TFK-1 cells. In contrast, promoter profiles of HOXA2, HOXA5, HOXA11, HOXB4 and HOXD13 were unmethylated in BEC. The graphical MeDIP-microarray data of the top 5 HOX genes is presented in Figure 2A.
Figure 2.
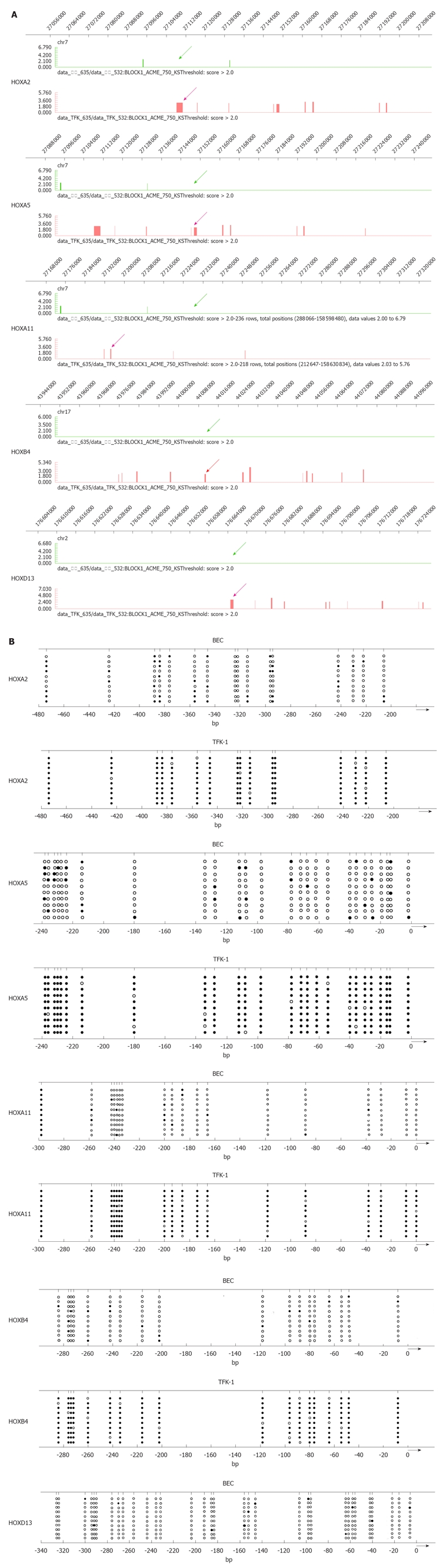
DNA methylation analyses of CpG islands of hypermethylated HOX genes. A: Graphical representation of Methylated DNA Immunoprecipitation microarray (Signalmap software, NimbleGen). The panels show the DNA methylation profile at homeobox cluster genes in normal epithelial cell of bile duct cell line bile duct epithelial cells (BEC) and cholangiocarcinoma cell line TFK-1. The methylated CpG islands are indicated by bar and arrow in TFK-1 (red) and BEC (green). Chromosomal location is indicated at the top of the diagram; B: Bisulfate-sequencing of HOXA2, HOXA5, HOXA11, HOXB4 and HOXD13. Each vertical line represents a single CpG site on the top. The regions were analyzed by bisulfate-sequencing. The transcription start site and location of exon 1 are shown by thick bars on the bottom. Each row represents an individual cloned allele. Circles represent CpG sites and their spacing accurately reflects the CpG density of the region. Black circles, methylated CpG site; white circles, unmethylated CpG site. Dense methylation at the promoters was found in TFK-1. In contrast, promoter hypomethylation found in BEC among all the genes.
Treatment with 5-Aza-2’-deoxycytidine
Methylation is a reversible process that DNA methyltransferase inhibitors can restore the original expression and function of epigenetically silenced genes in vitro and in vivo. To determine if the lack of transcription of these genes may be influenced by aberrant methylation, we treated the TFK-1 cells with demethylating agent 5-Aza-2’-deoxycytidine. Reverse transcription-PCR (RT-PCR) was carried out on the top 5 candidate HOX genes. The results showed that the expression of HOXA2, HOXA5, HOXA11, HOXB4 and HOXD13 was rare in TFK-1, whereas it was restored after the treatment with 5-Aza-2’-deoxycytidine (Figure 3).
Figure 3.

Reverse transcription-polymerase chain reaction analysis for expression profile of HOXA2, HOXA5, HOXA11, HOXB4 and HOXD13 in TFK-1 cells before and after the treatment with demethylating agent 5-aza-2-deoxycytidine (5-aza-dC). Loss of expression of genes was observed before the treatment in TFK-1. Re-expression of genes was observed upon the treatment with 1 μmol/L demethylating agent 5-aza-2-deoxycytidine (5-aza-dC). TFK-1 cells were subjected to the 1 μmol/L 5-aza-dC treatment for 3 d before RNA prepared for reverse transcription-polymerase chain reaction assessment. β-actin 700 bp.
Expression of candidate genes in fresh tumors
Genes can acquire extra DNA methylation changes in the cell culture process and methylation profile of cell lines may not accurately reflect the methylation profile of tumors in vivo[23]. Besides, only one ECC cell line TFK -1 was used in this research. To compensate for this limitation, we assayed the expression of target HOX genes in primary ECC and normal bile duct samples using immunofluorescence.
Representative immunofluorescemce photo images of the top 5 HOX genes which have been validated by BSP are shown in Figure 4. Strong positive staining for HOX genes was found in normal bile duct samples while tumor samples showed negative staining for HOX gene expression. The frequency of positive staining of HOXA2, HOXA5, HOXA11, HOXB4 and HOXD13 was 77.78% (7/9), 100% (9/9), 100% (9/9), 66.67% (6/9) and 88.89% (8/9), respectively in normal bile duct samples, while it was only 10% (3/30), 3.33% (1/30), 3.33% (1/30), 6.67% (2/30) and 6.67% (2/30), respectively in ECC samples (Table 3).
Figure 4.

Expression of HOXA2, HOXA5, HOXA11, HOAB4, HOXD13 in extra-hepatic cholangiocarcinoma by immunofluorescence. Green channel is nuclear staining by 4’,6-diamidiono-2-phenylindole (DAPI) (A), blue channel is the target gene (B), and overlay of DAPI and target genes (C). Arrows indicate the positive target genes in specimens.
Table 3.
HOXA2, HOXA5, HOXA11, HOXB4, and HOXD13 methylation status of 30 cholangiocarcinoma patients and 9 normal controls n (%)
| Genes | Age (n = 39) | Normal (n = 9) | Patients (n = 30) | P value1 | ||||
| < 60 yr (n = 11) | > 60 yr (n = 28) | P value | Tumor stageI/II (n = 3) | Tumor stage III/IV (n = 27) | P value | |||
| HOXA2 | 4 (7.69) | 6 (15.39) | 0.424 | 7 (77.78) | 1 (33.33) | 2 (7.41) | 0.511 | < 0.001 |
| HOXA5 | 3 (7.69) | 7 (17.95) | 1.0 | 9 (100) | 1 (33.33) | 0 (0) | 0.206 | < 0.001 |
| HOXA11 | 2 (5.13) | 7 (17.95) | 1.0 | 9 (100) | 1 (33.33) | 0 (0) | 0.206 | < 0.001 |
| HOXB 4 | 3 (7.69) | 5 (12.82) | 0.663 | 6 (66.67) | 2 (66.67) | 0 (0) | 0.007 | 0.001 |
| HOXD13 | 4 (7.69) | 6 (15.39) | 0.424 | 8 (88.89) | 1 (33.33) | 1 (3.70) | 0.193 | < 0.001 |
Fisher exact test was performed to identify differences in median expression values within the tissue samples.
Fisher test between normal controls and cholangiocarcinoma patients.
Aging is one of the most important risk factors in the development of neoplasia and methylation of genes increases with age[24]. There was no significant difference between the lower age group and advanced age group (age > 60 years) in our research. However, there was a tendency that HOX genes mostly expressed in early pathological stages but not in advanced pathological stages, the difference being not statistically significant.
DISCUSSION
The objective of this study was to identify novel genes inactivated by promoter methylation in ECC. We used a microarray-based strategy as an initial screening approach to identify the differential methylation pattern of ECC.
We then validated methylation status of candidate genes in cells and identified the expression of candidate genes in ECC samples. Kim et al[7] reported that there was a statistically significant difference between gene methylation frequencies of ECC and ICC. Yang et al[8] demonstrated that ICC and ECC tissues exhibited overlapping but distinct methylation profiles. Some hypermethylated genes that have been confirmed in these literatures were also identified in our study, including RASSF1A, P15INK4b, P16INK4a, NEUROG1, CDH1, IGF2, GSTP1, APC and RUNX3. However, previous studies have explored methylation profile of ECC, and only CpG islands that have been demonstrated to be hypermethylated in other human cancer tissue types were investigated in ECC later. Our present study applied microarray to investigate 28 226 CpG islands and demonstrated abundant differentially hypermethylated genes that were not previously known in ECC.
We found that 97 genes with hypermethylated promoter CpG islands belonged to homeobox gene clusters. Hox genes are one of the master regulators of morphogenesis and cell differentiation embryogenesis of animals[25]. Numerous examples of aberrant Hox gene expression have been found in cancer. Abate-Shen[26] proposed three mechanisms, including temporospatial deregulation, gene dominance and epigenetic deregulation, to classify aberrant changes of Hox genes[26]. The third mechanism, epigenetic deregulation, can modify or silence the expression of Hox genes in tumor tissues. However, lots of hypermethylated Hox genes have been identified at different stages of primary squamous cell carcinomas, such as lung carcinoma, ovarian carcinoma, breast carcinoma and cervical carcinoma[27,28], and little is known about methylation status of Hox gene in CCA.
We picked 11 hypermethylated homeobox genes according to the filter principles and mapped the biological relationship between the 11 HOX genes and their related genes to analyze in-depth their roles in tumorigenesis of ECC (Figure 5). EED-EZH2 and PRC2 complex had an intrinsic histone methyltransferase activity to H3K27 and silenced some HOX genes[29]. Besides, Hphf1 could associate with the core components of EED-EZH2 complex to modulate its enzymatic activity and HOX gene expression[30]. MeDIP microarray indicated that EZH2 was also methylated in TFK-1 cells and we therefore, speculated that the expression of HOX gene was down-regulated by epigenetically silenced core components of EED-EZH2 complex. However, more studies remain to be conducted to find out their relationship.
Figure 5.

Network analysis of known biological relationships between HOX genes and their related genes. Network analysis was performed and produced by Molecule Annotation System. Yellow icons indicate methylated Hox genes and colorless icons indicate methylated non-HOX genes, which have been demonstrated to be related to Hox genes in other studies. Green lines indicate high correlation and red lines indicate low correlation. All present hypermethylated status in the microarray results. Number between icons indicates the relationship mentioned in the reports.
BSP confirmed that HOXA2, HOXA5, HOXA11, HOXB4 and HOXD13 methylated by MeDIP microarray were densely hypermethylated in TFK-1 cells, but fully unmethylated in BEC cells. The 5-Aza-dC method is very useful for identifying the genes reactivated by treatment with DNA demethylating agent. One can also prioritize the reactivated genes by gene expression analysis to identify the genes that are normally expressed but undergo silencing in cancers[31]. In our research, TFK-1 cells showed loss of expression of HOXA2, HOXA5, HOXA11, HOXB4, and HOXD13. After the treatment with demethylating agent 5-aza-2-deoxycytidine, the expression was restored in TFK-1 cells. The association between DNA methylation and expression agreed with the theory that epigenetic deregulation would silence the expression of Hox genes in tumor tissues proposed by Abate-Shen[26]. Drugs that modulate DNA methylation are in clinical trials and have been shown to affect gene expression in vivo. Although there is considerable literature on the possible antitumor mode of action of DNA methylation, such as 5-aza-2-deoxycytidine drugs, their exact mechanism in CCA remains unclear and needs further investigations.
We have shown that the CpG islands hypermethylation of HOX genes is not a unique feature of TFK-1 cells, but common among ECC samples. However, the expression of HOX genes was detected more frequently in low-stage ECCs than in high-stage ECCs, the difference being not statistically significant. Our results were in line with the research published by Kim et al[7] that no association was found between CpG island hypermethylation and histologic classification in terms of hypermethylation of methylated CpG island locus or the total number of methylated CpG island loci. Jacinto et al[32] evaluated the aberrant hypermethylation changes in the benign colorectal adenomas, a precursor to invasive colorectal tumors, and suggested that hypermethylation of genes was an early event in the pathway to a full-blown tumor. To date, there have been few studies on the methylation patterns of precancerous lesions of CCA. We did not assess the association between DNA methylation changes and precancerous lesions of CCA in the present study.
As important members of the homeobox superfamily, HOX genes encode transcription factors and contribute to oncogenesis by allowing activation of anti-apoptotic pathways. HOXA5 has been identified to directly regulate the expression of p53 and hMLH1 genes as a transcription factor in breast cancer cells, and contribute to safeguarding the cells against malignant transformation. Moreover, its expression can induce the apoptosis in breast cancer through p53-independent apoptotic pathway mediated by caspases2 and 8[33]. Our results were in agreement with the results by others authors that the frequency of hypermethylated HOXA5 was high in ECC. However, additional studies are still needed before exact mechanism can be concluded.
DNA methylation of polycomb group target genes is an early step in tumorigenesis and can potentially be assayed to predict cancer risk. Fiegl et al[34] pointed out that DNA methylation of HOXA11 gene, a member of PGGT genes, was frequently present in ovarian cancer and HOXA11 methylation status was a prognostic marker. In our research, expression of HOXA11 was rare in TFK-1 cell lines and ECC. Silenced HOX genes in ECC, which might be induced by hypermethylation, might work as differential epigenetic biomarkers between malignant and benign biliary tissues.
HOXD13, binding origins primarily during G1 phase of the cell cycle, promotes the assembly of pre-replication complex proteins at replication origins and stimulates DNA synthesis in vivo in a transient DNA replication assay[35,36]. According to the literature, the expression of HOXD13 always increased in malignant tumors, such as brain, lung, and prostate carcinoma. However, our result that HOXD13 was methylated in TFK-1 cell lines with decreased expression, was opposite to the results from other studies. Besides, the expression frequency of HOXD13 was only 6.67% in ECC. Our results agreed with a recent work which demonstrated that HOXD13 expression decreased in pancreas and stomach tumor subtypes[37].
HOXA2 is regulated by growth hormone and growth factors. Kazuhiko Maeda has demonstrated that the expression frequency of HOXA2 in melanoma with distant metastasis was higher than that in melanoma without metastasis[38]. We gained an opposite result in ECC. The expression of HOXA2 was only 10% in ECC.
We speculated that tissue and cancer specificity might be responsible for the differential expression of HOXA2 and HOXD13 in ECC compared with other cancers. Several recent genome studies show that DNA methylation profiles in mammals are tissue specific[39,40]. Doi et al[41] demonstrated that differentially methylated regions in the reprogrammed cells completely distinguished brain from liver and spleen tissues and largely distinguished colon cancer from normal colon tissues. The studies also demonstrated that methylation pattern of HOX genes also differed greatly among various kinds of cancers, such as hypermethylated HOXA5, HOXA10 and HOXB7 in breast, hypermethylated HOXB13 and HOXC8 in prostate, and hypermethylated HOXD1, HOXD8, HOXC6 and HOXC11 in neuroblastoma. However, our understanding of tissue-specific DNA methylation of HOX genes in cancer is still limited and many questions remain to be answered.
HOXB4 is always considered as a catalyst for leukemia development[42]. To our knowledge, this is the first study to identify hypermethylated HOXB4 in the solid tumor. The expression frequency of HOXB4 was 6.67% in ECC samples.
Although MeDIP assay demonstrated that HOXC1, HOXD9, HOXB2, HOXB5 and HOXD1 were methylated in TFK-1 cells, BSP assay suggested that the hypermethylation was not significant. The artificial methylation induced during the culture might be responsible for the positive results of microarray. Besides, the discrepancy might be attributed to the different methods used in the methylation assay. BSP is a DNA sequencing approach, which has a higher sensitivity for detecting allelic hypermethylation in target sequences than the MeDIP.
In conclusion, we used the high throughput MeDIP microarray to investigate methylation profile of ECC. We identified 2013 differentially hypermethylated CpG islands that were involved in various cellular processes, such as cell-cell adhesion, cell migration, signal transduction and cell repair. The results provided a foundation for further studies about the mechanism of ECC. HOXA2, HOXA5, HOXA11, HOXB4 and HOXD13 showed a high frequency of hypermethylation in ECC cells and loss of expression in ECC samples, which raised the possibility that HOX genes might work as differential epigenetic biomarkers between malignant and benign biliary tissues.
COMMENTS
Background
Extrahepatic cholangiocarcinoma (ECC) is a malignant cancer with ineffective treatment and poor prognosis. It has been confirmed that aberrant epigenetic alterations contribute to cancer formation in multiple of cancers. DNA hypermethylation is the most common epigenetic abnormality in cancer.
Research frontiers
Aberrant promoter hypermethylation is an important mechanism of gene inactivation and contributes to the carcinogenesis of ECC. However, many epigenetically silenced genes have already been identified in cholangiocarcinoma, methylation file of ECC is still unclear. In this study, the authors compared differential methylation profile between normal bile duct epithelial cell and ECC cell lines by genome-wide CpG methylation profiling to discover candidate methylated genes.
Innovations and breakthroughs
The advent of microarray technologies that enables the analysis of a large number of DNA/RNA fragments in a high throughput way has opened new opportunities for epigenetic studies. This is the first study to utilize the high throughput Methylated DNA Immunoprecipitation (MeDIP) microarray to investigate methylation file of ECC. The authors identified 2013 differential hypermethylated CpG islands that were involved in various cellular processes. Furthermore, the authors validated that HOXA2, HOXA5, HOXA11, HOXB4 and HOXD13 showed high frequency of hypermethylation and loss expression in ECC.
Applications
By understanding the differential methylation profile of HOXA2, HOXA5, HOXA11, HOXB4 and HOXD13 in ECC, this study may raises the possibility that HOX genes may work as differential epigenetic biomarkers between malignant and benign biliary tissues for diagnosis and treatment.
Terminology
In mammals, 39 human HOX genes are located in four clusters (A-D) on different chromosomes at 7p15, 17q21.2, 12q13, and 2q31 respectively. HOX genes are evolutionarily high conserved. HOX proteins can function as monomers or homodimers to directly drive the transcription of downstream targets, and sequester other proteins to enhance or repress gene expression. HOX genes are integral to normal temporospatial limb and organ development along the anterior-posterior axis.
Peer review
The manuscript of Shu et al entitled “Identification of methylation profile and novel tumor marker of cholangiocarcinoma with MeDIP microarray” adds new evidence to the potential role of DNA methylation in ccholangiocarcinoma development and progression.
Footnotes
Supported by The grant for “Development of Novel Nano-Drug Delivery System Loaded with Traditional Chinese Anticancer Medicine for the Targeted Therapy of Malignant Tumors” issued by the Chinese Ministry of Science and Technology, Grant No. 2010DFA31870
Peer reviewers: Carlos J Pirola, PhD, FAHA, Medical Research Institute A Lanari, Combatientes de Malvinas 3150, Buenos Aires-1427, Argentina; Hiroki Sasaki, PhD, Genetics Division, National Cancer Center Research Institute, 1-1, Tsukiji 5-chome, Chuo-ku, Tokyo 104-0045, Japan; Shu Zheng, Professor, Scientific Director of Cancer Institute, Zhejiang University, Secondary Affiliated Hospital, Zhejiang University, 88# Jiefang Road, Hangzhou 310009, Zhejiang Province, China
S- Editor Tian L L- Editor Ma JY E- Editor Zheng XM
References
- 1.Mosconi S, Beretta GD, Labianca R, Zampino MG, Gatta G, Heinemann V. Cholangiocarcinoma. Crit Rev Oncol Hematol. 2009;69:259–270. doi: 10.1016/j.critrevonc.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Gatto M, Bragazzi MC, Semeraro R, Napoli C, Gentile R, Torrice A, Gaudio E, Alvaro D. Cholangiocarcinoma: update and future perspectives. Dig Liver Dis. 2010;42:253–260. doi: 10.1016/j.dld.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 3.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Costello JF, Frühwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomäki P, Lang JC, et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet. 2000;24:132–138. doi: 10.1038/72785. [DOI] [PubMed] [Google Scholar]
- 5.Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, Schübeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 6.Lee S, Kim WH, Jung HY, Yang MH, Kang GH. Aberrant CpG island methylation of multiple genes in intrahepatic cholangiocarcinoma. Am J Pathol. 2002;161:1015–1022. doi: 10.1016/S0002-9440(10)64262-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim BH, Cho NY, Choi M, Lee S, Jang JJ, Kang GH. Methylation profiles of multiple CpG island loci in extrahepatic cholangiocarcinoma versus those of intrahepatic cholangiocarcinomas. Arch Pathol Lab Med. 2007;131:923–930. doi: 10.5858/2007-131-923-MPOMCI. [DOI] [PubMed] [Google Scholar]
- 8.Yang B, House MG, Guo M, Herman JG, Clark DP. Promoter methylation profiles of tumor suppressor genes in intrahepatic and extrahepatic cholangiocarcinoma. Mod Pathol. 2005;18:412–420. doi: 10.1038/modpathol.3800287. [DOI] [PubMed] [Google Scholar]
- 9.Isomoto H. Epigenetic alterations associated with cholangiocarcinoma (review) Oncol Rep. 2009;22:227–232. [PubMed] [Google Scholar]
- 10.Corn PG. Genome-wide profiling of methylated promoters in pancreatic adenocarcinoma: defining the pancreatic cancer [corrected] epigenome. Cancer Biol Ther. 2008;7:1157–1159. doi: 10.4161/cbt.7.7.6616. [DOI] [PubMed] [Google Scholar]
- 11.Kamihira T, Shimoda S, Nakamura M, Yokoyama T, Takii Y, Kawano A, Handa M, Ishibashi H, Gershwin ME, Harada M. Biliary epithelial cells regulate autoreactive T cells: implications for biliary-specific diseases. Hepatology. 2005;41:151–159. doi: 10.1002/hep.20494. [DOI] [PubMed] [Google Scholar]
- 12.Jacinto FV, Ballestar E, Esteller M. Methyl-DNA immunoprecipitation (MeDIP): hunting down the DNA methylome. Biotechniques. 2008;44:35, 37, 39 passim. doi: 10.2144/000112708. [DOI] [PubMed] [Google Scholar]
- 13.Weng YI, Huang TH, Yan PS. Methylated DNA immunoprecipitation and microarray-based analysis: detection of DNA methylation in breast cancer cell lines. Methods Mol Biol. 2009;590:165–176. doi: 10.1007/978-1-60327-378-7_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruike Y, Imanaka Y, Sato F, Shimizu K, Tsujimoto G. Genome-wide analysis of aberrant methylation in human breast cancer cells using methyl-DNA immunoprecipitation combined with high-throughput sequencing. BMC Genomics. 2010;11:137. doi: 10.1186/1471-2164-11-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pälmke N, Santacruz D, Walter J. Comprehensive analysis of DNA-methylation in mammalian tissues using MeDIP-chip. Methods. 2011;53:175–184. doi: 10.1016/j.ymeth.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 16.Bullinger L, Ehrich M, Döhner K, Schlenk RF, Döhner H, Nelson MR, van den Boom D. Quantitative DNA methylation predicts survival in adult acute myeloid leukemia. Blood. 2010;115:636–642. doi: 10.1182/blood-2009-03-211003. [DOI] [PubMed] [Google Scholar]
- 17.Yasuda H, Soejima K, Nakayama S, Kawada I, Nakachi I, Yoda S, Satomi R, Ikemura S, Terai H, Sato T, et al. Bronchoscopic microsampling is a useful complementary diagnostic tool for detecting lung cancer. Lung Cancer. 2011;72:32–38. doi: 10.1016/j.lungcan.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 18.Marsit CJ, Houseman EA, Christensen BC, Gagne L, Wrensch MR, Nelson HH, Wiemels J, Zheng S, Wiencke JK, Andrew AS, et al. Identification of methylated genes associated with aggressive bladder cancer. PLoS One. 2010;5:e12334. doi: 10.1371/journal.pone.0012334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tischoff I, Wittekind C, Tannapfel A. Role of epigenetic alterations in cholangiocarcinoma. J Hepatobiliary Pancreat Surg. 2006;13:274–279. doi: 10.1007/s00534-005-1055-3. [DOI] [PubMed] [Google Scholar]
- 20.Hartmann O, Spyratos F, Harbeck N, Dietrich D, Fassbender A, Schmitt M, Eppenberger-Castori S, Vuaroqueaux V, Lerebours F, Welzel K, et al. DNA methylation markers predict outcome in node-positive, estrogen receptor-positive breast cancer with adjuvant anthracycline-based chemotherapy. Clin Cancer Res. 2009;15:315–323. doi: 10.1158/1078-0432.CCR-08-0166. [DOI] [PubMed] [Google Scholar]
- 21.Plowright L, Harrington KJ, Pandha HS, Morgan R. HOX transcription factors are potential therapeutic targets in non-small-cell lung cancer (targeting HOX genes in lung cancer) Br J Cancer. 2009;100:470–475. doi: 10.1038/sj.bjc.6604857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Widschwendter M, Apostolidou S, Jones AA, Fourkala EO, Arora R, Pearce CL, Frasco MA, Ayhan A, Zikan M, Cibula D, et al. HOXA methylation in normal endometrium from premenopausal women is associated with the presence of ovarian cancer: a proof of principle study. Int J Cancer. 2009;125:2214–2218. doi: 10.1002/ijc.24599. [DOI] [PubMed] [Google Scholar]
- 23.Shames DS, Girard L, Gao B, Sato M, Lewis CM, Shivapurkar N, Jiang A, Perou CM, Kim YH, Pollack JR, et al. A genome-wide screen for promoter methylation in lung cancer identifies novel methylation markers for multiple malignancies. PLoS Med. 2006;3:e486. doi: 10.1371/journal.pmed.0030486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calvanese V, Lara E, Kahn A, Fraga MF. The role of epigenetics in aging and age-related diseases. Ageing Res Rev. 2009;8:268–276. doi: 10.1016/j.arr.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 25.Grier DG, Thompson A, Kwasniewska A, McGonigle GJ, Halliday HL, Lappin TR. The pathophysiology of HOX genes and their role in cancer. J Pathol. 2005;205:154–171. doi: 10.1002/path.1710. [DOI] [PubMed] [Google Scholar]
- 26.Abate-Shen C. Deregulated homeobox gene expression in cancer: cause or consequence? Nat Rev Cancer. 2002;2:777–785. doi: 10.1038/nrc907. [DOI] [PubMed] [Google Scholar]
- 27.Soshnikova N, Duboule D. Epigenetic regulation of vertebrate Hox genes: a dynamic equilibrium. Epigenetics. 2009;4:537–540. doi: 10.4161/epi.4.8.10132. [DOI] [PubMed] [Google Scholar]
- 28.Tommasi S, Karm DL, Wu X, Yen Y, Pfeifer GP. Methylation of homeobox genes is a frequent and early epigenetic event in breast cancer. Breast Cancer Res. 2009;11:R14. doi: 10.1186/bcr2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao R, Tsukada Y, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell. 2005;20:845–854. doi: 10.1016/j.molcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Cao R, Wang H, He J, Erdjument-Bromage H, Tempst P, Zhang Y. Role of hPHF1 in H3K27 methylation and Hox gene silencing. Mol Cell Biol. 2008;28:1862–1872. doi: 10.1128/MCB.01589-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Omura N, Li CP, Li A, Hong SM, Walter K, Jimeno A, Hidalgo M, Goggins M. Genome-wide profiling of methylated promoters in pancreatic adenocarcinoma. Cancer Biol Ther. 2008;7:1146–1156. doi: 10.4161/cbt.7.7.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacinto FV, Ballestar E, Ropero S, Esteller M. Discovery of epigenetically silenced genes by methylated DNA immunoprecipitation in colon cancer cells. Cancer Res. 2007;67:11481–11486. doi: 10.1158/0008-5472.CAN-07-2687. [DOI] [PubMed] [Google Scholar]
- 33.Kim DS, Kim MJ, Lee JY, Lee SM, Choi JY, Yoon GS, Na YK, Hong HS, Kim SG, Choi JE, et al. Epigenetic inactivation of Homeobox A5 gene in nonsmall cell lung cancer and its relationship with clinicopathological features. Mol Carcinog. 2009;48:1109–1115. doi: 10.1002/mc.20561. [DOI] [PubMed] [Google Scholar]
- 34.Fiegl H, Windbichler G, Mueller-Holzner E, Goebel G, Lechner M, Jacobs IJ, Widschwendter M. HOXA11 DNA methylation--a novel prognostic biomarker in ovarian cancer. Int J Cancer. 2008;123:725–729. doi: 10.1002/ijc.23563. [DOI] [PubMed] [Google Scholar]
- 35.Salsi V, Ferrari S, Ferraresi R, Cossarizza A, Grande A, Zappavigna V. HOXD13 binds DNA replication origins to promote origin licensing and is inhibited by geminin. Mol Cell Biol. 2009;29:5775–5788. doi: 10.1128/MCB.00509-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hateboer G, Wobst A, Petersen BO, Le Cam L, Vigo E, Sardet C, Helin K. Cell cycle-regulated expression of mammalian CDC6 is dependent on E2F. Mol Cell Biol. 1998;18:6679–6697. doi: 10.1128/mcb.18.11.6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cantile M, Franco R, Tschan A, Baumhoer D, Zlobec I, Schiavo G, Forte I, Bihl M, Liguori G, Botti G, et al. HOX D13 expression across 79 tumor tissue types. Int J Cancer. 2009;125:1532–1541. doi: 10.1002/ijc.24438. [DOI] [PubMed] [Google Scholar]
- 38.Maeda K, Hamada J, Takahashi Y, Tada M, Yamamoto Y, Sugihara T, Moriuchi T. Altered expressions of HOX genes in human cutaneous malignant melanoma. Int J Cancer. 2005;114:436–441. doi: 10.1002/ijc.20706. [DOI] [PubMed] [Google Scholar]
- 39.Rakyan VK, Hildmann T, Novik KL, Lewin J, Tost J, Cox AV, Andrews TD, Howe KL, Otto T, Olek A, et al. DNA methylation profiling of the human major histocompatibility complex: a pilot study for the human epigenome project. PLoS Biol. 2004;2:e405. doi: 10.1371/journal.pbio.0020405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gordon-Keylock SA, Jackson M, Huang C, Samuel K, Axton RA, Oostendorp RA, Taylor H, Wilson J, Forrester LM. Induction of hematopoietic differentiation of mouse embryonic stem cells by an AGM-derived stromal cell line is not further enhanced by overexpression of HOXB4. Stem Cells Dev. 2010;19:1687–1698. doi: 10.1089/scd.2009.0467. [DOI] [PMC free article] [PubMed] [Google Scholar]