

Abstract
More than 2% of the adult U.S. population harbors a pancreatic cyst. These often pose a difficult management problem because conventional criteria cannot always distinguish cysts with malignant potential from those that are innocuous. One of the most common cystic neoplasms of the pancreas, and a bona fide precursor to invasive adenocarcinoma, is called intraductal papillary mucinous neoplasm (IPMN). To help reveal the pathogenesis of these lesions, we purified the DNA from IPMN cyst fluids from 19 patients and searched for mutations in 169 genes commonly altered in human cancers. In addition to the expected KRAS mutations, we identified recurrent mutations at codon 201 of GNAS. A larger number (113) of additional IPMNs were then analyzed to determine the prevalence of KRAS and GNAS mutations. In total, we found that GNAS mutations were present in 66% of IPMNs and that either KRAS or GNAS mutations could be identified in 96%. In eight cases, we could investigate invasive adenocarcinomas that developed in association with IPMNs containing GNAS mutations. In seven of these eight cases, the GNAS mutations present in the IPMNs were also found in the invasive lesion. GNAS mutations were not found in other types of cystic neoplasms of the pancreas or in invasive adenocarcinomas not associated with IPMNs. In addition to defining a new pathway for pancreatic neoplasia, these data suggest that GNAS mutations can inform the diagnosis and management of patients with cystic pancreatic lesions.
INTRODUCTION
Pancreatic cysts occur in more than 20% of patients evaluated at autopsy and in 2.4 to 13% of patients studied by abdominal imaging (computed tomography scan or magnetic resonance imaging) for reasons unrelated to pancreatic pathology (1–4). In view of the ever-increasing use of abdominal imaging and continuing improvements in the resolution of these technologies, the diagnosis of these lesions, often as incidental findings, can be expected to rise. They pose a challenging management problem because some cysts represent bona fide precursor lesions to invasive pancreatic ductal adenocarcinomas (PDAs), whereas others have minimal neoplastic potential (5). Yet, distinguishing cystic neoplasms from one another on the basis of clinical, imaging, and standard laboratory criteria is often not possible. Because of the diagnostic dilemmas posed by these lesions and the potentially lethal consequences of an incorrect clinical diagnosis, some patients with harmless cysts are overtreated by surgical resection of the cyst and part of the pancreas (6). These pancreatectomies are major surgical procedures associated with significant morbidity and even rare mortality, prompting most investigators to adopt a watchful waiting approach with frequent imaging, sometimes using endoscopic ultrasound (EUS) surveillance (7). New diagnostic modalities that will improve the management of the numerous patients with pancreatic cysts are therefore urgently needed.
There are three main types of pancreatic cystic neoplasms (8). Serous cystadenomas (SCAs) account for ~10% of surgically resected cystic lesions of the pancreas (8, 9). These tumors are thought to be totally benign with essentially no likelihood of becoming invasive (that is, of becoming PDA). Intraductal papillary mucinous neoplasms (IPMNs), representing ~20% of surgically resected cystic lesions of the pancreas (8, 9), occur in the main pancreatic duct or branch ducts. Those in the main ducts are most aggressive: ~45% are found to have progressed to invasive carcinoma at the time of surgical resection (10). Those in branch ducts are found to have progressed to invasive carcinoma in ~20% of patients (10). IPMNs can also be classified into three histologic subtypes—intestinal, pancreatobiliary, and gastric—on the basis of the resemblance of their epithelial cells to those of the corresponding normal tissues. The natural history of IPMNs, regardless of their histology or location within the pancreas, is not known with certainty because the lesions are often excised when they reach a certain size (3-cm diameter) or become symptomatic. However, based on the age of patients with resected lesions, there appears to be a 5-year lag time from noninvasive IPMN (average age, 63.2 years) to invasive IPMN (average age, 68.1 years) (11). The third type of pancreatic cystic neoplasms, representing 5 to 10% of surgically resected pancreatic cysts, is called mucinous cystic neoplasms (MCNs). These progress to invasive carcinomas less often than IPMNs: Only ~10% of MCNs contain an invasive component when surgically excised (12). Moreover, MCNs are less diagnostically challenging because most of these lesions occur in the body or tail of the pancreas in relatively young women. Women with MCNs are on average more than 20 years younger than patients with either SCAs or IPMNs (12). The remainder of surgically resected cystic lesions of the pancreas other than these three types is a heterogeneous group of retention cysts, congenital cysts, and other rarer lesions.
PDAs can be divided into two types: those that develop in the absence of cystic lesions and those that develop from a cystic lesion. Most PDAs that develop from cystic lesions do so from IPMNs, as SCAs do not give rise to PDAs and MCNs are less common than IPMNs. The prognoses of patients with PDAs that develop from IPMNs are different from those in patients with PDAs that develop in the apparent absence of IPMNs (11, 13). The survival of patients with surgically resected PDAs associated with IPMNs is as high as 45% at 5 years (11, 13). In contrast, survival is rare in other PDA patients (14). Although some of the difference in survival is due to differences in stage at diagnosis, investigators have suggested that “carcinoma arising in IPMNs is a different disease” from the more common type of PDA that develops without any associated cystic precursor (15).
What genetic features distinguish the PDAs that develop from IPMNs from those that develop in their absence? As described below, we have discovered that many PDAs developing in IPMNs contain GNAS mutations, whereas other PDAs do not. Moreover, these GNAS mutations generally occur early in IPMN development and can be used to distinguish IPMNs from other types of pancreatic cysts. Accordingly, GNAS mutations in cyst fluids provide a highly specific biomarker that has the capacity to improve the diagnosis and management of patients with cystic lesions of the pancreas.
RESULTS
Massively parallel sequencing of 169 genes in cyst fluid DNA
To initiate this study, we determined the sequences of 169 presumptive cancer genes in the cyst fluids of 19 IPMNs, each obtained from a different patient. Thirty-three of the 169 were oncogenes, and the remainder were tumor suppressor genes. Although only a tiny subset of these 169 genes were known to be mutated in PDAs, all were known to be frequently mutated in at least one solid tumor type (table S1). We additionally sequenced these genes in normal pancreatic, splenic, or intestinal tissues of the same patients to determine which of the alterations that we identified were the result of somatic mutation. We chose to use massively parallel sequencing rather than Sanger sequencing for this analysis because we did not know what fraction of DNA purified from the cyst fluid was derived from neoplastic cells. Massively parallel sequencing has the capacity to identify mutations present in 2% or more of the studied cells, whereas Sanger sequencing often requires >25% neoplastic cells. IPMNs are by definition connected with the pancreatic duct system, and the cyst fluid containing cellular debris and shed DNA from the neoplastic cells can be expected to be admixed with that of the cells and secretions derived from normal ductal epithelial cells.
We devised a strategy to capture sequences of the 169 genes from cyst fluid DNA (Fig. 1). In brief, 243,238 oligonucleotides, each 60 base pairs (bp) in length and in aggregate covering the exonic sequences of all 169 genes, were synthesized in parallel with phosphoramidite chemistry on a single chip synthesized by Agilent Technologies. After removal from the chip, the oligonucleotide sequences were amplified by polymerase chain reaction (PCR) and ligated. Multiple displacement amplification was then used to further amplify the oligonucleotides, which were then bound to a filter. Finally, the filter was used to capture complementary DNA sequences from the cyst fluids and corresponding normal samples, and the captured DNA was subjected to massively parallel sequencing.
Fig. 1.
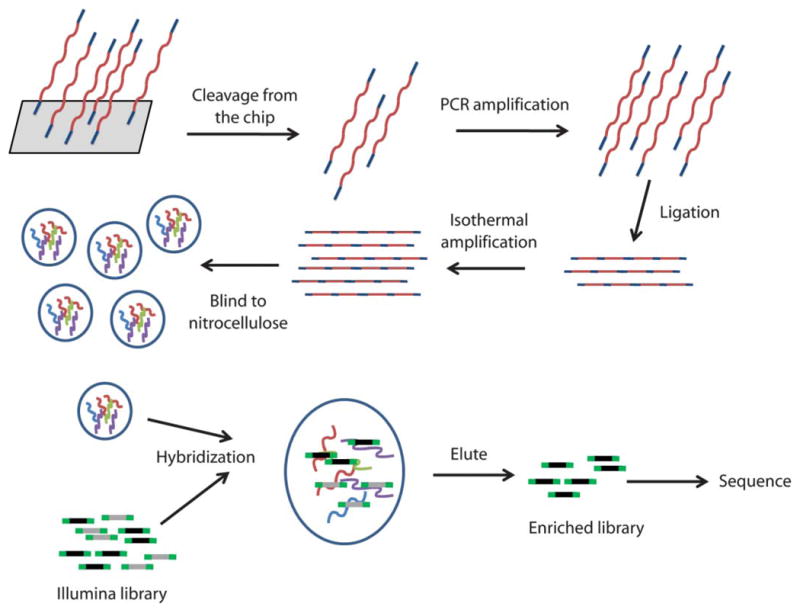
Schematic of capture strategy. Overlapping oligonucleotides flanked by universal sequences complimentary to the 169 genes listed in table S1 were synthesized on an array. The oligonucleotides were cleaved off the array, amplified by PCR with universal primers, ligated into concatamers, and amplified in an isothermal reaction. They were then bound to nitrocellulose filters and used as bait for capturing the desired fragments. An Illumina library was constructed from the sample DNA. The library was denatured and hybridized to the probes immobilized on nitrocellulose. The captured fragments were eluted, PCR-amplified, and sequenced on an Illumina GAIIX instrument.
The target region corresponding to the coding exons of the 169 genes encompassed 584,871 bp. These bases were redundantly sequenced, with 902 ± 411 (mean ± 1 SD) fold coverage in the 38 samples sequenced (19 IPMN cyst fluids plus 19 matched DNA samples from normal tissues of the same patients). This coverage allowed us to confidently detect somatic mutations present in >5% of the template molecules.
There were only two genes mutated in more than one IPMN—KRAS, which was mutated in 14 of the 19 IPMNs, and GNAS, which was mutated in 6 IPMNs. The mutations in GNAS all occurred at codon 201, resulting in either an R201H or an R201C substitution. GNAS is a well-known oncogene that is mutated in pituitary and other uncommon tumor types (16–19). However, such mutations have rarely been reported in common epithelial tumors (20–22). In pituitary tumors, mutations cluster at two positions—codons 201 and 227 (16, 19). This clustering provides extraordinary opportunities for diagnosis, similar to that of KRAS. For example, the clustering of KRAS mutations has facilitated the design of assays to detect mutations in tumors of colorectal cancer patients eligible for therapy with antibodies to epidermal growth factor receptor (EGFR) (23). All 12 KRAS mutations identified through massively parallel sequencing of cyst fluids were at codon 12, resulting in a G12D, G12V, or G12R amino acid change. KRAS mutations at codon 12 have previously been identified in most PDAs as well as in 40 to 60% of IPMNs (24–29). GNAS mutations have not previously been identified in pancreatic cysts or in PDAs.
Frequency of KRAS and GNAS mutations in pancreatic cyst fluid DNA
We next determined the frequency of KRAS codon 12 and GNAS codon 201 mutations in a larger set of IPMNs. The clinical characteristics of all IPMNs analyzed in this study are listed in table S2. To ensure that the analyses were performed robustly, we carried out preliminary experiments with cyst fluids from patients with known mutations based on the massively parallel sequencing experiments described above. We tested several methods for purifying DNA from often viscous cyst fluids and used the optimum method for subsequent experiments. Quantitative PCR was used to determine the number of amplifiable template molecules recovered with this procedure. In eight cases, we compared pelleted cells to supernatants derived from the same cyst fluid samples and found that the fraction of mutant templates in both compartments was similar. On the basis of these results, we purified DNA from 0.25 ml of whole cyst fluid (cells plus supernatant) and, as assessed by quantitative PCR, recovered an average of 670 ± 790 ng of usable DNA.
For each of 84 cyst fluid samples (an independent cohort of 65 patients plus the 19 patients whose fluids had been studied by massively parallel sequencing), we analyzed ~800 template molecules for five distinct mutations, three at KRAS codon 12 and two within GNAS codon 201 (see Materials and Methods). A PCR/ligation method that had the capacity to detect one mutant template molecule among 200 normal (wild-type) templates was used for these analyses (Fig. 2A). We identified GNAS and KRAS mutations in 61 and 82% of the IPMN fluids, respectively (representative examples in Fig. 2B). In those samples without GNAS codon 201 mutations, we searched for GNAS codon 227 mutations, but did not find any. We also analyzed macro- and microdissected frozen or paraffin-embedded cyst walls from an independent collection of 48 surgically resected IPMNs and similarly identified a high prevalence of GNAS (75%) and KRAS (79%) mutations. In aggregate, 66% of 132 IPMNs harbored a GNAS mutation, 81% harbored a KRAS mutation, slightly more than half (51%) harbored both GNAS and KRAS mutations, whereas at least one of the two genes was mutated in 96.2% (table S2). Given background mutation rates in tumors or normal tissues (30), the probability that either GNAS or KRAS mutations occurred by chance alone was less than 10−30. There were no significant correlations between the prevalence of KRAS or GNAS mutations and age, sex, or smoking history of the patients (P > 0.05) (Table 1). Small (<3 cm) as well as larger cysts had similar fractions of both KRAS and GNAS mutations, and the location of the IPMN (head, body, or tail) did not correlate with the presence of mutation in either gene (Table 1). GNAS and KRAS mutations were present in low-grade as well as in high-grade IPMNs. The prevalence of KRAS mutations was higher in lower-grade lesions (P = 0.03), whereas the prevalence of GNAS mutations was somewhat higher in more advanced lesions (P = 0.11) (Table 1). GNAS as well as KRAS mutations were present in each of the three major histologic types of IPMNs—intestinal, pancreatobiliary, and gastric. However, the prevalence of the mutations varied across histological types (P < 0.01 for both KRAS and GNAS). GNAS mutations were most prevalent in the intestinal subtype (100%) while KRAS mutations had the highest frequency (100%) in the pan-creatobiliary subtype and had the lowest frequency (42%) in the intestinal subtype (Table 1).
Fig. 2.

Ligation assays used to assess KRAS and GNAS mutations. (A) Schematic of the ligation assay. Oligonucleotide probes complementary to either the wild-type or the mutant sequences were incubated with a PCR product containing the sequence of interest. The wild-type– and mutant-specific probes were labeled with the fluorescent dyes 6-FAM and HEX, respectively, and the wild-type–specific probe was 11 bases longer than the mutant-specific probe. After ligation to a common anchoring primer, the ligation products were separated on a denaturing polyacrylamide slab gel. Further details of the assay are provided in Materials and Methods. (B) Examples of the results obtained with the ligation assay in the indicated patients. Templates were derived from DNA of normal duodenum or IPMN tissue. Each lane represents the results of ligation of one of four independent PCR products, each containing 200 template molecules. The probe in the left panel was specific to the GNAS R201H mutation, and the probe on the right panel was specific for the GNAS R201C mutation.
Table 1.
Patient information. Correlations between mutations and clinical and histopathologic parameters of IPMNs.
| N (total) |
KRAS mutation
|
P |
GNAS mutation
|
P | ||||
|---|---|---|---|---|---|---|---|---|
| n | % | n | % | |||||
| Age (years) | <65 | 29 | 22 | 75.9 | 0.42 | 18 | 62.1 | 0.62 |
| ≥65 | 103 | 85 | 82.5 | 69 | 67 | |||
| Gender | Male | 70 | 58 | 82.9 | 0.58 | 51 | 72.9 | 0.07 |
| Female | 62 | 49 | 79 | 36 | 58.1 | |||
| History of smoking | Yes | 25 | 21 | 84 | 0.77 | 17 | 68 | 0.85 |
| No | 37 | 30 | 81.1 | 26 | 70.3 | |||
| Grade | Low | 23 | 20 | 87 | 0.43 (low versus others) | 11 | 47.8 | 0.04 (low versus others) |
| Intermediate | 51 | 46 | 90.2 | 34 | 66.7 | |||
| High | 58 | 41 | 70.7 | 42 | 72.4 | |||
| Duct type | Main | 35 | 23 | 65.7 | 0.002 (main versus branch) | 24 | 68.6 | 0.37 (main versus branch) |
| Branch | 64 | 58 | 90.6 | 38 | 59.4 | |||
| Mixed | 28 | 21 | 75 | 20 | 71.4 | |||
| Subtype | Gastric | 52 | 45 | 86.5 | 0.02 (pancreatobiliary versus intestinal) | 34 | 65.4 | 0.002 (pancreatobiliary versus intestinal) |
| Pancreatobiliary | 7 | 7 | 100 | 3 | 42.9 | |||
| Intestinal | 13 | 6 | 46.2 | 13 | 100 | |||
| Diameter (cm) | <3 | 62 | 49 | 79 | 0.58 | 41 | 66.1 | 0.96 |
| ≥3 | 70 | 58 | 82.9 | 46 | 65.7 | |||
| Location | Proximal (head) | 77 | 64 | 83.1 | 0.44 (proximal versus distal) | 53 | 68.8 | 0.38 (proximal versus distal) |
| Distal (body, tail) | 49 | 38 | 77.6 | 30 | 61.2 | |||
| Proximal and distal | 6 | 5 | 83.3 | 4 | 66.7 | |||
| Associated cancer | Yes | 24 | 18 | 75 | 0.4 | 18 | 75 | 0.3 |
| No | 108 | 89 | 82.4 | 69 | 63.9 | |||
We then determined whether GNAS mutations were present in SCAs, a common but benign type of pancreatic cystic neoplasm. We examined a total of 44 surgically resected SCAs, each from a different patient (42 cyst fluids and 2 cyst walls). Many of these cysts were surgically resected because they clinically mimicked an IPMN. They would have likely not been surgically excised had they been known to be SCAs. The SCAs averaged 5.0 ± 2.8 cm in maximum diameter (table S3), similar to the IPMNs (4.4 ± 3.7 maximum diameter, table S2). There was little difference in the locations of the SCAs and IPMNs within the pancreas (tables S2 and S3). However, no GNAS or KRAS mutations were identified in the SCAs, in marked contrast to the IPMNs (P < 0.001, Fisher’s exact test). GNAS mutations were also not identified in any of 21 MCNs (P = 0.005 when compared to IPMNs, Fisher’s exact test), although KRAS mutations were found in 33% of MCNs (table S3). GNAS mutations were also not identified in five examples of an uncommon type of cyst, called intraductal oncocytic papillary neoplasm (IOPN), with characteristic oncocytic features (table S3).
IPMN polyclonality
KRAS G12D, G12V, and G12R mutations were found in 43, 39, and 13% of IPMNs, respectively (table S2). A small fraction (11%) of the IPMNs contained two different KRAS mutations and 2% contained three different mutations. Likewise, GNAS R201C and GNAS R201H mutations were present in 39 and 32% of the IPMNs, respectively, and 4% of the IPMNs had both mutations (table S2). More than one mutation in KRAS in IPMNs has been observed in previous studies of IPMNs (31–33), and the multiple KRAS and GNAS mutations are suggestive of a polyclonal origin of the tumor.
We investigated clonality in more detail by precisely quantifying the levels of mutations in the subset of cyst fluids containing more than one mutation of the same gene. To accomplish this, we used a technique called BEAMing (34). Through this method, individual template molecules are converted into individual magnetic beads attached to thousands of molecules with the identical sequence. The beads are then hybridized with mutation-specific probes and analyzed by flow cytometry (Fig. 3). The analysis of 17 IPMN cyst fluids, each with mutations in both KRAS and GNAS, showed that the fraction of mutant alleles varied widely, ranging from 0.8 to 45% of the templates analyzed. There was an average of 12.8 ± 12.2% mutant alleles of KRAS and an average of 24.4 ± 13.1% mutant alleles of GNAS in the 17 IPMN cyst fluids examined (table S4). In two of the seven IPMNs with more than one KRAS mutation, there was a predominant mutant that outnumbered the second KRAS mutant by >5:1 (table S4). Similarly, two of the four cases harboring two different GNAS mutations had a predominant mutant (table S4). In the other cases, the different mutations in KRAS (or GNAS) were distributed more evenly (table S4). These data support the idea that cells within a subset of IPMNs had undergone independent clonal expansions, giving rise to apparent polyclonality (35).
Fig. 3.

BEAMing assays used to quantify mutant representation. PCR was used to amplify KRAS or GNAS sequences containing the region of interest (KRAS codon 12 and GNAS codon 201). The PCR products were then used as templates for BEAMing, in which each template was converted to a bead containing thousands of identical copies of the templates (34). After hybridization to Cy3- or Cy5-labeled oligonucleotide probes specific for the indicated wild-type or mutant sequences, respectively, the beads were analyzed by flow cytometry. Scatter plots are shown for templates derived from the DNA of IPMN 130 or from normal spleen. Beads containing the wild-type or mutant sequences are widely separated in the scatter plots, and the fraction of mutant-containing beads are indicated. Beads whose fluorescence spectra lie between the wild-type– and the mutant-containing beads result from inclusion of both wild-type and mutant templates in the aqueous nanocompartments of the emulsion PCR. See Materials and Methods for details.
IPMNs are often multilocular or multifocal in nature, looking much like a bunch of grapes (Fig. 4, C and D) (36). To determine the relationship between cyst locules (individual grapes) and cyst fluid, we microdissected the walls from individual locules of each of 10 IPMNs from whom cyst fluid was available (example in Fig. 4, A and B). The individual locule walls generally appeared to be monoclonal, as more than one KRAS mutation was found in only 1 (4.5%) of the 22 locules examined. No locule wall contained more than one GNAS mutation, and two adjacent locules within a single grossly distinct IPMN were more likely to contain the same KRAS or GNAS mutation than the lining epithelium from two topographically different IPMNs (P < 0.05, Fisher’s exact test for KRAS G12D, KRAS G12R, and GNAS R201H mutations; P < 0.10 for KRAS G12V and GNAS R201H mutations). All of the 10 KRAS and 6 GNAS mutations identified in the cyst fluid could be identified in the corresponding locule walls. These data leave little doubt that the mutations in the cyst fluid are derived from the cyst locule walls and indicate that the cyst fluid provides an excellent representation of the neoplastic cells in an IPMN.
Fig. 4.

IPMN morphologies. (A and B) Typical IPMN before (A) and after (B) microdissection of the epithelium lining the cyst wall. (C and D) Examples of multiloculated IPMNs from two different patients. (E and F) IPMN (E) and associated invasive adenocarcinoma (F) from the same patient. (G and H) Another patient with an IPMN (G) and associated invasive adenocarcinoma (H). All photographs were of formalin-fixed, paraffin-embedded samples stained with hematoxylin and eosin. Scale bars, 500 μm [(A) to (C)], 200 μm [(D) and (E)], and 100 μm [(F) to (H)].
GNAS mutations in invasive cancers associated with IPMNs
Previous whole-exome sequencing had not revealed any GNAS mutations in 24 typical PDAs that occurred in the absence of an associated IPMN (29). We extended these data by examining 95 additional surgically resected PDAs in pancreata without evidence of IPMNs for mutations in GNAS R201H or R201C, using the ligation assay described above. Again, no GNAS mutations were identified in PDAs arising in the absence of IPMNs.
We suspected that IPMNs containing GNAS mutations had the potential to progress to an invasive carcinoma because fluids from IPMNs with high-grade dysplasia contained such mutations (Table 1). However, in light of the multilocular and multifocal nature of IPMNs described above, it was not clear whether the cells of the locule(s) that progress to an invasive carcinoma were those that contained GNAS mutations. To address this question, we purified DNA from invasive pancreatic adenocarcinomas that developed in association with IPMNs. In each case, the neoplastic cells of the IPMN and of the invasive adenocarcinoma were carefully microdissected. In seven of the eight patients, the identical GNAS mutation was found in the neoplastic cells of the IPMN and in the concurrent invasive adenocarcinoma (table S5). The KRAS mutational status of the PDA was consistent with that of the associated IPMN in the same seven cases. In the eighth case, the KRAS and GNAS mutations identified in the neoplastic cells of the IPMN were not found in the associated PDA, suggesting that this invasive cancer arose from a separate precursor lesion (table S5). Although KRAS mutations were found commonly in both types of PDAs, there was a marked difference between the prevalence of GNAS mutations in PDAs associated with IPMNs (7 of 8) and that in PDAs unassociated with IPMNs (0 of 116; P < 0.001, Fisher’s exact test).
DISCUSSION
The data document the existence of a heretofore unappreciated molecular pathway leading to pancreatic neoplasia. There is no doubt that GNAS mutations play a driving role in this IPMN-specific pathway: The mutations are remarkably common and they occur at a single codon (201), mutations of which are known to endow cells with extremely high adenyl cyclase activity and adenosine 3′,5′-monophosphate (cAMP) levels (37–39). Based on their rate of mutation and specificity (30), the probability that these mutations are passengers rather than drivers of IPMN development is negligible.
The data also demonstrated that >96% of IPMNs have either a GNAS or a KRAS mutation and more than half have both mutations. Which mutation—KRAS or GNAS—arises first? There were 20 cases in which GNAS mutations were identified in the absence of KRAS mutations and 6 additional cases in which GNAS mutations were at least five times more abundant than KRAS mutations in the same cyst fluid (table S4). The converse situation—KRAS mutations in the absence of GNAS mutations—was also observed in 40 cases (table S2). These data, in combination with the demonstration that more than one KRAS or more than one GNAS mutation could be identified in the same cyst (table S4), suggest two models for IPMN development. First, it is possible that IPMN locules represent independent entities whose evolution is unrelated to other locules within the same IPMN. This model is inconsistent with our data because two adjacent locules within a single grossly distinct IPMN were more likely to contain the same KRAS or GNAS mutation than the lining epithelium from two topographically different cysts, as noted in the Results section. Second, it is possible that all IPMNs are initiated by a single founding mutation in either GNAS or KRAS. Subsequent mutations of cells within the cystic lesion would lead to independent clonal expansions, perhaps represented by different locules. Such polyclonality has been observed in colorectal adenomas, which are initiated by mutations in adenomatous polyposis coli (APC) pathway genes but sometimes progress through heterogeneous KRAS mutations to a transient polyclonal stage (40). This stage is eventually replaced by subsequent clonal expansion of a cell with one of the KRAS mutations (40). A related possibility is that IPMNs are initiated by a genetic or epigenetic alteration in a gene other than KRAS or GNAS, and that we have observed subsequent clonal expansions of these initiated cells. Finally, it is possible that most IPMNs are indeed initiated by a mutation (in GNAS, KRAS, or another gene), but that occasionally two such IPMNs, initiated by completely different cells, develop adjacent to one another. Although these models are difficult to distinguish from one another, it is possible that lineage tracking can be accomplished by complete sequencing of IPMN locule genomes in the future (41).
Apart from its implications for understanding IPMN development, our data have potentially important practical ramifications. The appropriate management of a patient with a pancreatic cyst depends on the type of cyst (42). In particular, it is generally agreed that there is no need to remove asymptomatic SCAs because these lesions have a vanishingly small malignant potential (43). However, the distinction between SCA and mucinous cystic lesions (IPMN and MCN) of the pancreas is often not easy, even after extensive imaging and follow-up (6). One example of these difficulties is provided by the nature of the lesions in our study: Most of the 44 SCAs we examined were removed because they were pre-operatively believed to be cysts with malignant potential. Hence, many of these 44 surgical procedures were likely unnecessary.
These diagnostic difficulties have long been appreciated and have spurred attempts to develop biomarkers as adjuncts to clinical data, imaging, and cytology (44). Indeed, new protein and glycoprotein markers are showing promising results (45, 46). One conceptual disadvantage of these protein biomarkers is that they are simply associated with cyst development and do not play a pathogenic role. Alterations of oncogenes such as KRAS are attractive alternatives because they are intimately involved in pathogenesis (47–50). In the largest previous study to date on such alterations, 45% of the fluids from mucinous cysts were shown to contain KRAS mutations (25). Our data demonstrate that KRAS mutations are actually present in a larger fraction of IPMNs; this difference is probably a result of the more sensitive methods used in our study combined with optimization of procedures used to purify cyst fluid DNA (see Materials and Methods). Third, and most important, the combination of GNAS and KRAS mutation detection provides high sensitivity and specificity for distinguishing between SCAs and IPMNs. Most IPMNs had a GNAS and/or a KRAS [95% confidence interval (CI), 91 to 99%], whereas no SCAs had either mutation. These data indicate a sensitivity of 0.96 (95% CI, 0.91 to 0.99) and a specificity of 1.0 (97.5% one-sided CI, 0.92 to 1) for distinguishing between these two lesions. In addition, although not as sensitive, the presence of a GNAS mutation in cyst fluid can also distinguish IPMNs from MCNs (table S3). The assay involves just two amplicons (GNAS and KRAS) and can be performed with as little as 250 μl of cyst fluid.
Several caveats to the potential utility of such tests should be noted. First, the analysis of cyst fluid obtained through EUS, although safe, is an invasive procedure. Complications include bleeding, infection, and pancreatitis; are reversible; and are generally observed in <1% of patients [reviewed in (51)]. Second, neither KRAS nor GNAS mutations can distinguish high-grade or invasive from low-grade IPMNs. The supplementation of KRAS and GNAS mutational analyses with other markers indicative of grade would be useful (11). Third, we cannot yet reliably distinguish IPMNs from MCNs through the analysis of cyst fluid. Although MCNs do not contain GNAS mutations, a third of them contain KRAS mutations (table S3). MCN-specific mutations may be identified in the future through a strategy similar to the one we used to identify mutations in IPMNs.
Astute clinicians and pathologists have long suspected that adenocarcinomas of the pancreas arising in IPMNs are a “different disease” from those arising locally distant or in the absence of an IPMN (15, 52). We here provide evidence in support of this hypothesis and identify a key molecular component that underlies this difference.
MATERIALS AND METHODS
Patients and specimens
The present study was approved by the Institutional Review Boards of Johns Hopkins Medical Institutions, Memorial Sloan-Kettering Cancer Center, and the University of Indiana. We included individuals in whom pancreatic cyst fluid samples from pancreatectomy specimens and/or fresh-frozen tumor tissues were available for molecular analysis. Relevant demographic, clinicopathologic data were obtained from prospectively maintained clinical databases and correlated with mutational status.
Pancreatic cyst fluids were harvested in the Surgical Pathology suite from surgically resected pancreatectomy specimens with a sterile syringe. Aspirated fluids were stored at −80°C within 30 min of resection. Fresh-frozen tissue specimens of surgically resected cystic neoplasms of the pancreas were obtained through a prospectively maintained Johns Hopkins Surgical Pathology Tumor Bank. These lesions as well as normal tissues were macrodissected with serial frozen sections to guide the trimming of optimal cutting temperature (OCT) compound–embedded tissue blocks to obtain a minimum neoplastic cellularity of 80%. Formalin-fixed and paraffin-embedded archival tissues from surgically resected pancreata were sectioned at 6 μm, stained with hematoxylin and eosin, and dissected with a sterile needle on an SMZ1500 stereomicroscope (Nikon). An estimated 5000 to 10,000 cells were microdissected from each lesion. Lesions were classified as IPMNs, MCNs, or SCAs with standard criteria (53). IPMNs were subtyped by internationally accepted criteria (54).
DNA purification
DNA was purified from frozen cyst walls with an AllPrep kit (Qiagen) and from formalin-fixed, paraffin-embedded sections with the QIAamp DNA FFPE tissue kit (Qiagen) according to the manufacturer’s instructions. DNA was purified from 250 μl of cyst fluid by adding 3 ml of RLTM buffer (Qiagen) and then binding to an AllPrep DNA column (Qiagen) following the manufacturer’s protocol. DNA was quantified in all cases with quantitative PCR with primers and conditions as described (55).
Illumina library preparation
Cyst fluid DNA was first quantified through real-time PCR with primers specific for repeated sequences in DNA [long interspersed nuclear element (LINE)] as described (56). A minimum of 100 ng of DNA from cyst fluid was used to make Illumina libraries according to the manufacturer’s protocol, with the exception that the amount of adapters was decreased in proportional fashion when a lower amount of template DNA was used. The number of PCR cycles used to amplify the library after ligation of adapters was varied to ensure a yield of ~5 μg of the final library product for capture.
Target DNA enrichment
The targeted region included all of the 3386 exons of 169 cancer-related genes and was enriched with custom-made oligonucleotide probes. The design of each oligonucleotide was as follows: 5′-TCCCGCGACGAC–genomic region of interest–GCTGGAGTCGCG-3′. Probes were designed to capture both the plus and the minus strand of the DNA and had a 33-base overlap. The probes were custom-synthesized by Agilent Technologies on a chip. The oligonucleotides were cleaved from the chip by treatment for 5 hours with 3 ml of 35% ammonium hydroxide at room temperate. The solution was transferred to two 2-ml tubes, dried under vacuum, and redissolved in 400 μl of ribonuclease (RNase)– and deoxyribonuclease (DNase)–free water. Five microliters of the solution was used for PCR amplification with primers complementary to the 12-base sequence common to all probes: 5′-TGATCCCGCGACGA*C-3′ and 5′-GACCGCGACTCCAG*C-3′, with * indicating a phosphorothioate bond. The PCR mix contained 27 μl of H2O, 5 μl of template DNA, 2 μl of forward primer (25 μM), 2 μl of reverse primer (25 μM), 4 μl of MgCl2 (50 mM), 5 μl of 10× Platinum Taq buffer (Life Technologies), 4 μl of deoxyribonucleotide triphosphates (dNTPs) (10 mM each), and 1 μl of Platinum Taq (5 U/μl, Life Technologies). The cycling conditions were as follows: 1 cycle of 98°C for 30 s; 35 cycles of 98°C for 30 s, 40°C for 30 s, 60°C for 15 s, 72°C for 45 s; 1 cycle of 72°C for 5 min. The PCR product was purified with a MinElute Purification Column (Qiagen) and end-repaired with End-IT DNA End-Repair Kit (Epicentre) as follows: 34 μl of DNA, 5 μl of 10× End-Repair Buffer, 5 μl of dNTP Mix, 5 μl of adenosine 5′-triphosphate (ATP), and 1 μl of End-Repair Enzyme Mix. The mix was incubated at room temperature for 45 min and then purified with a MinElute Purification Column (Qiagen). The PCR products were ligated to form concatamers with the following protocol: 35 μl of End-Repaired DNA product, 40 μl of 2× T4 DNA ligase buffer, and 5 μl of T4 DNA ligase (3000 U; Enzymatics Inc.). The mix was incubated at room temperature for 4 hours, then purified with QIAquick Purification Column (Qiagen), and quantified by absorption at 260 nm.
Replicates of 50 ng of concatenated PCR product were amplified in 25 μl of solution with the REPLI-g midi whole genome amplification kit (Qiagen) according to the manufacturer’s protocol. The REPLI-g–amplified DNA (20 μg) was then bound to a nitrocellulose membrane and used to capture DNA libraries as described (57). In general, 5 μg of library DNA was used per capture. After washing, the captured libraries were ethanol-precipitated and redissolved in 20 μl of tris-EDTA (TE) buffer. The DNA was then amplified in a PCR mix containing 51 μl of distilled water (dH2O), 20 μl of 5× Phusion buffer, 5 μl of dimethyl sulfoxide (DMSO), 2 μl of 10 mM dNTPs, 50 pmol of Illumina forward and reverse primers, and 1 μl of HotStart Phusion enzyme (New England Biolabs) with the following cycling program: 98°C for 30 s; 15 cycles of 98°C for 25 s, 65°C for 30 s, 72°C for 30 s; and 72°C for 5 min. The amplified PCR product was purified with a NucleoSpin column (Macherey Nagel Inc.) according to the manufacturer’s suggested protocol, except that the NT buffer was not diluted and the DNA bound to the column was eluted in 35 μl of elution buffer. The captured library was quantified with real-time PCR with the primers used for grafting to the Illumina sequencing chip.
Ligation assay
PCR products containing codon 12 of KRAS and codon 201 of GNAS were amplified with the primers described in table S6. Each 10-μl PCR contained 200 template molecules in 5 μl of 2× Phusion Flash PCR Master Mix (New England Biolabs) and final concentrations of 0.25 μM forward and 1.5 μM reverse primers. Note that the mutant-specific probes sometimes included locked nucleic acid residues (table S6; Exiqon). The following cycling conditions were used: 98°C for 2 min; 3 cycles of 98°C for 10 s, 69°C for 15 s, 72°C for 15 s; 3 cycles of 98°C for 10 s, 66°C for 15 s, 72°C for 15 s; 3 cycles of 98°C for 10 s, 63°C for 15 s, 72°C for 15 s; 41 cycles of 98°C for 10 s, 60°C for 60 s. Reactions were performed in at least quadruplicate, and each was evaluated independently. Five microliters of a solution containing 0.5 μl of proteinase K (18.8 mg/ml, Roche) and 4.5 μl of dH2O was added to each well and incubated at 60°C for 30 min to inactivate the Phusion polymerase and then for 10 min at 98°C to inactivate the proteinase K.
The ligation assay was based on techniques described previously, with thermotolerant DNA ligases (58–61). Each 10-μl reaction contained 2 μl of PCR product (unpurified), 1 μl of 10× Ampligase buffer (Epicentre), 0.5 μl of Ampligase (5 U/μl, Epicentre), anchoring primer (final concentration, 2 μM), wild-type–specific primer (final concentration, 0.1 μM), and mutant-specific primer (final concentration, 0.025 μM). The sequences of these primers are listed in table S6. The following cycling conditions were used: 95°C for 3 min; 35 cycles of 95°C for 10 s, 37°C for 30 s, 45°C for 60 s. Five microliters of each reaction was added to 5 μl of formamide, and the ligation products were separated on a 10% urea-tris-borate-EDTA gel (Invitrogen) and imaged with an Amersham-GE Typhoon instrument (GE Healthcare).
BEAMing assays
BEAMing assays were performed as described (62) with the PCR products generated for the ligation assay as templates and the oligonucleotides listed in table S6 as hybridization probes.
Statistical analysis
Fisher’s exact tests were used to compare the differences between proportions, and Wilcoxon rank sum tests were used to compare differences in mutational status by age. Confidence intervals for the prevalence of mutations were estimated with the binomial distribution. To compare the prevalence of mutations in grossly distinct IPMNs to adjacent locules within a single grossly distinct IPMN, we compared the probability of observing given KRAS or GNAS mutation in the 111 distinct IPMNs to conditional probability that, given the first locule sequenced contained a specific KRAS or GNAS mutation, all other locules contained the same KRAS or GNAS mutations. The probabilities of GNAS or KRAS mutations occurring by chance were calculated with a binomial distribution and the previously estimated mutation rates of tumors or normal cells (30). STATA version 11 was used for all statistical analysis (63).
Supplementary Material
Acknowledgments
We thank J. Schaeffer, J. Ptak, N. Silliman, L. Dobbyn, J. A. Waters, H. Wu, and M. T. Yip-Schneider.
Funding: This work was supported by the Lustgarten Foundation for Pancreatic Cancer Research; the Virginia and D. K. Ludwig Fund for Cancer Research; the Sol Goldman Center for Pancreatic Cancer Research; the Joseph L. Rabinowitz Fund; the Michael Rolfe Foundation; the Indiana Genomics Initiative of Indiana University (which is supported in part by Lilly Endowment Inc.); the J. C. Monastra Foundation; Swim Across America; NIH grants CA 62924, CA 43460, and CA 134292; and National Cancer Institute contract N01-CN-43302. H.M. is a Mildred Scheel Scholar of the German Cancer Aid (Deutsche Krebshilfe e.V.).
Footnotes
Author contributions: J.W., H.M., A.M., M.D.M., L.D.W., and B.V. performed the experiments; J.W., H.M., A.M., J.R.E., M.G., R.D.S., B.H.E., C.L.W., L.A.D., P.J.A., C.M.S., K.W.K., N.P., R.H.H., and B.V. designed the experiments; A.P.K. performed the statistical analysis; A.M., R.H.H., and B.V. wrote the first draft of the paper.
Competing interests: Johns Hopkins University has filed patent applications on inventions described in this manuscript, with inventors including J.W., H.M., A.M., L.A.D., K.W.K., N.P., R.H.H., and B.V.
REFERENCES AND NOTES
- 1.Laffan TA, Horton KM, Klein AP, Berlanstein B, Siegelman SS, Kawamoto S, Johnson PT, Fishman EK, Hruban RH. Prevalence of unsuspected pancreatic cysts on MDCT. AJR Am J Roentgenol. 2008;191:802–807. doi: 10.2214/AJR.07.3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Jong K, Nio CY, Hermans JJ, Dijkgraaf MG, Gouma DJ, van Eijck CH, van Heel E, Klass G, Fockens P, Bruno MJ. High prevalence of pancreatic cysts detected by screening magnetic resonance imaging examinations. Clin Gastroenterol Hepatol. 2010;8:806–811. doi: 10.1016/j.cgh.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 3.Kimura W, Nagai H, Kuroda A, Muto T, Esaki Y. Analysis of small cystic lesions of the pancreas. Int J Pancreatol. 1995;18:197–206. doi: 10.1007/BF02784942. [DOI] [PubMed] [Google Scholar]
- 4.Lee KS, Sekhar A, Rofsky NM, Pedrosa I. Prevalence of incidental pancreatic cysts in the adult population on MR imaging. Am J Gastroenterol. 2010;105:2079–2084. doi: 10.1038/ajg.2010.122. [DOI] [PubMed] [Google Scholar]
- 5.Matthaei H, Schulick RD, Hruban RH, Maitra A. Medscape, Cystic precursors to invasive pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2011;8:141–150. doi: 10.1038/nrgastro.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katz MH, Mortenson MM, Wang H, Hwang R, Tamm EP, Staerkel G, Lee JH, Evans DB, Fleming JB. Diagnosis and management of cystic neoplasms of the pancreas: An evidence-based approach. J Am Coll Surg. 2008;207:106–120. doi: 10.1016/j.jamcollsurg.2007.12.048. [DOI] [PubMed] [Google Scholar]
- 7.Tanaka M. Controversies in the management of pancreatic IPMN. Nat Rev Gastroenterol Hepatol. 2011;8:56–60. doi: 10.1038/nrgastro.2010.193. [DOI] [PubMed] [Google Scholar]
- 8.Hruban RH, Pitman MB, Klimstra DS. Atlas of Tumor Pathology, Series IV. American Registry of Pathology and Armed Forces Institute of Pathology; Washington, DC: 2007. Tumors of the pancreas. [Google Scholar]
- 9.Klöppel G, Kosmahl M. Cystic lesions and neoplasms of the pancreas. The features are becoming clearer. Pancreatology. 2001;1:648–655. doi: 10.1159/000055876. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka M, Chari S, Adsay V, Fernandez-del Castillo C, Falconi M, Shimizu M, Yamaguchi K, Yamao K, Matsuno S. International Association of Pancreatology, International consensus guidelines for management of intraductal papillary mucinous neoplasms and mucinous cystic neoplasms of the pancreas. Pancreatology. 2006;6:17–32. doi: 10.1159/000090023. [DOI] [PubMed] [Google Scholar]
- 11.Sohn TA, Yeo CJ, Cameron JL, Hruban RH, Fukushima N, Campbell KA, Lillemoe KD. Intraductal papillary mucinous neoplasms of the pancreas: An updated experience. Ann Surg. 2004;239:788–797. doi: 10.1097/01.sla.0000128306.90650.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crippa S, Fernández-del Castillo C, Salvia R, Finkelstein D, Bassi C, Domínguez I, Muzikansky A, Thayer SP, Falconi M, Mino-Kenudson M, Capelli P, Lauwers GY, Partelli S, Pederzoli P, Warshaw AL. Mucin-producing neoplasms of the pancreas: An analysis of distinguishing clinical and epidemiologic characteristics. Clin Gastroenterol Hepatol. 2010;8:213–219. doi: 10.1016/j.cgh.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poultsides GA, Reddy S, Cameron JL, Hruban RH, Pawlik TM, Ahuja N, Jain A, Edil BH, Iacobuzio-Donahue CA, Schulick RD, Wolfgang CL. Histopathologic basis for the favorable survival after resection of intraductal papillary mucinous neoplasm-associated invasive adenocarcinoma of the pancreas. Ann Surg. 2010;251:470–476. doi: 10.1097/SLA.0b013e3181cf8a19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sohn TA, Yeo CJ, Cameron JL, Koniaris L, Kaushal S, Abrams RA, Sauter PK, Coleman J, Hruban RH, Lillemoe KD. Resected adenocarcinoma of the pancreas—616 patients: Results, outcomes, and prognostic indicators. J Gastrointest Surg. 2000;4:567–579. doi: 10.1016/s1091-255x(00)80105-5. [DOI] [PubMed] [Google Scholar]
- 15.Salvia R, Fernández-del Castillo C, Bassi C, Thayer SP, Falconi M, Mantovani W, Pederzoli P, Warshaw AL. Main-duct intraductal papillary mucinous neoplasms of the pancreas: Clinical predictors of malignancy and long-term survival following resection. Ann Surg. 2004;239:678–685. doi: 10.1097/01.sla.0000124386.54496.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freda PU, Chung WK, Matsuoka N, Walsh JE, Kanibir MN, Kleinman G, Wang Y, Bruce JN, Post KD. Analysis of GNAS mutations in 60 growth hormone secreting pituitary tumors: Correlation with clinical and pathological characteristics and surgical outcome based on highly sensitive GH and IGF-I criteria for remission. Pituitary. 2007;10:275–282. doi: 10.1007/s11102-007-0058-2. [DOI] [PubMed] [Google Scholar]
- 17.Kalfa N, Ecochard A, Patte C, Duvillard P, Audran F, Pienkowski C, Thibaud E, Brauner R, Lecointre C, Plantaz D, Guedj AM, Paris F, Baldet P, Lumbroso S, Sultan C. Activating mutations of the stimulatory G protein in juvenile ovarian granulosa cell tumors: A new prognostic factor? J Clin Endocrinol Metab. 2006;91:1842–1847. doi: 10.1210/jc.2005-2710. [DOI] [PubMed] [Google Scholar]
- 18.Fragoso MC, Latronico AC, Carvalho FM, Zerbini MC, Marcondes JA, Araujo LM, Lando VS, Frazzatto ET, Mendonca BB, Villares SM. Activating mutation of the stimulatory G protein (gsp) as a putative cause of ovarian and testicular human stromal Leydig cell tumors. J Clin Endocrinol Metab. 1998;83:2074–2078. doi: 10.1210/jcem.83.6.4847. [DOI] [PubMed] [Google Scholar]
- 19.Yamasaki H, Mizusawa N, Nagahiro S, Yamada S, Sano T, Itakura M, Yoshimoto K. GH-secreting pituitary adenomas infrequently contain inactivating mutations of PRKAR1A and LOH of 17q23–24. Clin Endocrinol. 2003;58:464–470. doi: 10.1046/j.1365-2265.2003.01740.x. [DOI] [PubMed] [Google Scholar]
- 20.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JKV, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PVK, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 21.Idziaszczyk S, Wilson CH, Smith CG, Adams DJ, Cheadle JP. Analysis of the frequency of GNAS codon 201 mutations in advanced colorectal cancer. Cancer Genet Cytogenet. 2010;202:67–69. doi: 10.1016/j.cancergencyto.2010.04.023. [DOI] [PubMed] [Google Scholar]
- 22.Shin JS, Spillane A, Wills E, Cooper WA. PEComa of the retroperitoneum. Pathology. 2008;40:93–95. doi: 10.1080/00313020701716300. [DOI] [PubMed] [Google Scholar]
- 23.Dahabreh IJ, Terasawa T, Castaldi PJ, Trikalinos TA. Systematic review: Anti–epidermal growth factor receptor treatment effect modification by KRAS mutations in advanced colorectal cancer. Ann Intern Med. 2011;154:37–49. doi: 10.7326/0003-4819-154-1-201101040-00006. [DOI] [PubMed] [Google Scholar]
- 24.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 25.Fritz S, Fernandez-del Castillo C, Mino-Kenudson M, Crippa S, Deshpande V, Lauwers GY, Warshaw AL, Thayer SP, Iafrate AJ. Global genomic analysis of intraductal papillary mucinous neoplasms of the pancreas reveals significant molecular differences compared to ductal adenocarcinoma. Ann Surg. 2009;249:440–447. doi: 10.1097/SLA.0b013e31819a6e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soldini D, Gugger M, Burckhardt E, Kappeler A, Laissue JA, Mazzucchelli L. Progressive genomic alterations in intraductal papillary mucinous tumours of the pancreas and morphologically similar lesions of the pancreatic ducts. J Pathol. 2003;199:453–461. doi: 10.1002/path.1301. [DOI] [PubMed] [Google Scholar]
- 27.Schönleben F, Qiu W, Bruckman KC, Ciau NT, Li X, Lauerman MH, Frucht H, Chabot JA, Allendorf JD, Remotti HE, Su GH. BRAF and KRAS gene mutations in intraductal papillary mucinous neoplasm/carcinoma (IPMN/IPMC) of the pancreas. Cancer Lett. 2007;249:242–248. doi: 10.1016/j.canlet.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wada K, Takada T, Yasuda H, Amano H, Yoshida M, Sugimoto M, Irie H. Does “clonal progression” relate to the development of intraductal papillary mucinous tumors of the pancreas? J Gastrointest Surg. 2004;8:289–296. doi: 10.1016/j.gassur.2003.09.027. [DOI] [PubMed] [Google Scholar]
- 29.Jones S, Zhang X, Parsons DW, Lin JCH, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parmigiani G, Boca S, Lin J, Kinzler KW, Velculescu V, Vogelstein B. Design and analysis issues in genome-wide somatic mutation studies of cancer. Genomics. 2009;93:17–21. doi: 10.1016/j.ygeno.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schonleben F, Allendorf JD, Qiu W, Li X, Ho DJ, Ciau NT, Fine RL, Chabot JA, Remotti HE, Su GH. Mutational analyses of multiple oncogenic pathways in intraductal papillary mucinous neoplasms of the pancreas. Pancreas. 2008;36:168–172. doi: 10.1097/MPA.0b013e318158a4d2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kitago M, Ueda M, Aiura K, Suzuki K, Hoshimoto S, Takahashi S, Mukai M, Kitajima M. Comparison of K-ras point mutation distributions in intraductal papillary-mucinous tumors and ductal adenocarcinoma of the pancreas. Int J Cancer. 2004;110:177–182. doi: 10.1002/ijc.20084. [DOI] [PubMed] [Google Scholar]
- 33.Izawa T, Obara T, Tanno S, Mizukami Y, Yanagawa N, Kohgo Y. Clonality and field cancerization in intraductal papillary-mucinous tumors of the pancreas. Cancer. 2001;92:1807–1817. doi: 10.1002/1097-0142(20011001)92:7<1807::aid-cncr1697>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 34.Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D. BEAMing: Single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods. 2006;3:551–559. doi: 10.1038/nmeth898. [DOI] [PubMed] [Google Scholar]
- 35.Fujii H, lnagaki M, Kasai S, Miyokawa N, Tokusashi Y, Gabrielson E, Hruban RH. Genetic progression and heterogeneity in intraductal papillary-mucinous neoplasms of the pancreas. Am J Pathol. 1997;151:1447–1454. [PMC free article] [PubMed] [Google Scholar]
- 36.Taouli B, Vilgrain V, Vullierme MP, Terris B, Denys A, Sauvanet A, Hammel P, Menu Y. Intraductal papillary mucinous tumors of the pancreas: Helical CT with histopathologic correlation. Radiology. 2000;217:757–764. doi: 10.1148/radiology.217.3.r00dc24757. [DOI] [PubMed] [Google Scholar]
- 37.Diaz A, Danon M, Crawford J. McCune-Albright syndrome and disorders due to activating mutations of GNAS1. J Pediatr Endocrinol Metab. 2007;20:853–880. doi: 10.1515/jpem.2007.20.8.853. [DOI] [PubMed] [Google Scholar]
- 38.Lania A, Spada A. G-protein and signalling in pituitary tumours. Horm Res. 2009;71(Suppl 2):95–100. doi: 10.1159/000192446. [DOI] [PubMed] [Google Scholar]
- 39.Lania AG, Mantovani G, Spada A. Mechanisms of disease: Mutations of G proteins and G-protein-coupled receptors in endocrine diseases. Nat Clin Pract Endocrinol Metab. 2006;2:681–693. doi: 10.1038/ncpendmet0324. [DOI] [PubMed] [Google Scholar]
- 40.Shibata D, Schaeffer J, Li ZH, Capella G, Perucho M. Genetic heterogeneity of the c-K-ras locus in colorectal adenomas but not in adenocarcinomas. J Natl Cancer Inst. 1993;85:1058–1063. doi: 10.1093/jnci/85.13.1058. [DOI] [PubMed] [Google Scholar]
- 41.Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, Traulsen A, Nowak MA, Siegel C, Velculescu VE, Kinzler KW, Vogelstein B, Willis J, Markowitz SD. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci USA. 2008;105:4283–4288. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Correa-Gallego C, Ferrone CR, Thayer SP, Wargo JA, Warshaw AL, Fernández-del Castillo C. Incidental pancreatic cysts: Do we really know what we are watching? Pancreatology. 2010;10:144–150. doi: 10.1159/000243733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tseng JF, Warshaw AL, Sahani DV, Lauwers GY, Rattner DW, Fernandez-del Castillo C. Serous cystadenoma of the pancreas: Tumor growth rates and recommendations for treatment. Ann Surg. 2005;242:413–419. doi: 10.1097/01.sla.0000179651.21193.2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hong SM, Kelly D, Griffith M, Omura N, Li A, Li CP, Hruban RH, Goggins M. Multiple genes are hypermethylated in intraductal papillary mucinous neoplasms of the pancreas. Mod Pathol. 2008;21:1499–1507. doi: 10.1038/modpathol.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allen PJ, Qin LX, Tang L, Klimstra D, Brennan MF, Lokshin A. Pancreatic cyst fluid protein expression profiling for discriminating between serous cystadenoma and intraductal papillary mucinous neoplasm. Ann Surg. 2009;250:754–760. doi: 10.1097/SLA.0b013e3181bd7f20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ke E, Patel BB, Liu T, Li XM, Haluszka O, Hoffman JP, Ehya H, Young NA, Watson JC, Weinberg DS, Nguyen MT, Cohen SJ, Meropol NJ, Litwin S, Tokar JL, Yeung AT. Proteomic analyses of pancreatic cyst fluids. Pancreas. 2009;38:e33–e42. doi: 10.1097/MPA.0b013e318193a08f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khalid A, Zahid M, Finkelstein SD, LeBlanc JK, Kaushik N, Ahmad N, Brugge WR, Edmundowicz SA, Hawes RH, McGrath KM. Pancreatic cyst fluid DNA analysis in evaluating pancreatic cysts: A report of the PANDA study. Gastrointest Endosc. 2009;69:1095–1102. doi: 10.1016/j.gie.2008.07.033. [DOI] [PubMed] [Google Scholar]
- 48.Sawhney MS, Devarajan S, O’Farrel P, Cury MS, Kundu R, Vollmer CM, Brown A, Chuttani R, Pleskow DK. Comparison of carcinoembryonic antigen and molecular analysis in pancreatic cyst fluid. Gastrointest Endosc. 2009;69:1106–1110. doi: 10.1016/j.gie.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 49.Schoedel KE, Finkelstein SD, Ohori NP. K-Ras and microsatellite marker analysis of fine-needle aspirates from intraductal papillary mucinous neoplasms of the pancreas. Diagn Cytopathol. 2006;34:605–608. doi: 10.1002/dc.20511. [DOI] [PubMed] [Google Scholar]
- 50.Bartsch D, Bastian D, Barth P, Schudy A, Nies C, Kisker O, Wagner HJ, Rothmund M. K-ras oncogene mutations indicate malignancy in cystic tumors of the pancreas. Ann Surg. 1998;228:79–86. doi: 10.1097/00000658-199807000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Al-Haddad M, Wallace MB, Woodward TA, Gross SA, Hodgens CM, Toton RD, Raimondo M. The safety of fine-needle aspiration guided by endoscopic ultrasound: A prospective study. Endoscopy. 2008;40:204–208. doi: 10.1055/s-2007-995336. [DOI] [PubMed] [Google Scholar]
- 52.Sahani DV, Kadavigere R, Saokar A, Fernandez-del Castillo C, Brugge WR, Hahn PF. Cystic pancreatic lesions: A simple imaging-based classification system for guiding management. Radiographics. 2005;25:1471–1484. doi: 10.1148/rg.256045161. [DOI] [PubMed] [Google Scholar]
- 53.Bosman FT, Carneiro F, Hruban RH, Thiese ND. WHO Classification of Tumours of the Digestive System. 4. Vol. 3 IARC Press; Lyon, France: 2010. [Google Scholar]
- 54.Furukawa T, Klöppel G, Volkan Adsay N, Albores-Saavedra J, Fukushima N, Horii A, Hruban RH, Kato Y, Klimstra DS, Longnecker DS, Lüttges J, Offerhaus GJ, Shimizu M, Sunamura M, Suriawinata A, Takaori K, Yonezawa S. Classification of types of intraductal papillary-mucinous neoplasm of the pancreas: A consensus study. Virchows Arch. 2005;447:794–799. doi: 10.1007/s00428-005-0039-7. [DOI] [PubMed] [Google Scholar]
- 55.Rago C, Huso DL, Diehl F, Karim B, Liu G, Papadopoulos N, Samuels Y, Velculescu VE, Vogelstein B, Kinzler KW, Diaz LA., Jr Serial assessment of human tumor burdens in mice by the analysis of circulating DNA. Cancer Res. 2007;67:9364–9370. doi: 10.1158/0008-5472.CAN-07-0605. [DOI] [PubMed] [Google Scholar]
- 56.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA., Jr Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Herman DS, Hovingh GK, Iartchouk O, Rehm HL, Kucherlapati R, Seidman JG, Seidman CE. Filter-based hybridization capture of subgenomes enables resequencing and copy-number detection. Nat Methods. 2009;6:507–510. doi: 10.1038/nmeth.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fouquet C, Antoine M, Tisserand P, Favis R, Wislez M, Commo F, Rabbe N, Carette MF, Milleron B, Barany F, Cadranel J, Zalcman G, Soussi T. Rapid and sensitive p53 alteration analysis in biopsies from lung cancer patients using a functional assay and a universal oligonucleotide array: A prospective study. Clin Cancer Res. 2004;10:3479–3489. doi: 10.1158/1078-0432.CCR-0994-03. [DOI] [PubMed] [Google Scholar]
- 59.Dong SM, Traverso G, Johnson C, Geng L, Favis R, Boynton K, Hibi K, Goodman SN, D’Allessio M, Paty P, Hamilton SR, Sidransky D, Barany F, Levin B, Shuber A, Kinzler KW, Vogelstein B, Jen J. Detecting colorectal cancer in stool with the use of multiple genetic targets. J Natl Cancer Inst. 2001;93:858–865. doi: 10.1093/jnci/93.11.858. [DOI] [PubMed] [Google Scholar]
- 60.Luo J, Bergstrom DE, Barany F. Improving the fidelity of Thermus thermophilus DNA ligase. Nucleic Acids Res. 1996;24:3071–3078. doi: 10.1093/nar/24.15.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shi C, Eshleman SH, Jones D, Fukushima N, Hua L, Parker AR, Yeo CJ, Hruban RH, Goggins MG, Eshleman JR. LigAmp for sensitive detection of single-nucleotide differences. Nat Methods. 2004;1:141–147. doi: 10.1038/nmeth713. [DOI] [PubMed] [Google Scholar]
- 62.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA., Jr Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stata Statistical Software: Release 11. StataCorp; College Station, TX: 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.