

SUMMARY
Frequent co-directional collisions between the replisome and RNA polymerase (RNAP) are inevitable because the rate of replication is much faster than that of transcription. Here we show that the outcome of such collisions depends on the productive state of transcription elongation complexes (ECs). Co-directional collisions with backtracked (arrested) ECs lead to DNA double strand breaks (DSBs), whereas head-on collisions do not. A mechanistic model is proposed to explain backtracking-mediated DSBs. We further show that bacteria employ various strategies to evade replisome collisions with backtracked RNAP, the most general of which is translation that prevents RNAP backtracking. If translation is abrogated, DSBs are suppressed by elongation factors that either prevent backtracking or reactivate backtracked ECs. Finally, termination factors also contribute to genomic stability by removing arrested ECs. Our results establish RNAP backtracking as the intrinsic hazard to chromosomal integrity and implicate active ribosomes and other anti-backtracking mechanisms in genome maintenance.
INTRODUCTION
Survival and propagation of living cells require simultaneous DNA replication and transcription. During RNA synthesis RNA polymerase (RNAP) forms one of the most stable protein-DNA complexes, the elongation complex (EC). ECs, however, must frequently be dislodged by the replication machinery (replisome) (Figure S1). RNAP and the replisome share the same DNA track, but the replisome moves ~20 times more rapidly than RNAP (Kornberg and Baker, 1992). Therefore, even though transcription of the most active and essential bacterial genes tend to occur with the same polarity as replication fork progression (Brewer, 1988; Blattner et al., 1997; Kunst et al., 1997; Rocha and Danchin, 2003), frequent collisions between the replisome and ECs are destined to occur. In vivo studies in bacteria and eukaryotes suggest that co-directional collisions are less detrimental than head-on collisions, which often result in replication fork arrest and DNA recombination (Mirkin and Mirkin, 2007; Rudolph et al., 2007; Wang et al., 2007; Prado and Aguilera, 2005; French, 1992; Deshpande and Newlon, 1996; Vilette et al., 1996). Recent in vitro studies support these views, demonstrating that during a co-directional collision the replisome rapidly dislocates RNAP while bypassing the EC roadblock, whereas head-on collisions result in replisome stalling followed by slow EC displacement (Pomerantz and O’Donnell, 2008; 2010). Notably, co-directional collision with a halted EC can result in discontinuous replication in which the DNA polymerase (PolIII) terminates the leading strand to restart synthesis using the nascent transcript as a primer (Pomerantz and O’Donnell, 2008). In contrast to this view, a recent ChIP-chip interrogation of transcription-replication conflicts in exponentially growing Bacillus subtilis indicates that codirectional collisions at highly expressed ribosomal genes can lead to replication disruption (Merrikh et al., 2011). Since RNAP alternates between different conformational states (productive and backtracked) at numerous sites and interacts with multiple elongation/termination factors in vivo (Nudler et al., 1997; Komissarova and Kashlev, 1997; Epshtein and Nudler, 2003; Roberts et al., 2008; Nudler, 2009), the actual outcome and mechanism of replication fork progression through the EC remains unknown.
RESULTS
Collisions between permanently arrested ECs and replisome induce DSBs in vivo
To examine this fundamental issue, we designed a plasmid-based system to monitor co-directional and head-on collisions between the replisome and RNAP in living cells. One plasmid carried the phage λ cI857 pL-nutL-RBSN-UTR cassette oriented co-directionally with the ColE1 origin of replication (pCODIR); in a second plasmid, the origin and the transcription cassette were oriented head-on (pHDON) (Figure 1A). The temperature sensitive cI857 repressor allows for rapid activation of the pL promoter at 42°C. Other features of the cassette make it possible to modulate EC processivity. The nutL site in the RNA can be used to recruit λ N or phage HK022 Nun factors, which stimulate or inhibit elongation, respectively (Greive et al., 2005; Washburn et al., 2003). Nun protein arrests ECs downstream of nutL in vitro and in vivo (Washburn et al., 2003). Finally, it is possible to insert an ATG downstream to RBSN, which allows us to determine the effect of translating ribosomes, which stimulate transcription elongation (Proshkin et al., 2010), on EC-replisome collisions.
Figure 1. Collisions between replication and permanently arrested ECs lead to DNA damage in vivo.
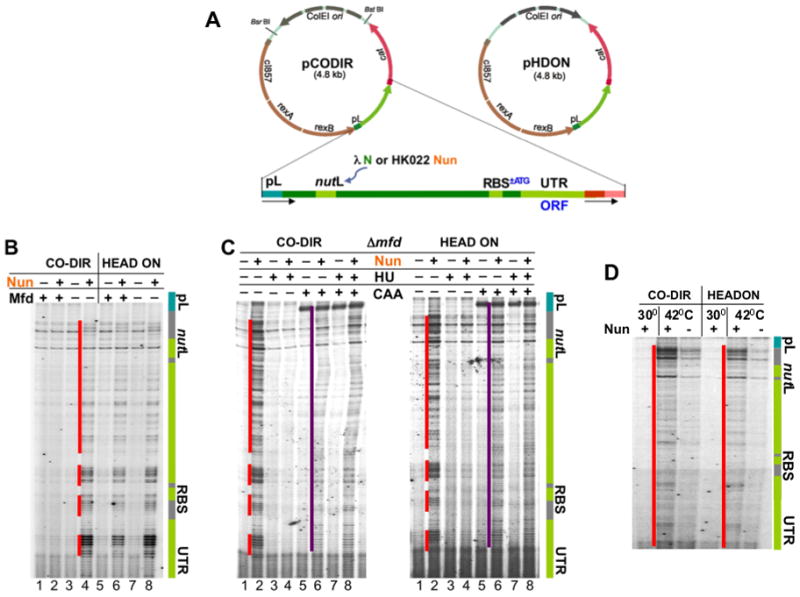
(A) Plasmids pCODIR and pHDON used to monitor co-directional and head-on collisions between the replisome and RNAP. The gray arrow indicates the direction of replication. The phage λ cassette containing the pL promoter and N utilization site (nutL) is shown in green. NutL is also used to recruit Nun. The downstream part of the cassette can be converted to ORF upon insertion of ATG linked to preexisting RBS. ColE1 replication is similar to that of E. coli chromosome. Although Pol I synthesizes the first ~400 nt of the leading strand during the early phase of ColE1 replication, the remainder of the plasmid is replicated by the pol III replisome, as is the E. coli chromosome (del Solar et al., 1998). Thus the DNA polymerase that collides with arrested ECs is pol III, mimicking the natural replication of the chromosome.
(B) Nun-arrested ECs cause DNA breaks, which are prevented by Mfd in instances of co-directional but not in head-on transcription and replication. Sequencing gels demonstrate primer extension analyses of the nontemplate strand in pCODIR (lanes 1–4) and pHDON (lanes 5–8). Where indicated (+) Nun and/or Mfd were expressed. The green bar shows the elements of the λ cassette. Red lines show the positions of DNA lesions. See also Supplemental Figure S2.
(C) Nun-mediated DNA breaks depend on replication. Where indicated (+), the replication inhibitor HU and/or the single strand DNA probe CAA was added. The purple line shows CAA modifications of the nontemplate strand corresponding to queuing transcription bubbles of initiation and elongation complexes.
(D) Nun-mediated DNA breaks depend on transcription. Where indicated cells were shifted to 42°C to induce transcription from pL promoter. Red lines show the positions of DNA lesions.
In the first series of experiments we probed the effect of Nun-arrested ECs on chromosome stability. Wild-type (wt) and Mfd-deficient (Δmfd) cells carrying pCODIR or pHDON plasmids were shifted to 42°C for 1 hr to induce the pL promoter in the presence or absence of HK022 nun+ prophage. Nun-arrested ECs are resistant to Rho-dependent termination and can be cleared from DNA only by the powerful ATP-driven Mfd helicase (Washburn et al., 2003). Primer extension performed on pCODIR and pHDON isolated from Δmfd cells revealed a characteristic pattern of bands that appeared only in the presence of Nun (Figure 1B). The breaks were downstream of nutL, as expected if they were induced by Nun-mediated transcriptional arrest. Because the pattern of breaks was similar for both DNA strands regardless of promoter orientation (Figures 1B and S2A), and the clusters of breaks always overlapped between strands (Figure S2B), we conclude that Nun-arrested ECs caused double strand breaks (DSBs) in both co-directional and head-on orientations. Treatment with the replication inhibitor hydroxyurea (HU) just prior to induction of pL eliminated DSBs (Figure 1C), supporting the idea that DNA damage depended on replication fork collision with arrested ECs. Also, no breaks were detected without promoter induction (Figure 1D), confirming that DSBs required both transcription and replication to occur at the same time.
The location of the DSBs coincided precisely with the positions of the Nun-arrested ECs. The pattern of in situ chloroacetaldehyde (CAA) footprinting, which probes transcription bubbles (Proshkin et al., 2010) was very similar to that of the DSBs (Figure 1C). Taken together these results demonstrate that both co-directional and head-on collisions between Nun-arrested ECs and the replisome induce DSBs. Mfd relieved co-directional DNA damage by releasing Nun-arrested ECs (Figure 1B). In contrast, head-on transcription in the presence of Nun induced equivalent DNA damage in both wt and Δmfd mutant cells. The inability of Mfd in vivo to release Nun-arrested ECs ahead of replisomes approaching head-on contrasts with a recent report indicating that head-on collisions between replication and transcription can be resolved by Mfd in vitro (Pomerantz and O’Donnell, 2010).
Co-directional collisions between spontaneously backtracked ECs and replication forks induce DSBs
The experiments with Nun demonstrate directly that transcriptional arrest leads to DNA damage and validate our experimental system to monitor co-directional and head-on collisions in vivo. To determine whether spontaneously arrested ECs (i.e. ECs arrested in the absence of Nun) also interfere with replication, we assayed for DSBs under conditions that compromise the elongation process. Transcription elongation is frequently interrupted by pauses and arrests. The majority of such interruptions result from RNAP backtracking, the reverse sliding of an EC along DNA and RNA that occurs at many different DNA sequences (Nudler et al., 1997; Komissarova and Kashlev, 1997; Epshtein and Nudler, 2003; Nudler, 2009). During RNAP backtracking, the 3′-OH end of the RNA detaches from the catalytic site and is extruded into the RNAP substrate-binding pore (secondary channel), thereby causing transient or permanent EC inactivation (Nudler et al., 1997; Komissarova and Kashlev, 1997; Wang et al., 2009; Cheung and Cramer, 2011). The bacterial cell employs several mechanisms to minimize backtracking. GreA and GreB elongation factors reactivate backtracked ECs by stimulating a transcript cleavage reaction in the RNAP catalytic site to generate a new 3′-OH terminus (Nudler, 2009; Borukhov et al., 2005). Additionally, we recently showed that active ribosomes enhance the rate of transcription elongation by suppressing RNAP backtracking (Proshkin et al., 2010).
To determine the role of anti-backtracking mechanisms in genome stability, we monitored DSBs downstream of pL in the UTR region of pCODIR and pHDON. No significant DNA lesions were observed in wt cells (Figure 2A, lane 1). However, deletion of greA or greB resulted in a prominent cluster of DSBs ~30 nucleotides downstream of RBSN in pCODIR (Figure 2A, lanes 2 and 3). In contrast, no DSBs were seen in head-on transcription and replication (pHDON; Figure 2A, lanes 5, 6). The lesions depended on both DNA replication and transcription, because they didn’t appear without promoter activation (Figure 2B) and were entirely eliminated by HU (Figure 2C). The breaks occurred in both DNA strands (Figure S2) and were associated with plasmid linearization (Figure 2D, lanes 2 and 3), confirming that they were DSBs.
Figure 2. Anti-backtracking factors eliminate DNA damage associated with co-directional collisions of the replisome with naturally arrested ECs.
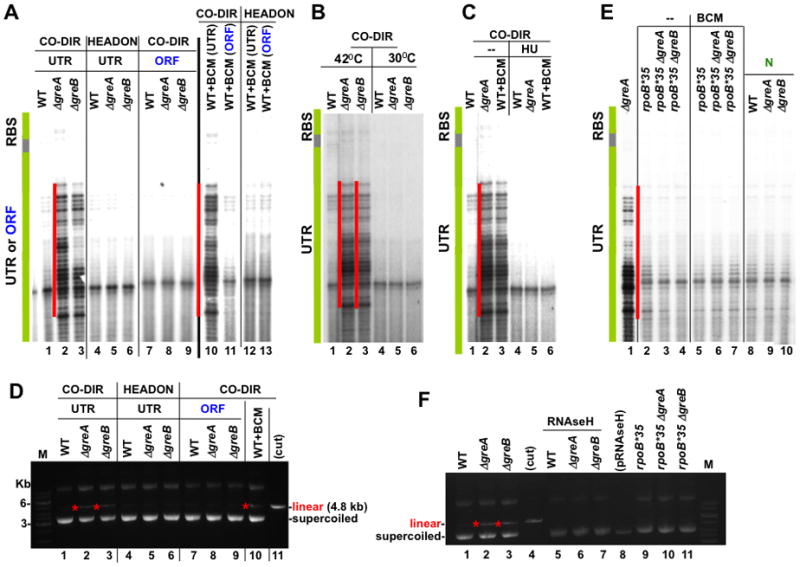
(A) Active ribosomes, GreA, GreB, and Rho protect against DNA breaks. Panels demonstrate primer extension analyses of the non-template strand of pCODIR (lanes 1–3 and 7–11) and pHDON (lanes 4–6 and 12, 13). UTR and ORF specify conditions when translation was prohibited or allowed, respectively, from RBSN. The green bar shows the location of RBSN. The red lines show the positions of major DNA lesions, which appear co-directionally only in the absence of translation and without GreA or GreB (lanes 2 and 3). The same lesions also appear when Rho termination was inhibited by BCM in the absence of translation (lane 10). See also Supplemental Figure S2.
(B) DSBs depend on transcription. Primer extension analysis of the pCODIR plasmids isolated from wild type, ΔgreA and ΔgreB strains shows that DNA lesions do not appear at 30°C, i.e. without pL promoter activation.
(C) DSBs are the result of co-directional collisions between the replisome and naturally arrested ECs. HU inhibition of replication eliminates DSBs (compare lanes 2–4 with 5–7).
(D) The agarose gel analysis of the plasmids used in (A). Red stars indicate linearized species that appear in the absence of translation and without GreA or GreB (lanes 2 and 3) and also when Rho termination was inhibited by BCM in the absence of translation (lane 10).
(E) Backtracking-resistant RNAP mutant rpoB*35 (lanes 2–7) and λN (lanes 8–10) suppress DSBs within the UTR of pCODIR. See also Supplemental Figure S3.
(F) rpoB*35 and RNAse H suppress plasmid linearization associated with Gre deficiency. The agarose gel shows linear and supercoiled pCODIR species. The low copy plasmid expressing RNAse H from the Tac promoter (pRNAseH) was present in pCODIR-transformed cells (lanes 5–7).
Remarkably, the insertion of an ATG linked to RBSN, i.e. the conversion of the UTR to an ORF, eliminated all breaks and plasmid linearization associated with Gre deficiency (Figure 2A, lanes 7–9; Figure 2D, lanes 7–9). We conclude that naturally backtracked ECs cause DSBs in vivo due to co-directional collisions with the replisome. The transcript cleavage factors and actively translating ribosomes suppress RNAP backtracking, thereby preventing DNA damage. Translation dominates this process because no damage was detected in Gre-deficient cells when ribosomes were allowed to initiate translation.
Unlike Nun-arrested ECs, naturally paused or arrested ECs should be sensitive to Rho-dependent termination. Consistent with this idea, addition of the antibiotic bicyclomycin (BCM), which specifically targets Rho (Zwiefka et al., 1993; Cardinale et al., 2008), induced prominent DNA breaks within the UTR. These DSBs were located at the same sites as those observed in Δgre cells (Figure 2A, lane 10) and were also associated with plasmid linearization (Figure 2D, lane 10). BCM-mediated DNA breaks could be detected only in the case of co-directional transcription and replication (Figure 2A, lanes 10–13). HU suppressed the BCM-induced breaks (Figure 2C, lane 6), as did conversion of the UTR to an ORF (Figure 2A, lane 11). Taken together, the results with BCM and Gre-deficient cells indicate that in the absence of translation, Rho and the transcript cleavage factors, together, remove arrested ECs. Failure to remove naturally arrested ECs induces DNA damage upon co-directional collisions with the replisome. Importantly, removal of arrested EC from head-on replication clearly does not depend on these factors.
The transcription bubble of the major arrested EC in Δgre or BCM-treated cells in the presence of HU was located by in situ CAA footprinting. The arrested EC was ~285 nt downstream of the transcription start site, near the beginning of the UTR (Figure 3A, lanes 2 and 3, orange line #1). To confirm that this EC was indeed backtracked, we transcribed a PCR-generated DNA template containing the pL-nutL- RBSN-UTR sequence in vitro using purified His6-tagged E. coli RNAP. After a single round of transcription with a physiological concentration of NTPs, approximately 40% of ECs halted at position +298 (Figure 3B, lane 1). In the presence of GreA or GreB, however, most RNAPs progressed through this major arrest site (Figure 3B, lanes 2 and 3). Because RNAP was immobilized on Co++-chelating beads, we could wash away unincorporated NTPs and then treat EC298 with GreA or GreB. EC298 was highly sensitive to both factors (Figure 3B, lanes 6 and 7). The sizes of the GreB cleavage products were consistent with backtracking of as much as 15 nt (Figures 3B, lane 7 and 3C). These in vitro results confirm that the CAA footprint in Gre-deficient or BCM-treated cells (Figure 3A) corresponds to spontaneously backtracked EC298. Notably, the cluster of major DSB breaks (~18 nt long) begins close to the upstream edge of the DNA bubble formed by fully backtracked EC298 (Figure 3C).
Figure 3. Characterization of ECs that lead to DSBs.
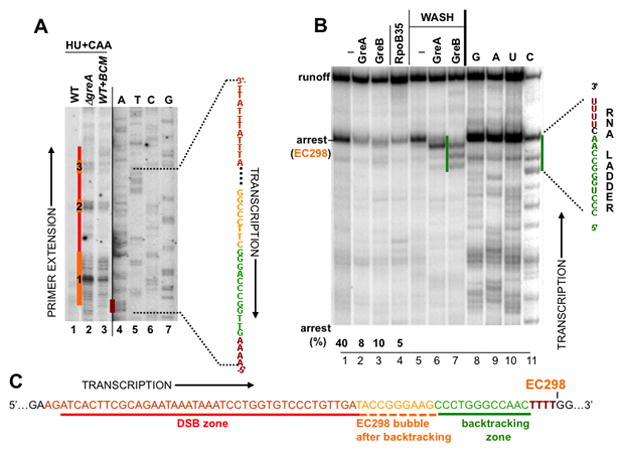
(A) Mapping arrested ECs in vivo. HU + CAA indicates that HU-treated cells were were treated with CAA. The CAA footprint reveals the position of the transcription bubble of EC298, which was spontaneously arrested within the UTR (orange line #1; see B and C) and two other ECs arrayed behind EC298 (orange line #2 and 3). These ECs could be detected only in the absence of either of two Gre factors (lanes 2) or in the presence of BCM (lane 3) (compare to lane 1).
(B) A single-round runoff assay utilizing a PCR-generated λ pL template from pCODIR. Wt (lane 1–3) and RpoB*35 (lane 4) RNAPs were immobilized on metal-chelating beads and chased in the presence or absence of GreA or GreB. The major arrest site at position +298, which was mapped by RNA sequencing, is indicated (lanes 8–11). To determine the sensitivity of EC298 to GreA and GreB and the size of their cleavage products, beads were washed after the chase reaction followed by incubation with GreA/GreB (see Experimental Procedures) (lanes 5–7). The green line shows the size of cleavage products (up to 16 nt) generated by GreB, reflecting the maximum backtracking distance (Nudler et al., 1997).
(C) EC298 positioning before and after backtracking with respect to DSBs detected in vivo. Schematics show the sequence of a UTR segment encompassing major DSBs (red lines in A). The position of the transcription bubble was mapped in vivo (orange line #1 in A). It corresponds to spontaneously backtracked and arrested EC298. The four Ts from which EC298 backtracked (dark red) correspond to the dark red line in (A) (“A” sequencing on the non-template strand).
A mechanistic model of DNA damage due to transcription-replication collisions
DNA lesions resulting from conflicts between replication and transcription are thought to occur because the replisome stalls at stable RNAP complexes and then collapses (Mirkin and Mirkin, 2007; Rudolph et al., 2007; French, 1992; Vilette et al., 1996; Trautinger et al., 2005). RNAP mutants that reduce the frequency of stalled replication forks have been described (McGlynn et al., 2000; Trautinger and Lloyd, 2002). It has been suggested that the mutants form less stable complexes with template DNA, thus decreasing the probability of collisions with replisomes (McGlynn and Lloyd, 2000; Trautinger and Lloyd, 2002). The rpoB*35 mutation (β H1244Q), suppresses the UV sensitivity of ruv and mfd strains and the temperature sensitivity of greA/greB deletion mutants (McGlynn and Lloyd, 2000; Trautinger and Lloyd, 2002). Indeed, we found that rpoB*35 suppressed the formation of DSBs formation in Gre-deficient cells (Figures 2E, lanes 2–4 and 2F, lanes 9–11) as well as in cells treated with BCM (Figure 2E, lanes 5–7). This effect was not due to EC instability, however, since the ECs formed in vitro with RpoB*35 RNAP were as stable as ECs formed with wt RNAP (Figure S3). Instead, RpoB*35 polymerase largely ignored the +298 arrest site (Figure 3B, lane 4). Thus, failure of the mutant polymerase to pause and backtrack explains its ability to suppress the formation of DSBs formation. These results further support the idea that the backtracked state of the EC determines the conflict between replication and transcription with subsequent DNA damage.
Taken together the above observations demonstrate that DSBs result from clashes between the replisome and backtracked RNAP (Figure 4A and S4). Rapid re-annealing of RNA after RNAP has been dislodged by the replisome can generate R-loops (RNA:DNA hybrids) because the DNA nontemplate strand is not readily available to displace RNA after the collision (Figures 4A and S1). Stable R-loops are possible with backtracked ECs, because they carry longer segments of RNA available for re-annealing (the extruded 3′ portion) (Figure 4A). In contrast, active ECs can generate only ~8 bp hybrids (Nudler et al., 1997) that are unstable without RNAP (Figure 4A). Extended R-loops from backtracked ECs provide accessible 3′-OH termini that could serve as primers for DNA synthesis (Figure 4A). Indeed, in vitro experiments show a replisome involved in a co-directional collision with RNAP can restart DNA synthesis using nascent RNA as a replication primer (Pomerantz and O’Donnell, 2008). Such a switch leads to a break in the leading DNA strand, which must be repaired before the next replisome converts it to a DSB (Kuzminov, 1995; Figure 4A). Our results suggest that the repair process may not be sufficiently fast. It may be complicated by a 5′ RNA overhang (Figure 4A) or by approaching ECs, which could obstruct gap filling (Selby and Sancar, 1994). Approaching ECs are suggested by in situ footprinting of RNAP in the ΔgreA mutant (Figure 3A, orange lines #2 and 3). The location and size of the DSB zone (~20 nt) promoter-proximal to the backtracked EC298 supports this model of DSBs formation.
Figure 4. The proposed mechanism of DSB formation as a result of codirectional collisions with backtracked ECs.
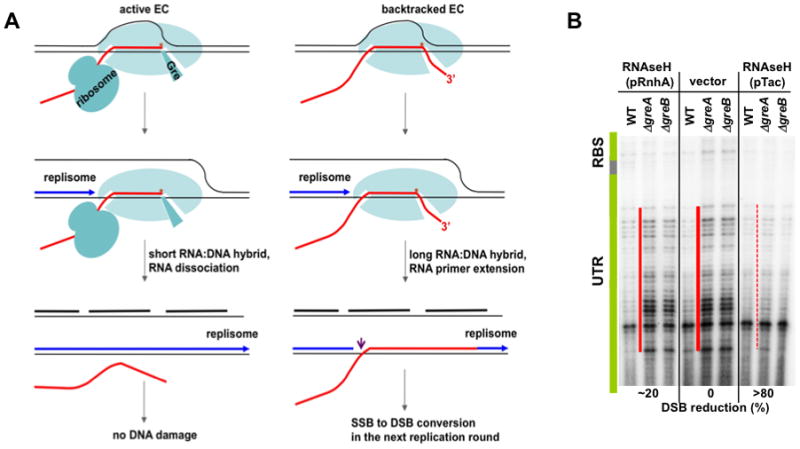
(A) The pink arrow indicates the single strand break (SSB) due to replisome switching from the leading DNA strand (blue) to the RNA primer (red) (Pomerantz and O’Donnell. 2008). Transcript cleavage factor (Gre) is shown bound in the secondary channel (left), through which the RNA 3′-OH end is extruded during backtracking (right). See the text for details.
(B) R-loop-mediated DSBs formation. RNAse H suppresses DSBs in Gre-deficient cells in a dose-dependent manner. The conditions were as in Figure 2A, except that pCODIR-bearing cells were also transformed with a low-copy plasmid expressing RNAse H (rnhA) from its own promoter (lanes 1–3) or the Tac promoter (lanes 7–9). See also Supplemental Figure S4.
To determine whether extended R-loops were indeed the cause of DSBs, we examined the effect of the hybrid-specific RNAse H on backtracking-induced DSBs. Wild-type and Gre-deficient cells bearing pCODIR were transformed with a compatible low copy plasmid expressing RNAse H from its own rnhA promoter or the very strong tac promoter. Highly expressed RNAse H (pTac) almost completely suppressed DSBs detected by both primer extension (Figure 4B, lanes 7–9) and plasmid linearization assays (Figure 2F, lanes 5–7). RNAse H expressed from its own weaker promoter caused only a partial suppression (Figure 4B, lanes 1–3). The dose-dependent effect of RNAse H confirms that R-loops induce DSBs and that cellular RNAse H concentrations are insufficient to eliminate R-loops when anti-backtracking mechanisms are compromised.
In head-on collisions, the replisome cannot prime from a nascent transcript, thus avoiding breaks that result from strand-jumping (Figures S1 and 2A). Nevertheless, head-on collisions are thought to be more deleterious than co-directional collisions (Pomerantz and O’Donnell, 2010). The former are resolved by the dedicated helicases DinG, Rep, and UvrD (Boubakri et al., 2010). These helicases must be efficient, since we observed DSBs in the head-on collision plasmid only when we generated highly stable Nun-arrested ECs (Figure 1). The fact that the seven E. coli ribosomal RNA genes are aligned co-directionally with replication suggests that the helicases, likewise, cannot resolve head-on collisions with antitermination complexes or with tightly arrayed ECs (Brewer, 1988; Blattner et al.,1997; Kunst et al., 1997; Rocha and Danchin, 2003).
Active ribosomes and other anti-backtracking mechanisms assure overall genome stability
To test whether anti-backtracking mechanisms contribute to genome stability, we examined the effects of GreA/GreB, the translation elongation inhibitor chloramphenicol (Cm), and BCM on chromosomal DNA integrity and the SOS response. Sub-lethal concentrations of Cm decelerate ribosomes, reducing their ability to “push” elongating RNAP (Proshkin et al., 2010). In principle, this should increase the probability of RNAP backtracking (Proshkin et al., 2010) and, therefore, DSB formation. Lack of compensatory anti-backtracking mechanisms (Gre and Rho) should further increase the level of DNA damage. The relative frequency of genomic DSBs can be monitored directly by PCR of an arbitrarily chosen segment of the bacterial chromosome. Equal amounts of genomic DNA from wt and mutant cells grown with or without exposure to antibiotics were isolated for PCR. PCR conditions were selected to ensure that the yield of a final product (~10 kB) would be reduced by chromosomal DSBs (Park and Imlay, 2003; Gusarov and Nudler, 2005). Treatment of wt bacteria with a sub-lethal dose of Cm or BCM only slightly decreased the yield of the expected 10 kB PCR product (Figure 5A, lanes 1–3). Similarly, deletion of GreB had little effect on recovery of the PCR product (Figure 5A, lane 4). However, treatment of ΔgreB cells with Cm or BCM substantially reduced the yield of PCR product, indicating extensive DSBs formation (Figure 5A, lanes 5, 6). Overexpression of GreB largely eliminated most of the DNA damage induced in ΔgreB cells exposed to Cm (Figure 5A, lane 7). These results confirm the generality of chromosomal DSB formation induced by RNAP backtracking.
Figure 5. Chromosomal damage as a function of RNAP backtracking.
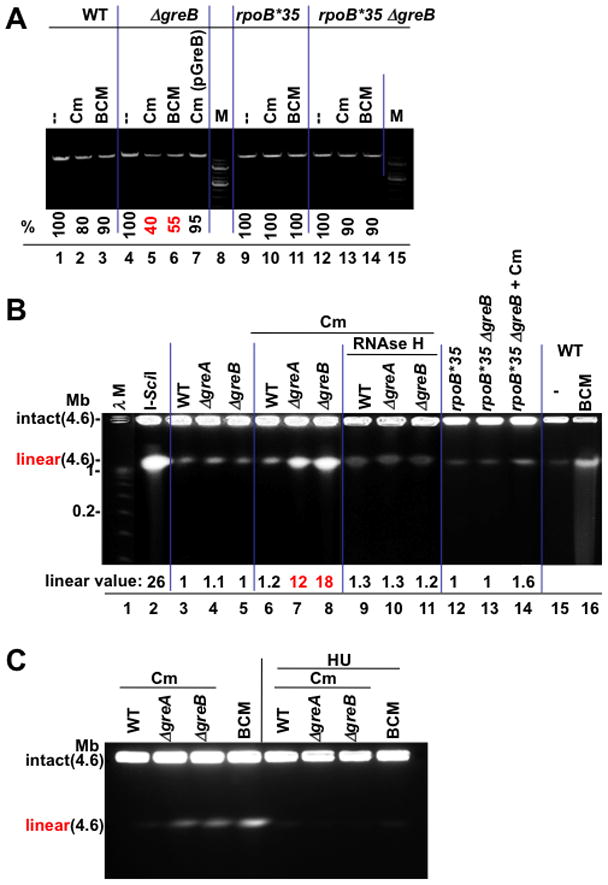
(A) Sub-lethal amounts of the translation inhibitor chloramphenicol (Cm) and BCM produce greater chromosome damage in GreB-deficient cells. The integrity of chromosomal DNA was monitored by PCR. A representative agarose gel shows a 10 kB fragment amplified from equal amounts of genomic DNA isolated from wt (lanes 1–3), ΔgreB (lane 4–6), ΔgreB(pGreB) (lane 7), and rpoB*35 (lanes 9–11), and rpoB*35 ΔgreB (lane 12–14) cells (see Methods). M, 1 kb DNA marker. % indicates the fraction of the full-length PCR products. Values are the average numbers from three experiments with the error margin of less than 5%.
(B) Pulse field gel analysis of chromosomal DSBs; lane 1: λ concatemers from 0.05–1.0 Mb; lane 2: 4.6 Mb linearized E. coli chromosomes (I-SceI); lane 3–5: DNA from wild type (WT) and Gre-deficient cells; lanes 6–8: DNA from wt and Gre-deficient cells after treatment with 4 μg/ml Cm; lanes 9–11: DNA from RNAse H expressing cells after Cm treatment; lanes 12–14: DNA from rpoB*35 and rpoB*35 Gre-deficient cells before and after Cm treatment; lane 16: BCM-treated cells. “Linear value” indicates the fold-increase in linear DNA after Cm or BCM treatment. The values are the average of two or more independent experiments. Note that due to low resolution of PFGE, the species indicated as linear may result from more than one random DSB.
(C) Chromosomal DSBs depend on replication. Pulse field gel analysis of chromosomal DSBs shows that inhibition of replication by HU eliminates all DSBs in wild type and Gre-deficient cells exposed to sub-lethal doses of Cm or BCM.
To corroborate the above results and to extend them to a non-plasmid template, we utilized pulse field gel electrophoresis (PFGE) to directly visualize chromosomal DSBs (Figure 5B). The intact circular E. coli chromosome and its replication and recombination intermediates remain at the origin and do not migrate into the agarose gel (Birren and Lai, 1993; Azvolinsky et al., 2006). Linear chromosomes with a single DSB migrate as 4.6 Mb species (Figure 5B, lane 2). Without antibiotic challenge, DNA isolated from wt and Δgre cells remained almost entirely at the origin (lanes 3–5). However, treatment of cells with a sub-lethal dose of Cm (4 μg/ml) induced significant chromosome linearization in Δgre cells (lanes 6–8). These DSBs required replication, because they could not be detected after HU treatment (Figure 5C). Overexpression of RNAse H suppressed DSB formation, as did the rpoB*35 allele (Figure 5C, lanes 9–14). These results indicate the generality of our model linking backtracked RNAP and DSBs (Figure 4A).
We also evaluated overall genomic damage by monitoring the SOS response with a chromosomal recA-gfp fusion (Kostrzynska et al., 2002). Addition of sub-lethal amounts of Cm to log-phase cells induced significant fluorescence (Figure 6A), indicating that ribosome deceleration yields a SOS response. Induction of RecA by Cm was more pronounced in GreB-deficient cells than in wt cells (Figure 6A). A sub-lethal concentration of BCM (6 μg/ml), which by itself had little effect on RecA induction, enhanced SOS activation by Cm, particularly in GreB-deficient cells (Figure 6A). Finally, GreB overexpression not only suppressed hyper-induction of SOS by Cm in ΔgreB mutants, but, reduced GFP fluorescence below that of untreated wt cells (Figure 6A).
Figure 6. Cell survival under conditions of excessive backtracking depends on error-prone DSBs repair.
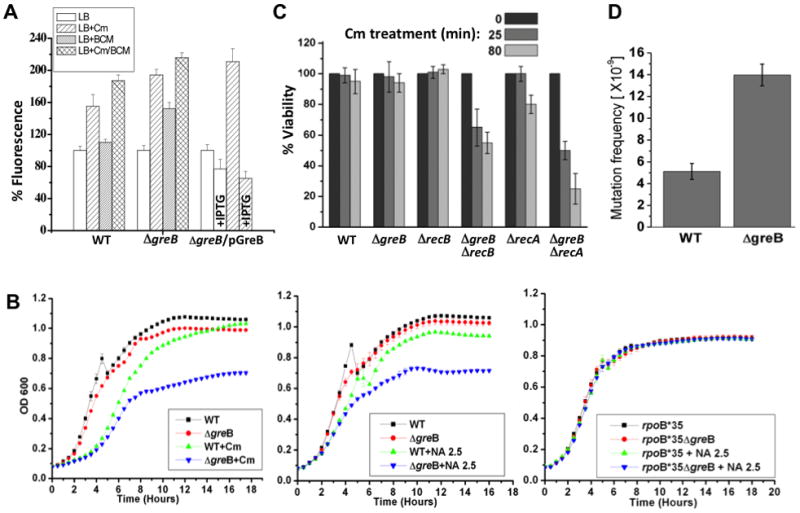
(A) SOS response activation by sub-lethal amounts of Cm and BCM in the presence or absence of GreB. Cm induced fluorescence in recA’::gfp (wt), and both Cm and BCM induced in recA’::gfp ΔgreB. SOS caused by Cm was suppressed by IPTG-induced overexpression of GreB (recA’::gfp ΔgreB(pGreB) cells). In each case, the basal level of fluorescence before induction was taken as 100%. Values are the mean ±SD from four experiments.
(B) GreB-deficient cells are more sensitive to Cm (top) and NA (bottom). Numbers indicate the antibiotic concentrations (μg/ml).
(C) Survival of RNAP backtracking-prone cells depends on the DSBs repair machinery. Exponentially grown wild type and mutant E. coli cells were challenged with Cm (100 μg/ml) for indicated time periods, washed, and then plated for overnight incubation to determine CFUs. The fraction of surviving cells (%) in relation to wild type is shown as the mean ±SD from three independent experiments.
(D) Mutation frequency increases in GreB deficient cells. Wild type and mutant strains grew to OD 0.6 and then spread over LB-agar surface containing 30 μg/ml of rifampicin (rif). Plates were incubated at 30°C for 24 hours to detect RifR colonies. CFU/ml was calculated by spreading serially diluted cultures over LB-agar plates. The estimated mutation frequency is shown as the mean ±SD from three independent experiments.
Taken together these results confirm the importance of translation in the maintenance of chromosome integrity. If translation is inefficient, specific anti-backtracking factors (GreA and GreB) as well as Rho-dependent termination become indispensible to mitigate DNA damage. In fact, ΔgreB mutants were more sensitive to Cm and to the genotoxic agent, nalidixic acid (NA), than were wt cells (Figure 6B). This sensitivity was observed during late stages of growth, when translation is compromised due to nutrient deficiency (Figure 6B). Notably, ΔgreB sensitivity to NA was suppressed in rpoB*35 mutant cells, further implicating EC backtracking in chromosome instability (Figure 6B).
To further address the relationship between RNAP backtracking and genome instability, we generated recA greB and recB greB double deletion strains and examined their viability under conditions of excessive backtracking (inhibition of translation with Cm). Cm is a bacteriostatic antibiotic, i.e. it does not kill bacterial cells within hours of exposure to potentially lethal concentrations, but only arrests their growth. Indeed, neither wt nor single mutants were significantly affected by 25 and 80 min of incubation with Cm. However, both recA greB and recB greB double mutants showed a substantial loss of viability (e.g. more than 70% in the case of recA greB) (Figure 6C). These results demonstrate that failure to repair backtracking-associated DSBs, which depends critically on RecA and RecBCD (Dillingham and Kowalczykowski, 2008), compromises cell survival.
DISCUSSION
The results described above in conjunction with our recent study (Proshkin et al., 2010) argue that actively translating ribosomes play a key role in maintaining genome stability by preventing RNAP backtracking. Because the ribosome is a primary sensor of cellular metabolism and various stresses, these results demonstrate the functional and mechanistic link between growth conditions and genome instability. Nutrient deprivation, proteotoxic stress, and many natural antibiotics inhibit translation, resulting in an increased probability of RNAP backtracking and risk of DSBs. Stress-induced mutagenesis is activated in response to adverse growth conditions, such as starvation or exposure to antibiotics (Galhardo et al., 2007). Such mutagenesis depends on error-prone DSB repair and the SOS response, thereby accelerating adaptation to environmental changes, e.g. acquisition of antibiotic resistance (Galhardo et al., 2007). In this respect, backtracking-induced DSBs may account, at least in part, for stress-driven evolution. Indeed, the mutation rate in backtracking-prone GreB deficient cells is significantly higher than that in wild-type (Figure 6D). Moreover, the probability of backtracking depends on codon usage, since the frequency of rare codons modulates the rate of ribosome movement (Proshkin et al., 2010). Therefore, our model also explains why the mutation rate is significantly higher for rare codons than for common codons (Alff-Steinberger, 2000) and predicts that many mutational hot spots are associated with local decreases in the rate of ribosome transit.
Cells employ multiple mechanisms to prevent excessive RNAP backtracking when translation is inefficient or absent (e.g. within UTRs) (Table 1). Rho factor, which travels with RNAP (Epshtein et. al., 2010), suppresses backtracking by terminating transcription that becomes uncoupled from translation (Richardson, 1991; Roberts et al., 2008; Greive and von Hippel, 2005; Epshtein et. al., 2010). Rho has been recently implicated in genome maintenance (Washburn and Gottesman, 2010). The increased levels of DSBs and SOS induced by BCM in ΔgreB mutants argue that Rho contributes to DNA stability primarily by eliminating backtracked ECs (Figure 5). When Rho is not successful, as, for example, in Nun-arrested ECs or promoter-proximal arrests, cells also rely on the backup termination factor Mfd, which is capable of disrupting (albeit slowly) ECs arrested by several different mechanisms (Roberts et al., 2008; Washburn et al., 2003; Selby and Sancar, 1994; Park et al., 2002).
Table 1.
Anti-backtracking mechanisms that contribute to genome stability
| anti-backtracking factors | conditions |
|---|---|
| active ribosomes | ORFs |
| antitermination complexes (e.g. N, S4) | stable RNA and phage genes |
| “secondary channel” factors (e.g. GreA, GreB) | UTRs, poor translation, starvation |
| termination factors (e.g. Rho, Mfd) | UTRs, poor translation, unusually stable ECs |
Factors studied in this work are underlined (see text for discussion).
An additional backup mechanism employs the transcript cleavage factors GreA and GreB, which suppress DSBs by restarting backtracked ECs (Borukhov et al., 2005) (Figure 2). Finally, DksA has been implicated in replication fork progression and the DNA damage response during starvation (Trautinger et al., 2005; Tehranchi et al., 2010). Like Gre factors, DksA binds RNAP in the secondary channel (Perederina et al., 2004) and could potentially suppress backtracking by plugging the exit for the RNA 3′-OH terminus. DksA may represent an emergency tool to compensate for the lack of efficient translation during starvation, although its ability to bind the EC and prevent backtracking has not been demonstrated.
Genes for stable RNAs – ribosomal and transfer RNA – are the most active in bacteria and are aligned co-directionally with replication (Brewer, 1988; Blattner et al.,1997; Kunst et al., 1997). RNAPs that transcribe those genes have the highest rate of elongation (Condon et al., 1995), implying that they are less susceptible to backtracking. At least two anti-backtracking mechanisms could explain the high elongation rate of these genes. One depends on cooperation between multiple RNAP molecules that suppresses each one’s backtracking (Epshtein and Nudler, 2003). The second mechanism depends on the S4 (“ribosomal”) antitermination complex, which accelerates transcription elongation in vitro and in vivo (Condon et al., 1995). To determine whether processive antitermination contributes to genome stability, we expressed λN protein in cells bearing pCODIR. Like the S4 antitermination system, λN accelerates transcription elongation (Rees et al., 1997). Importantly, induction of λN abolished DSBs within the UTR in GreA-deficient cells (Figure 2E, lanes 8–10). This provides the first evidence that λN can suppress EC backtracking. The similarities between the λN and S4 antitermination systems (Torres et al, 2001; Greive et al., 2005) suggests that the antitermination complex protects stable RNA genes from potential DNA damage associated with co-directional transcription/replication collisions.
In summary, this study demonstrates that spontaneous backtracking of RNAP in vivo leads to DNA damage due to co-directional collision with the replisome. Active ribosomes and multiple transcription elongation and termination factors decrease the probability of backtracking events, thereby maintaining genome stability. Because the basic structural organization of cellular replisomes and RNAPs is preserved in evolution (Yao and O’Donnell, 2010; Kettenberger et al., 2004; Nudler, 2009), and backtracking is a ubiquitous phenomenon (Nudler et al., 1997; Komissarova and Kashlev, 1997; Epshtein and Nudler, 2003; Nudler, 2009; Sigurdsson et al., 2010), the mechanism of transcription-driven genome instability reported here for E. coli is likely to be applicable to other organisms as well.
EXPERIMENTAL PROCEDURES
Strains
Strains, 7723 (WT), 28357 (WT), 9939 (=7723 with HK022 prophage), 9199 (=Δmfd) and 9948 (=9199 with HK022 prophage), 32645 (=28357, ΔgreA), 32646 (=28357, ΔgreB), 10989 (=28357, pNAS200), 10990 (=32645, pNAS200), 10991 (=32646, pNAS200) were transformed with pCODIR or pHDON plasmids for in situ DNA breaks and footprint studies. Strains, 10970 (=28357, rpoB*35), 10972 (=32646, rpoB*35) were constructed by P1 transduction and transformed with pCODIR or pHDON plasmids (see Supplemental Experimental Procedures). Similarly, 10973 (=28357, recA:gfp), 10975 (=32646, recA:gfp) strains were constructed by P1 transduction to study the SOS response (Kostrzynska et al., 2002). Strain, SMR8476 (=MG1655 codA21::miniTn7kan(I-SceI) was used to prepare linearized chromosome by I-SceI endonuclease (Pennington and Rosenberg, 2007).
Plasmid preparation, primer extension, and in situ DNA footprinting
E. coli strains bearing either pCODIR or pHDON were grown in M9 minimal medium at 30°C until OD600 =0.6. Then cells were incubated at 42°C for an hour to de-repress the pL promoter. After heat induction, cells were harvested by centrifugation at 7000 rpm for 2 min at 4°C. Plasmids were isolated using a Qiagen Miniprep Kit following the manufacturer’s protocol. BCM (100 μg/ml) and HU (3.8 mg/ml) were added to the cultures 0 min and 25 min before the heat induction, respectively. Primer extension was performed with [32P]-labeled primers as described in Supplementary Experimental Procedures.
Footprinting was performed using 3% CAA as described (Proshkin et al., 2010). After 1 hour of heat induction, CAA was added for 5 min with shaking. Cells were harvested and washed once with a volume of M9 media equal to the volume of the culture media. Plasmids were isolated immediately as described above, and primer extension was performed, also as described above.
DNA sequencing ladders were generated using Sequenase Version 2.0 DNA Sequencing Kit (USB). To detect linearized plasmid bands, 250 ng of plasmid DNA was loaded onto a 1% agarose gel and resolved with ethidium bromide staining.
Generation of growth curves
Growth curves were obtained on a Bioscreen C automated growth analysis system. Subcultures of specified strains were grown until OD~0.8 in M9 minimal medium at 30°C. 10 μl of cultures were diluted in 1 ml ice cold M9 minimal media with appropriate antibiotics or reagents as described in the text or figure legends. 200 μl of each mixture was pippetted into the honeycomb wells in triplicate and grown at 37°C with shaking on the platform of the Bioscreen C instrument. OD values were recorded automatically at different times and plotted.
Assay of chromosomal DNA damage
The assay was done as described previously (Gusarov a, 2005). Cells were grown to OD = 1.0 in M9 minimal media and treated with Cm (10 μg/ml) or BCM (25 μg/ml) for 4 hours. Cells were equalized to OD600=1.0 by addition of M9 minimal media and the cells from 2 ml of the equalized cultures were harvested by centrifugation. Total genomic DNA was isolated from the bacteria pellets with the Qiagen Kit. DNA was extracted with phenol/chloroform and quantified with the PicoGreen dsDNA quantitation reagent (Molecular Probes) and λ phage DNA as a standard. An arbitrarily chosen 10-kb fragment was used for qPCR. Primer sequences were as follows: 5′-TTCCATTGGGATGTAGATGCTG-3′ (forward) and 5′-GGTAAAAGAGTCAAGGGAAGAACC-3′ (reverse). PCR was performed with Phusion DNA polymerase (Finnzymes). The 50-μl PCR mixture contained 5 ng of genomic DNA as a template, 1.5 μM primers, 200 μM dNTPs (Fermentas), 5μl Phusion GC PCR buffer, and 0.5 μl of DNA polymerase. DNA was subjected to 23 cycles of PCR (98°C for 30 s, 53°C for 30 s, 72°C for 9 min). PCR products were separated by electrophoresis in an 0.8% agarose gel, stained with ethidium bromide, scanned, and quantified with an AlphaImager (Imgen Technologies).
Pulse Field Gel Electrophoresis (PFGE)
E. coli cells were grown until OD600~0.8 at 32°C. 4 μg/ml of Cm or 100 μg/ml BCM were added as indicated and cells were allowed to grow at 42°C for 4 more hours. The agarose plugs were prepared using 3×108 cells/plug and treated with lysozyme and Proteinase K according to the BioRad CHEF protocol. Linearized 4.6 Mb E. coli chromosomal DNA marker was made by digesting the genomic DNA plug of SMR8476 strain with I-SceI endonuclease. DNA fragments were separated on a 1% agarose gel in 0.5x TBE at 14°C for 24 hours at 6 V/cm using a BioRad CHEF-DR II angle system with a 2.8–26.3 second linear switch time ramp. Gels were stained with EtBr and visualized with UV-trans-illumination. DNA was quantified (integrated density of the linear products) using ImageJ software.
Assays of the SOS response
Overnight cultures of strains 10973 and 10975 harboring a recA’-gfp chromosomal fusion (Kostrzynska et al., 2002) were grown in Luria broth with kanamycin (20 μg/ml). The cultures were diluted in LB to OD600 = 0.1 and grown at 37°C to an OD ~1.0. Cm (3 μg/ml) or BCM (10 μg/ml) were added at OD600 = 0.5. Fluorescence was measured (Ex. wave length: 480 nm and Em. wavelength: 520 nm) in a Perkin Elmer LS55 spectrofluorimeter. Fluorescence values were normalized to OD values and plotted.
Cell viability and mutagenesis assay
E. coli cells were grown in M9 minimal media to the mid log phase. Cm was added to 100 μg/ml for indicated time intervals. A cell aliquot was precipitated, washed, resuspended and then serially diluted in M9 media, and plated on LB agar for overnight incubation at 37°C. CFUs were counted and normalized against that of untreated cells.
To determine the mutation frequency, the wild type and GreB deficient cells were grown to OD ~0.6 at 30°C and then shifted at 42°C for 2 hours. Cells were directly spread over LB-agar surface containing 30 μg/ml rifampicin and incubated at 30°C for 24 hours. RifR colonies were counted. Number of CFUs/per ml culture was calculated by spreading serially diluted cultures over LB-agar plates.
In vitro transcription
His6-tagged wild type and RpoB*35 (H1244Q) RNAPs were purified and immobilized on Talon Co2+ NTA affinity beads (Clontech) as described (Nudler et al., 2003). The pL-nutL-rIII-RBS-UTR template was generated by PCR using DD41 (5′-TCACCTA CCAAACAATGC-3′) and DD28 primers. 20 nM of template and 20 nM of RNAP holoenzyme were mixed in TB50 [40 mM Tris*HCl pH=8.0; 10 mM MgCl2; 50 mM NaCl] containing ApUpC (10 μM, Oligos Etc.) ATP, GTP (25 μM, each) and 2 μL [α-P32]CTP (3000Ci/mmol; NEN Life Sciences Products). The samples were incubated for 10 min at 37°C to form the initial radiolabeled EC15. 50 nM of GreA or GreB protein was added where indicated. The reaction was chased with unlabeled rNTPs (250 μM, each) for 3 min. In another experiment, EC15 was immobilized on 20 μl Talon Co2+ beads and chased as described above. The beads were washed 4 times with TB followed by addition of GreA or GreB for 3 min at 37°C.
To assess the stability of ECs (Figure S3), the immobilized radiolabeled EC32 was prepared on the T7A1-tR2 template as described (Nudler et al., 2003). Briefly, His6-tagged wild type or RpoB*35 RNAP (~2 pmol) was mixed with a 2-fold molar excess of DNA in 20 μl of TB50 (10 mM MgCl2, 40 mM Tris-HCl, pH 7.9, 50 mM KCl) for 5 min at 37° followed by addition of ApUpC (10 μM), GTP and ATP (25 μM) for 7 min. Next, 5 μl TB50-equilibrated Talon Co++ bead suspension were added for 5 min at room temperature followed by 2 washes with 1.5 ml of TB200 (200 mM KCl). ATP, GTP (5 μM) and 1 μl of [α-P32] CTP (3000 Ci/mmol) were then added for 5 min at room temperature followed by CTP (5 μM) for another 2 min. To measure the dissociation rate of EC32 and readthrough ECs, beads were incubated in TB1000 (1 M NaCl, 40 mM Tris HCl pH 8.0, 10 mM MgCl2) followed by 2 washes. The efficiency of dissociation (%) was calculated by comparing the total amount of radioactivity in the EC32 band and all read-through bands after the TB1000 washes before the TB1000 incubation (taken as 100%).
The RNA sequencing ladder was prepared using wt RNAP and 25 μM of NTPs doped with any of the four 3′ dNTPs (3′dGTP, 3′dATP, 3′dUTP or 3′dCTP) at 10:1 ratio in four different transcription reactions for 10 min.
All reactions were stopped by addition of an equal volume of stop buffer (SB: 8M Urea, TBE buffer, 20 mM EDTA, 0.125% bromophenol blue, 0.125% xylene cyanol). The samples were loaded in a 6% Urea-PAGE gel after preheating at 90°C for 3 min.
Supplementary Material
Acknowledgments
We thank Yuri Nedyalkov, Linqi Wang, Catherine Potenski, Grigory Mogilnitskiy, and Robert Washburn for technical assistance, Lynn Thomason for plasmid pCODIR (pHW27), Susan Rosenberg for SMR8476 strain, and Hanna Klein and Danny Reinberg for critical reading of the manuscript. This work was supported by grants from the NIH R01 GM58750 (E.N.) and R01 GM32719 (M.E.G.). We also thank Timur Artemyev and BGRF for continuous support.
Footnotes
Supplemental information includes four figures and can be found with this article online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alff-Steinberger C. Comparative study of mutations in Escherichia coli and Salmonella typhimurium shows that codon conservation is strongly correlated with codon usage. J Theor Biol. 2000;206:307–311. doi: 10.1006/jtbi.2000.2126. [DOI] [PubMed] [Google Scholar]
- Azvolinsky A, Dunaway S, Torres JZ, Bessler JB, Zakian VA. The S. cerevisiae Rrm3p DNA helicase moves with the replication fork and affects replication of all yeast chromosomes. Genes Dev. 2006;20:3104–3116. doi: 10.1101/gad.1478906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birren B, Lai E. Pulse Field Gel Electrophoresis, a Practical Guide. Academic Press; New York: 1993. [Google Scholar]
- Blattner FR, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Borukhov S, Lee J, Laptenko O. Bacterial transcription elongation factors: new insights into molecular mechanism of action. Mol Microbiol. 2005;55:1315–1324. doi: 10.1111/j.1365-2958.2004.04481.x. [DOI] [PubMed] [Google Scholar]
- Boubakri H, de Septenville AL, Viguera E, Michel B. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J. 2010;29:145–57. doi: 10.1038/emboj.2009.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer BJ. When polymerases collide: replication and the transcriptional organization of the E. coli chromosome. Cell. 1988;53:679–686. doi: 10.1016/0092-8674(88)90086-4. [DOI] [PubMed] [Google Scholar]
- Cardinale CJ, Washburn RS, Tadigotla VR, Brown LM, Gottesman ME, Nudler E. Termination factor Rho and its cofactors NusA and NusG silence foreign DNA in E. coli. Science. 2008;320:935–938. doi: 10.1126/science.1152763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung AC, Cramer P. Structural basis of RNA polymerase II backtracking, arrest and reactivation. Nature. 2011;471:249–253. doi: 10.1038/nature09785. [DOI] [PubMed] [Google Scholar]
- Condon C, Squires C, Squires CL. Control of rRNA transcription in Escherichia coli. Microbiol Rev. 1995;59:623–645. doi: 10.1128/mr.59.4.623-645.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Solar G, Giraldo R, Ruiz-Echevarría MJ, Espinosa M, Díaz-Orejas R. Replication and control of circular bacterial plasmids. Microbiol Mol Biol Rev. 1998;62:434–464. doi: 10.1128/mmbr.62.2.434-464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande AM, Newlon CS. DNA replication fork pause sites dependent on transcription. Science. 1996;272:1030–1033. doi: 10.1126/science.272.5264.1030. [DOI] [PubMed] [Google Scholar]
- Dillingham MS, Kowalczykowski SC. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol Mol Biol Rev. 2008;72:642–671. doi: 10.1128/MMBR.00020-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epshtein V, Dutta D, Wade J, Nudler E. An allosteric mechanism of Rho-depended transcription termination. Nature. 2010;463:245–249. doi: 10.1038/nature08669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epshtein V, Nudler E. Cooperation between RNA polymerase molecules in transcription elongation. Science. 2003;300:801–805. doi: 10.1126/science.1083219. [DOI] [PubMed] [Google Scholar]
- French S. Consequences of replication fork movement through transcription units in vivo. Science. 1992;258:1362–1365. doi: 10.1126/science.1455232. [DOI] [PubMed] [Google Scholar]
- Galhardo RS, Hastings PJ, Rosenberg SM. Mutation as a stress response and the regulation of evolvability. Crit Rev Biochem Mol Biol. 2007;42:399–435. doi: 10.1080/10409230701648502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greive SJ, Lins AF, von Hippel PH. Assembly of an RNA-protein complex. Binding of NusB and NusE (S10) proteins to boxA RNA nucleates the formation of the antitermination complex involved in controlling rRNA transcription in Escherichia coli. J Biol Chem. 2005;280:36397–36408. doi: 10.1074/jbc.M507146200. [DOI] [PubMed] [Google Scholar]
- Gusarov I, Nudler E. NO-mediated cytoprotection: instant adaptation to oxidative stress in bacteria. Proc Natl Acad Sci USA. 2005;102:13855–13860. doi: 10.1073/pnas.0504307102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenberger H, Armache K, Cramer P. Complete RNA polymerase II elongation complex structure and its interactions with NTP and TFIIS. Mol Cell. 2004;16:955–965. doi: 10.1016/j.molcel.2004.11.040. [DOI] [PubMed] [Google Scholar]
- Komissarova N, Kashlev M. RNA polymerase switches between inactivated and activated states by translocating back and forth along the DNA and the RNA. J Biol Chem. 1997;272:15329–15338. doi: 10.1074/jbc.272.24.15329. [DOI] [PubMed] [Google Scholar]
- Kornberg A, Baker T. DNA replication. 2. W. H. Freeman and Co; New York, NY: 1992. [Google Scholar]
- Kostrzynska M, Leung KT, Lee H, Trevors JT. Green fluorescent protein-based biosensor for detecting SOS-inducing activity of genotoxic compounds. J Microbiol Methods. 2002;48:43–51. doi: 10.1016/s0167-7012(01)00335-9. [DOI] [PubMed] [Google Scholar]
- Kunst F, et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- Kuzminov A. Collapse and repair of replication forks in Escherichia coli. Mol Microbiol. 1995;16:373–384. doi: 10.1111/j.1365-2958.1995.tb02403.x. [DOI] [PubMed] [Google Scholar]
- McGlynn P, Lloyd RG. Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell. 2000;101:35–45. doi: 10.1016/S0092-8674(00)80621-2. [DOI] [PubMed] [Google Scholar]
- Merrikh H, Machón C, Grainger WH, Grossman AD, Soultanas P. Co-directional replication-transcription conflicts lead to replication restart. Nature. 2011;470:554–557. doi: 10.1038/nature09758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin EV, Mirkin SM. Replication fork stalling at natural impediments. Microbiol Mol Biol Rev. 2007;71:13–35. doi: 10.1128/MMBR.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudler E. RNA polymerase active center: the molecular engine of transcription. Annu Rev Biochem. 2009;78:335–361. doi: 10.1146/annurev.biochem.76.052705.164655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudler E, Gusarov I, Bar-Nahum G. Methods of walking with the RNA polymerase. Methods Enzymol. 2003;371:160–169. doi: 10.1016/S0076-6879(03)71011-8. [DOI] [PubMed] [Google Scholar]
- Nudler E, Mustaev A, Lukhtanov E, Goldfarb A. The RNA/DNA hybrid maintains the register of transcription by preventing backtracking of RNA polymerase. Cell. 1997;89:33–41. doi: 10.1016/s0092-8674(00)80180-4. [DOI] [PubMed] [Google Scholar]
- Park JS, Marr MT, Roberts JW. E. coli Transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell. 2002;109:757–767. doi: 10.1016/s0092-8674(02)00769-9. [DOI] [PubMed] [Google Scholar]
- Park S, Imlay JA. High levels of intracellular cysteine promote oxidative DNA damage by driving the fenton reaction. J Bacteriol. 2003;185:1942–1950. doi: 10.1128/JB.185.6.1942-1950.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington JM, Rosenberg SM. Spontaneous DNA breakage in single living Escherichia coli cells. Nat Genet. 2007;39:797–802. doi: 10.1038/ng2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perederina A, Svetlov V, Vassylyeva MN, Tahirov TH, Yokoyama S, Artsimovitch I, Vassylyev DG. Regulation through the secondary channel–structural framework for ppGpp-DksA synergism during transcription. Cell. 2004;118:297–309. doi: 10.1016/j.cell.2004.06.030. [DOI] [PubMed] [Google Scholar]
- Pomerantz RT, O’Donnell M. Direct restart of a replication fork stalled by a head-on RNA polymerase. Science. 2010;327:590–592. doi: 10.1126/science.1179595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz RT, O’Donnell M. The replisome uses mRNA as a primer after colliding with RNA polymerase. Nature. 2008;456:762–766. doi: 10.1038/nature07527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado F, Aguilera A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 2005;24:1267–1276. doi: 10.1038/sj.emboj.7600602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proshkin S, Rahmouni AR, Mironov A, Nudler E. Cooperation between translating ribosomes and RNA polymerase in transcription elongation. Science. 2010;328:504–508. doi: 10.1126/science.1184939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees WA, Weitzel SE, Das A, von Hippel PH. Regulation of the elongation-termination decision at intrinsic terminators by antitermination protein N of phage lambda. J Mol Biol. 1997;273:797–813. doi: 10.1006/jmbi.1997.1327. [DOI] [PubMed] [Google Scholar]
- Richardson JP. Preventing the synthesis of unused transcripts by Rho factor. Cell. 1991;64:1047–1049. doi: 10.1016/0092-8674(91)90257-y. [DOI] [PubMed] [Google Scholar]
- Roberts JW, Shankar S, Filter JJ. RNA polymerase elongation factors. Annu Rev Microbiol. 2008;62:211–233. doi: 10.1146/annurev.micro.61.080706.093422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EP, Danchin A. Essentiality, not expressiveness, drives gene-strand bias in bacteria. Nat Genet. 2003;34:377–378. doi: 10.1038/ng1209. [DOI] [PubMed] [Google Scholar]
- Rudolph CJ, Dhillon P, Moore T, Lloyd RG. Avoiding and resolving conflicts between DNA replication and transcription. DNA Repair (Amst) 2007;6:981–993. doi: 10.1016/j.dnarep.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Selby CP, Sancar A. Mechanisms of transcription-repair coupling and mutation frequency decline. Microbiol Rev. 1994;58:317–329. doi: 10.1128/mr.58.3.317-329.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson S, Dirac-Svejstrup AB, Svejstrup JQ. Evidence that transcript cleavage is essential for RNA polymerase II transcription and cell viability. Mol Cell. 2010;38:202–210. doi: 10.1016/j.molcel.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tehranchi AK, Blankschien MD, Zhang Y, Halliday JA, Srivatsan A, Peng J, Herman C, Wang JD. The transcription factor DksA prevents conflicts between DNA replication and transcription machinery. Cell. 2010;141:595–605. doi: 10.1016/j.cell.2010.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres M, Condon C, Balada JM, Squires C, Squires CL. Ribosomal protein S4 is a transcription factor with properties remarkably similar to NusA, a protein involved in both non-ribosomal and ribosomal RNA antitermination. EMBO J. 2001;20:3811–3820. doi: 10.1093/emboj/20.14.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautinger BW, Lloyd RG. Modulation of DNA repair by mutations flanking the DNA channel through RNA polymerase. EMBO J. 2002;21(24):6944–6953. doi: 10.1093/emboj/cdf654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautinger BW, Jaktaji RP, Rusakova E, Lloyd RG. RNA polymerase modulators and DNA repair activities resolve con icts between DNA replication and transcription. Mol Cell. 2005;19:247–258. doi: 10.1016/j.molcel.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Vilette D, Ehrlich SD, Michel B. Transcription-induced deletions in plasmid vectors: M13 DNA replication as a source of instability. Mol Gen Genet. 1996;252:398–403. doi: 10.1007/BF02173004. [DOI] [PubMed] [Google Scholar]
- Wang JD, Berkmen MB, Grossman AD. Genome-wide coorientation of replication and transcription reduces adverse effects on replication in Bacillus subtilis. Proc Natl Acad Sci USA. 2007;104:5608–5613. doi: 10.1073/pnas.0608999104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Bushnell DA, Huang X, Westover KD, Levitt M, Kornberg RD. Structural basis of transcription: backtracked RNA polymerase II at 3.4 angstrom resolution. Science. 2009;324:1203–1206. doi: 10.1126/science.1168729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn RS, Gottesman ME. Transcription termination maintains chromosome integrity. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.1009564108. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn RS, Wang Y, Gottesman ME. Role of E. coli transcription-repair coupling factor Mfd in Nun-mediated transcription termination. J Mol Biol. 2003;329:655–662. doi: 10.1016/s0022-2836(03)00465-0. [DOI] [PubMed] [Google Scholar]
- Yao NY, O’Donnell M. SnapShot: The Replisome. Cell. 2010;141:1088–1088. doi: 10.1016/j.cell.2010.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwiefka A, Kohn H, Widger WR. Transcription termination factor Rho: the site of bicyclomycin inhibition in Escherichia coli. Biochemistry. 1993;32:3564–3570. doi: 10.1021/bi00065a007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.