

Abstract
Background
Anopheles lesteri is a primary vector of Plasmodium spp. in central China. A complete understanding of vector population structure and the processes responsible for the differentiation is important to the vector-based malaria control programmes and for identifying heterogeneity in disease transmission as a result of discrete vector populations. There is no adequate An. lesteri population genetic data available.
Methods
Polymorphism of sequence variations in mitochondrial COII and Cytb genes were assessed to explore the level of genetic variability and differentiation among six populations of An. lesteri from China.
Results
There were 30 (4.37%) and 21 (5.33%) polymorphic sites for mtDNA-COII and Cytb gene, respectively. Totally 31 COII and 30 Cytb haplotypes were obtained. The range of FST values was from 0.101 to 0.655 by mtDNA-COII, and 0.029 to 0.231 by Cytb gene. The analysis of molecular variance (AMOVA) showed that the percentage of variation within populations (65.83%, 88.48%) was greater than that among populations (34.17%, 11.52%) using both genes. The Tajima's D and Fu's Fs values were all negative, except Tajima's D values of YN and HNB populations, which suggest a large number of low-frequency mutations in populations and the populations were in expansion proceeding.
Conclusions
Levels of genetic variation within An. lesteri populations were higher than among them. While these results may suggest considerable levels of gene flow, other explanations, such as the effect of historical population perturbations can also be hypothesized.
Background
Anopheles lesteri, which belongs to the Hyrcanus group of the genus Anopheles is a primary vector of malaria in central China [1]. Genetically-based methods have been proposed for malaria vector control. These methods focus mainly in altering vectorial capacity through the genetic modification of natural vector populations by means of introducing refractoriness genes or by sterile insect technologies [2]. Knowledge of the genetic structure of vector species is, therefore, an essential requirement as it should contribute not only to predict the spread of genes of interest, such as insecticide resistance or refractory genes, but also to identify heterogeneities in disease transmission due to distinct vector populations [3]. A complete understanding of vector population structure and the processes responsible for the distribution of differentiation is important to vector-based malaria control programmes and for identifying heterogeneity in disease transmission as a result of discrete vector populations [4]. Susceptibility to Plasmodium infection, survival and reproductive rates, degree of anthropophily, and the epidemiology of malaria in the human host may all be affected by genetic variation in vector populations [5].
Anopheles lesteri is almost morphologically undistinguishable from its sibling species because of lacking the objective and stable identification characters, so the taxonomic status on An. lesteri in China has revised many times. Xu and Feng [6] regarded the Chinese "An. lesteri" as a new subspecies An. lesteri anthropophagus because it was distinct from both An. lesteri lesteri from the Philippines and An. lesteri paraliae [7] from Malaysia in bionomics as well as morphology. The subspecies was later elevated to a full species rank [8]. However, the second internal transcribed spacer (ITS2) of ribosomal DNA (rDNA) of An. anthropophagus in China was similar to that of An. lesteri from the Philippines, South Korea, Guam and Japan [9,10]. The molecular evidence strongly support that An. anthropophagus is the synonym of An. lesteri.
Anopheles lesteri exhibits variation in ecology [11], morphology [12], chromosomes [12], and random amplified polymorphic DNA (RAPD) markers [13]. Furthermore, An. lesteri was not considered as malaria vector in Guam and Philippines, but had high transmission capacity of malaria in central China [11,14], and a certain transmission capacity in South Korea and Japan [15,16]. Despite its significance in malaria transmission, only a few studies on population genetics have been conducted [13]. Many genes of mtDNA were used to analyse the genetic variation and population structure of the Anopheline mosquitoes, such as cytochrome subunit I (COI) [17-20], cytochrome subunit II (COII) [21,22], control region [23], NADH dehydrogenase subunit 4 [24] and subunit 5 [4,25-29]. The present study aimed to estimate genetic variability and population structure and to infer the extent of gene flow among An. lesteri populations from China based on mtDNA-COII and cytochrome B (Cytb) genes sequences.
Methods
Mosquito collections and species identification
Wild adult An. lesteri were collected from 2004 to 2007, by using indoor light traps and human landing catches at human living room and livestock corrals. The eight collection sites in China were located from 22°17'N to 39°58'N, and 103°29'E to 123°50'E (Table 1 Figure 1). The HNB and YN populations consisted of specimens pools from two or three sites in proximity to each other, as stated in Table 1. The distances between sites were below 50 km. There were total five field populations and a laboratory colony, with JS population in this study.
Table 1.
The collecting data of Anopheles lesteri mosquito populations in this study
| Population Code | Collecting site | Collecting date | Sample size | Latitude (N) | Longitude (E) |
|---|---|---|---|---|---|
| GD | Zhuhai, Guangdong | Oct. 2007 | 22 | 22°17' | 113°30' |
| YN | Yanjing, Yunnan | June 2006 | 9 | 28°60' | 104°13' |
| Junlian, Sichuan | June 2006 | 4 | 28°10' | 104°34' | |
| SC | Pujiang, Sichuan | June 2006 | 23 | 30°14' | 103°29' |
| HNB | Guangshui & Suizhou, Hubei | June 2007 | 8 | 31°41'-31°52' | 113°15'-113°47' |
| Tongbai, Heinan | June 2007 | 5 | 32°29' | 113°23' | |
| JS* | Xuyi, Jiangsu | June 1985 | 17 | 32°54' | 118°34' |
| LN | Donggang, Liaoning | June 2004 | 28 | 39°58' | 123°50' |
* The collecting site and date of laboratory colony were original information. The mosquitoes were kept at 26 ± 1°C and 65 ± 5% (RH), under a 12: 12 hr (light: dark) photoperiod.
Figure 1.
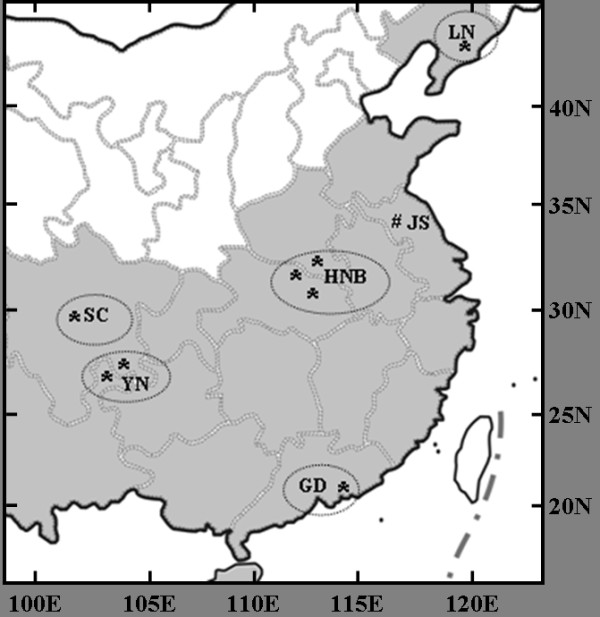
Localities of collections for populations of Anopheles lesteri in China. The places in grey indicate the distribution areas. The five field populations are enclosed in separate circles. The well code was the original collection site of the JS lab colony.
Adult mosquitoes of An. hyrcanus group were identified by morphology using the identification keys of Lu et al [14]. Specimens were kept individually in silica gel filled tubes at 4°C, until DNA extraction was performed according to Collins et al [30]. Anopheles lesteri species identification was done by a PCR assay based on rDNA-ITS2 markers previously described in Ma et al [31].
mtDNA-COII and Cytb genes amplification and sequencing
Sequence variation was examined in the mtDNA-COII and the Cytb genes. The COII and Cytb regions were amplified in 50 μL reaction mixtures containing 1 × reaction buffer (QIAGEN, Courtaboeuf, France), 0.1 mM of each dNTP (Eurogentec, Angers, France), 1 unit of Taq DNA polymerase, 0.1 μM each of the forward and reverse primers and 1.5 μL genomic DNA. The COII gene was amplified using primers COIIF (5'- TCT AAT ATG GCA GAT TAG TGC A -3', forward) and COIIR (5'- ACT TGC TTT CAG TCA TCT AAT G -3', reverse), and the Cytb gene using primers CytbF (5'- GGA CAA ATA TCA TTT TGA GGA GCA ACA G-3', forward) and CytbR (5'- ATT ACT CCT CCT AGC TTA TTA GGA ATT G -3', reverse). The cycle conditions in PTC-100 Peltier Thermal Cycler included an initial denaturation step at 94°C for 2 min, followed by 30 cycles at 94°C for 30 s, 50°C for 30 s and 72°C for 30 s, with a final extension step at 72°C for 8 min. After electrophoresis, PCR products were purified and used for sequencing in both directions with the previous primers, on an ABI 3730 automatic sequencer (Applied Biosystems). Sequences were inspected and corrected, where necessary, using SEQSCAPE software (Applied Biosystems).
Data analyses
Multiple sequence alignments for each gene were performed using MEGA 4.0 [32] and CLUSTAL × [33]. The sequences polymorphism was assessed with MEGA 4.0. A haplotype networks and outgroup probability of the haplotypes were constructed based on statistical parsimony using TCS 1.21 [34].
The parameters θπ equivalent to the average pairwise number of differences between sequences [35], θs equivalent to the number of segregating nucleotide sites per sequence [36], and haplotypes diversity (h) were estimated for COII and Cytb polymorphism within populations. The population genetic structure was analysed with 5 field populations, and assessed by analyzing molecular variance with ARLEQUIN 3.11 [37]. The percentage of sequence divergence within and between populations was calculated based on Nei and Li [38], and pairwise FST values for short-term genetic distance between populations were estimated with the methods of Slatkin (1995) [39] and tested for significance by permutation. Mismatch distributions were calculated using ARLEQUIN 3.11, and the neutrality tests were evaluated by Tajima's D and Fu's Fs. Isolation by geographical distance was assessed by GENEPOP 4.0.10 [40] using Mantel test.
Results
Sequences characteristics of mtDNA-COII
One hundred and sixteen An. lesteri mosquitoes were distinguished by PCR assay from China (Table 1). A 686 bp COII sequence was determined in 88 mosquitoes, and a Cytb fragment of 394 bp was obtained from 112 mosquitoes. All segregating sites and the sequence variants (haplotypes) are shown in Figures 2 and 3. The summary statistics for both genes are given in Table 2. Across the whole dataset, there were 30 (4.37%) and 21 (5.33%) polymorphic sites for COII and Cytb, respectively. This low number of variable sites resulted in low nucleotide diversity and low haplotype diversity across samples. The θS of overall field populations was from 0.581 ± 0.435SD to 4.285 ± 1.709SD for COII, and 0.274 ± 0.274SD to 3.545 ± 1.655SD for Cytb; θπ was from 0.477 ± 0.485SD to 2.598 ± 1.606SD for COII, 0.091 ± 0.188SD to 2.231 ± 1.476SD for Cytb and h was from 0.005 ± 0.003SD to 0.000 ± 0.000SD (Table 2).
Figure 2.

Variable bases of the mtDNA-COII gene for the haplotypes of Anopheles lesteri population.
Figure 3.

Variable bases of the mtDNA-Cytb gene for the haplotypes of Anopheles lesteri population.
Table 2.
Data summary for populations, haplotypes and nucleotide diversity of Anopheles lesteri
| Population | Gene | N | Haplotypes (n) | S | h (± SD) | θs (± SD) | θπ (± SD) |
|---|---|---|---|---|---|---|---|
| YN | COII | 8 | 1(2), 3(2), 4(2), 5(1), 6(1) | 4 | 0.002 ± 0.002 | 1.543 ± 0.961 | 1.643 ± 1.227 |
| Cytb | 13 | 1(2), 2(5), 4(3), 5(1), 6(1), 7(1) | 5 | 0.004 ± 0.003 | 1.611 ± 0.899 | 1.615 ± 1.147 | |
| HNB | COII | 8 | 1(1), 8(1), 9(1), 11(1), 12(2), 13(1), 14(1) | 8 | 0.004 ± 0.002 | 2.314 ± 1.308 | 2.464 ± 1.692 |
| Cytb | 12 | 1(3), 2(2), 4(1), 8(1), 10(1), 11(1), 12(1), 13(1), 14(1) | 13 | 0.006 ± 0.004 | 3.545 ± 1.655 | 2.231 ± 1.476 | |
| JS | COII | 12 | 19(2), 20(10) | 1 | 0.000 ± 0.002 | 0.331 ± 0.331 | 0.303 ± 0.379 |
| Cytb | 17 | 2(17) | 0 | 0.000 ± 0.000 | 0.000 ± 0.000 | 0.000 ± 0.000 | |
| SC | COII | 18 | 6(3), 21(14), 22(1) | 2 | 0.001 ± 0.001 | 0.581 ± 0.435 | 0.477 ± 0.485 |
| Cytb | 22 | 2(21), 4(1) | 1 | 0.000 ± 0.000 | 0.274 ± 0.274 | 0.091 ± 0.188 | |
| GD | COII | 18 | 1(9), 23(1), 24(3), 25(1), 26(1), 27(1), 28(1), 29(1) | 7 | 0.002 ± 0.001 | 2.035 ± 1.006 | 1.288 ± 0.946 |
| Cytb | 21 | 2(6), 4(2), 20(1), 21(2), 24(2), 26(1), 27(3), 28(1), 29(1), 30(1), 31(1) | 10 | 0.005 ± 0.003 | 2.780 ± 1.239 | 2.076 ± 1.350 | |
| LN | COII | 24 | 1(4), 5(1), 7(1), 20(8), 30(1), 31(1), 32(3), 33(1), 34(1), 35(1), 36(1), 37(1) | 15 | 0.005 ± 0.002 | 4.285 ± 1.709 | 2.598 ± 1.606 |
| Cytb | 27 | 2(14), 4(1), 32(1), 33(1), 34(1), 35(3), 36(1), 37(1), 38(2), 39(1), 40(1) | 9 | 0.005 ± 0.003 | 2.037 ± 0.928 | 1.926 ± 1.255 |
h is haplotype diversity, S is the number of segregating sites and θs and θπ are the estimates of nucleotide diversity. The bold haplotypes occur in more than one population and the number in parentheses indicates the frequency of the haplotype.
Among the 88 COII sequences, 31 haplotypes were found. Four haplotypes of COII_1, COII_5, COII_6 and COII_20 occurred in more than one population, the frequency was 12.90% (4/31). Thirty of 112 Cytb haplotypes were observed. Three haplotypes of Cytb_1, Cytb_2 and Cytb_4 were shared, especially; Cytb_2 occurred in all populations (Table 2). Haplotype networks showed that An. lesteri haplotypes derived from a single common ancestral COII haplotype and two ancestral Cytb haplotypes (Figure 4).
Figure 4.
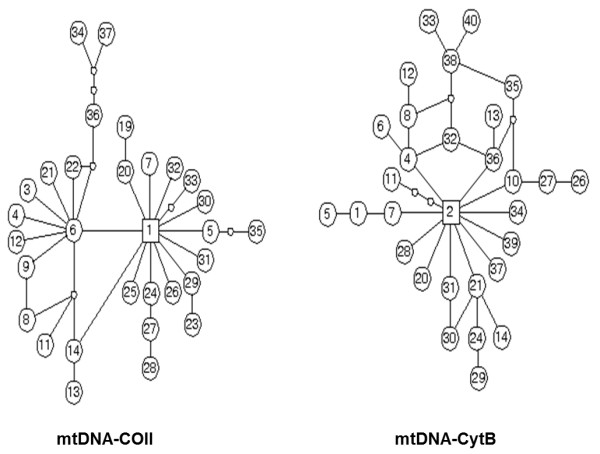
Genealogical relationships among haplotypes of two mtDNA genes estimated by TCS. The circle indicates one of haplotypes and the number in circle represents the type of haplotype. A unit branch represents one mutation. The empty circles indicate haplotypes that were not observed.
Population genetic structure of An. lesteri population
The genetic structure was analysed with GD, LN, YN, SC and HNB populations. The range values of paiwise FST was from 0.101 (GD/LN) to 0.655 (GD/SC) with mtDNA-COII, and 0.029 (HNB/LN) to 0.231 (YN/SC) with Cytb (Table 3). A Mantel test was carried out, and the correlation coefficient for the FST with geographical distance was 0.271 by COII (P ≥ 0.803) and 0.089 by Cytb (P ≥ 0.400), which was not significance based on 1,000 permutations.
Table 3.
Pairwise genetic distance (FST) for populations of Anopheles lesteri
| YN | HNB | SC | GD | LN | |
|---|---|---|---|---|---|
| YN | 0.345/0.114 | 0.055 | 0.231* | 0.128* | 0.153* |
| HNB | 0.124 | 0.324/0.096 | 0.125* | 0.055* | 0.029 |
| SC | 0.500* | 0.501* | 0.374/0.159 | 0.109* | 0.172* |
| GD | 0.272* | 0.319* | 0.655* | 0.252/0.099 | 0.117* |
| LN | 0.135* | 0.194* | 0.455* | 0.101* | 0.315/0.103 |
The pairwise values calculated by mtDNA-COII and Cytb gene are below and above the diagonal. The numbers along the diagonal are FST values within population. The bold values are by COII gene. *P < 0.05.
In the hierarchical AMOVE, both the 'among populations' and 'within populations' variance components were considerable high, the latter was more contribution to total variances than the former (Table 4). The mean genetic divergence among populations was greater by COII (0.342) than Cytb (0.115).
Table 4.
AMOVA analysis of genetic variation in Anopheles lesteri populations by mtDNA genes
| Source of variation | Degree of freedom | Variance components | Percentage of variation | |||
|---|---|---|---|---|---|---|
| COII | Cytb | COII | Cytb | COII | Cytb | |
| Among populations | 4 | 4 | 0.433 | 0.109 | 34.17 | 11.52 |
| Within populations | 71 | 93 | 0.835 | 0.772 | 65.83 | 88.48 |
| Total | 75 | 97 | 1.268 | 0.872 | 100 | 100 |
FST = 0.342(COII), 0.115 (Cytb)
The simulated mismatch distribution among the mtDNA-COII and Cytb haplotypes was smooth and unimodal peak, which coincide with the population expansion model. Although, observed value appeared multimodal, the result of variance test indicated the degree of coincidence between them was not significance (P ≥ 0.00 with COII, P ≥ 0.15 with Cytb) [41]. The Tajima's D and Fu's Fs values were all negative, except Tajima's D values of YN and HNB populations (Table 5), which suggested a large number of low-frequency mutations in populations and the populations were in expansion proceeding. The strongly negative values for Fu's Fs suggested population growth and this is supported by the estimated values using COII gene from the rapid expansion model fitted in ARLEQUIN (τ = 2ut = 2.615, θ0 = 0.00-0.39, θ1 = 99 999, u = per sequence mutation rate, t = time since expansion, N = effective number of females). With a mutation rate of 1 × 10-8 per site per generation [42], these values suggested a change in population size from a few thousand females to 108 females, in the range of 3970 years ago based on two generations of Anopheline mosquitoes in one month.
Table 5.
Values of neutrality test for Anopheles lesteri populations by mtDNA genes
| YN | HNB | SC | GD | LN | ||
|---|---|---|---|---|---|---|
| Tajima's D | COII | 0.283 | 0.303 | -0.438 | -1.246* | -1.402* |
| Cytb | 0.009 | -1.498* | -1.162 | -0.873 | -0.167 | |
| Fu's Fs | COII | -8.139 | -∝ | -28.504 | -26.580 | -26.580 |
| Cytb | -18.044 | -15.265 | -∝ | -27.052 | -27.112 |
*P < 0.1.
Discussion
Sampling strategy and geographic coverage greatly influence the analysis and interpretation of the data generated from the samples. In China, An. lesteri was distributed in a range as the east of 100° E, and from 19° N to 42° N [14]. In this study, An. lesteri mosquitoes were collected from most localities across its range. Although field An. lesteri specimen was difficult to collect due to usage of insecticide and environment changes, our sampling still covered geographic span of An. lesteri distribution. The LN was at the most northern limit, and GD was at the most southern limit of the distribution basically.
In this study, both level of mtDNA- OII and Cytb gene nucleotide diversity in field populations were greater than JS laboratory colony, such as all Cytb sequences in JS population were the same, which was similar to other gene on mitochondrial DNA, as COI (An. dirus, An. darlingi, An. stephensi) [17-20] and COII (An. jeyporiensis, An. minimus ) [21,22]. Thus, they are useful marker for exploring An. lesteri population genetic structure.
The pairwise genetic distance using mtDNA-COII gene (0.101-0.655) was higher than Cytb (0.029-0.231). In theory, it was hard to prevent genetic divergence caused by genetic drift if the gene flow [Nm= (1- FST)/4 FST]) value was less than one [43]. The level of gene flow in these An. lesteri pairwise populations was below one, except YN/HNB, YN/LN, HNB/LN and LN/GD using mtDNA-COII gene, but all more than one except SC/YN using Cytb. The shallow population genetic structure was showed by Cytb gene. But the results by COII gene suggested that there was an apparent segregation from LN with the other populations, which is in agreement with the previous investigations with RAPD markers [13]. So, the level of An. lesteri population genetic divergence using mtDNA-COII gene should represent wild populations.
The factors responsible for population genetic structure should be analysed related with the climate, geography and the behaviour of mosquitoes. Yunnan is a highly complex region topographically due to its transitional position from tropical southern Himalayas to eastern Asia and from tropical Southeast Asia to sub-tropical China as well as at the junction of the India and Burmese plates, derived from Gondwanaland, and the Eurasian plate [44]. It is a noted centre of biodiversity [45-47]. It could have retained sufficiently mesic habitats for mosquitoes during the glaciations, when drier, more open habitats were spread widely [48]. If YN population of An. lesteri was the ancestor and the other region populations spread from Yunnan in the late stage of glaciations. The haplotype network suggested that An. lesteri migrated and spread from Yunnan towards the North and the East China, and occurred colonization and expansion during migration proceeding. They were the same as the An. lesteri population patterns with An. dirus complex in Southeast Asia by mtDNA-COI and microsatellite DNA [17,49], An. jeyporiensis in South China by mtDNA-COII [21]. If the migrating and expansion route was true, the An. lesteri samples in south of Yunnan should be increase to further investigation. An. lesteri is widespread in Palaearctic and Oriental region, and there is different climate, breeding habitation and blood preference, such as An. lesteri in southern and central China mainly is anthropophagic, but in Liaoning preferred animal's blood [11]. The above should be the key factors of influencing population genetic structure of An. lesteri in China.
Conclusion
Levels of genetic variation within An. lesteri populations were higher than among them. There was an apparent segregation from Liaoning with the other populations using mtDNA-COII gene. The results of neutrality test suggested a large number of low-frequency mutations in populations and the populations were in expansion proceeding. While these results may suggest considerable levels of gene flow, other explanations such as the effect of historical population perturbations can also be hypothesized.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
YM conceived and designed the experiments. MY performed the experiments. MY and YM analyzed the data. MY and YM wrote the paper. JW Provided part of the material. All authors read and approved the manuscript.
Contributor Information
Manni Yang, Email: yangmanni568@yahoo.com.cn.
Yajun Ma, Email: yajunm@yahoo.com.cn.
Jing Wu, Email: wujing0801@yahoo.com.cn.
Acknowledgements
The authors thank Zhe Chen, Huayun Zhou, Xintian Lei, Yunan Gu, Guangquan Huang, Guoying Chen, Peng Li, Yaohua Shi, Liqun Huang and Yong Fan for their assistant with specimen collection. This work was funded by National Natural Science Foundation of China-Yunnan Joint Fund (U0932604), National Institutes of Health (NIH) grants R03 AI 067366 and National Natural Science Foundation of China (30571624).
References
- Baisas FE, Hu SMK. Anopheles hyrcanus var. sinensis of the Philippines and certain parts of China, with some comments on Anopheles hyrcanus var. nigerrimus of the Philippines. Mon Bull Bur Health. 1936;16:205–242. [Google Scholar]
- Christophides GK. Transgenic mosquitoes and malaria transmission. Cell Microbiol. 2005;7:325–333. doi: 10.1111/j.1462-5822.2005.00495.x. [DOI] [PubMed] [Google Scholar]
- Lehmann T, Licht M, Elissa N, Maega BTA, Chimumbwa JM, Watsenga F T, Wondji CS, Simard F, Hawley WA. Population structure of Anopheles gambiae in Africa. J Heredity. 2003;94:133–147. doi: 10.1093/jhered/esg024. [DOI] [PubMed] [Google Scholar]
- Michel AP, Guelbeogo WM, Grushko O, Schemerhorn BJ, Kern M, Willard MB, Sagnon NF, Costantini C, Besansky NJ. Molecular differentiation between chromosomally defined incipient species of Anopheles funestus. Insect Mol Biol. 2005;14:375–387. doi: 10.1111/j.1365-2583.2005.00568.x. [DOI] [PubMed] [Google Scholar]
- Donnelly MJ, Simard F, Lehmann T. Evolutionary studies of malaria vectors. Trends Parasitol. 2002;18:75–80. doi: 10.1016/s1471-4922(01)02198-5. [DOI] [PubMed] [Google Scholar]
- Xu JJ, Feng LC. Studies on the Anopheles hyrcanus group in mosquitoes in China. Acta Entomol Sin. 1975;18:77–104. [Google Scholar]
- Sandosham AA. Malariology with special reference to Malaya Singapore. Singapore, University of Malay Press; 1959. [Google Scholar]
- Ma SF. Studies on the Anopheles (A.) sinensis group of mosquitoes in China, including four new sibling species. Sinozool. 1981;1:59–70. [Google Scholar]
- Wilkerson RC, Li C, Rueda LM, Kim HC, Klein TA, Song GH, Strickman D. Molecular confirmation of Anopheles (Anopheles) lesteri from the Republic of South Korea and its genetic identity with An. (Ano.) anthropophagus from China (Diptera: Culicidae) Zootaxa. 2003;378:1–14. [Google Scholar]
- Hwang UW, Tang LH, Kobayashi M, Yong TS, Ree HI. Molecular evidence supports that Anopheles anthropophagus from China and Anopheles lesteri from Japan are the same species. J Am Mosq Control Assoc. 2006;22:324–326. doi: 10.2987/8756-971X(2006)22[324:MESTAA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Tang LH. Anopheles anthropoptagus in China: biology and control. Shanghai, Shanghai Scientific & Technical Publishers; 2008. [Google Scholar]
- Ma YJ, Yang P, Xu JN, Chen Z, Pan B. Identification of Anopheles lesteri in China (Diptera: Culicidae): morphologic characters, chromosome karyotype and molecular markers. Entomotaxonomia. 2005;27:199–208. [Google Scholar]
- Ma YJ, Song GH, Li XY. Study on population genetic divergence of Anopheles anthropophagus between Liaoning and other distributions in China. Chin J Parasitic Dis Control. 2002;15:321–324. [Google Scholar]
- Lu BL. Fauna Sinica, Insect Vol.9 Diptera: Culicidae II. Beijing. Science Press; 1997. [Google Scholar]
- Shin EH, Kim TS, Lee HW, Lee JS, Lee WJ. Vector competence of Anopheles lesteri Baisas and Hu (Diptera: Culicidae) to Plasmodium vivax in Korea. Korean J Parasitol. 2002;40:41–44. doi: 10.3347/kjp.2002.40.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Mizusawa K, Saugstad ES. A revision of the adult and larval mosquitoes of Japan (including the Ryukyu Archipelago and the Ogasawara Islands) and Korea (Diptera: Culicidae) Contr Amer Ent Inst. 1979;16:1–987. [Google Scholar]
- Walton C, Handley JM, Tun-lin W, Collins FH, Harbach RE, Baimai V, Butlin RK. Population structure and population history of Anopheles dirus mosquitoes in Southeast Asia. Mol Bio Evol. 2000;17:962–974. doi: 10.1093/oxfordjournals.molbev.a026377. [DOI] [PubMed] [Google Scholar]
- Wang D, Ma YJ, Zhou HN. Genetic variation of Anopheles dirus A and D (Diptera: Culicidae) in China: inferred by mtDNA-COI gene sequences. Chin J Parasitol Parasit Dis. 2007;5:368–371. [PubMed] [Google Scholar]
- Gutiérrez LA, Gómez GF, González JJ, Castro MI, Luckhart S, Conn JE, Correa MM. Microgeograpic genetic variation of the malaria vector Anopheles darlingi Root (Diptera: Culicidae) from Córdoba and Antioquia, Colombia. Am J Trop Med Hyg. 2010;83:38–47. doi: 10.4269/ajtmh.2010.09-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali N, Hume JCC, Dadzie SK, Donnelly MJ. Molecular genetic studies of Anopheles stephensi in Pakistan. Med Vet Entomol. 2007;21:265–269. doi: 10.1111/j.1365-2915.2007.00691.x. [DOI] [PubMed] [Google Scholar]
- Chen B, Harbach RE, Butlin RK. Genetic variation and population structure of the mosquito Anopheles jeyporiensis in southern China. Mol Ecol. 2004;13:3051–3056. doi: 10.1111/j.1365-294X.2004.02309.x. [DOI] [PubMed] [Google Scholar]
- Chen B, Pedro PM, Harbach RE, Somboon O, Walton C, Butlin RK. Mitochondrial DNA variation in the malaria vector Anopheles minimus across China, Thailand and Vietnam: evolutionary hypothesis, population structure and population history. Heredity. 2010. pp. 1–12. [DOI] [PMC free article] [PubMed]
- Jung J, Jung Y, Min GS, Kim W. Analysis of the population genetic structure of the malaria vector Anopheles sinensis in South Korea based on mitochondrial sequences. Am J Trop Med Hyg. 2007;77:310–315. [PubMed] [Google Scholar]
- Ndo C, Antonio-Nkondjio C, Cohuet A, Ayala D, Kengne P, Morlais I, Awono-Ambene PH, Couret D, Ngassam P, Fontenille D, Simard F. Population genetic structure of the malaria vector Anopheles nili in sub-Saharan Africa. Malar J. 2010;9:161. doi: 10.1186/1475-2875-9-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AP, Ingrasci MJ, Schemerhorn BJ, Kern M, Le Goff G, Coetzee M, Elissa N, Fontenille D, Vulule J, Lehmann T, Sagnon F, Costantini C, Besansky NJ. Rangewide population genetic structure of the African malaria vector Anopheles funestus. Mol Ecol. 2005;14:4235–4248. doi: 10.1111/j.1365-294X.2005.02754.x. [DOI] [PubMed] [Google Scholar]
- Michel AP, Grushko O, Guelbeogo WM, Lobo NF, Sagnon N, Costantini C, Besansky NJ. Divergence with gene flow in Anopheles funestus from the Sudan Savanna of Burkina Faso, West Africa. Genetics. 2006;173:1389–1395. doi: 10.1534/genetics.106.059667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AP, Grushkol O, Guelbeogo WM, Sagnon N, Costantini C, Besansky NJ. Effective population size of Anopheles funestus chromosomal in Burkina Faso. Malar J. 2006;5:115. doi: 10.1186/1475-2875-5-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temu EA, Yan G. Microsatellites and mitochondrial genetic differentiation of Anopheles arabiensis (Diptera: Culicidae) from Western Kenya, the great rift valley, and coastal Kenya. Am J Trop Med Hyg. 2005;73:726–733. [PubMed] [Google Scholar]
- Donnelly MJ, Licht MC, Lehmann T. Evidence for recent population expansion in the evolutionary history of the malaria vector Anopheles arabiensis and Anopheles gambiae. Mol Biol Evol. 2001;8:1353–1364. doi: 10.1093/oxfordjournals.molbev.a003919. [DOI] [PubMed] [Google Scholar]
- Collins FH, Mendez MA, Rasmussen MO, Mehaffey PC, Besansky NJ, Finnerty V. A ribosomal RNA gene probe differentiates member species of the Anopheles gambiae complex. Am J Trop Med Hyg. 1987;37:37–41. doi: 10.4269/ajtmh.1987.37.37. [DOI] [PubMed] [Google Scholar]
- Ma YJ, Qu FY, Cao YC, Yang BJ. On molecular identification and taxonomic status of Anopheles lesteri and Anopheles anthropophagus in China (Diptera: Culicidae) Chin J Parasitol Parasit Dis. 2000;18:325–328. [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The Clustal × windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;24:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–1660. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- Tajima F. Evolutionary relationship of DNA sequences in finite population. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson GA. On the number of segregation sites in genetic models within recombination. Theor Popul Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- Schneider S, Roeddli D, Excoffier L. ARLEQUIN, Version 2.000: a software for population genetics data analysis. Genetics and Biometry Laboratory. Switzerland, University of Geneva; 2000. [Google Scholar]
- Nei M, Li WH. Mathematical model for studying genetic variation in terms of restriction endonuclases. Proc Natl Acad Sci USA. 1979;76:5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M. A measure of population subdivision based on microsatellite allele frequencies. Genetics. 1995;139:457–462. doi: 10.1093/genetics/139.1.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond M, Rousset F. GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Heredity. 1995;86:248–249. [Google Scholar]
- Schneider S, Excoffier L. Estimation of past demographic parameters from the distribution of the pariwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics. 1999;152:1079–1089. doi: 10.1093/genetics/152.3.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JR, Caccone A, Amato GD, Yoon C. Rates of nucleotide substitution in Drosophila mitochondrial DNA and nuclear DNA are similar. Proc Natl Acad Sci USA. 1986;83:9090–9093. doi: 10.1073/pnas.83.23.9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M. Gene flow and the geographic structure of natural population. Science. 1987;236:787–792. doi: 10.1126/science.3576198. [DOI] [PubMed] [Google Scholar]
- Audley-Charles. In: MGDispersal of Gondwanaland: relevance to evolution of the angiosperms. Biogeographical Evolution of the Malay Archipelago. Whitemore TC, editor. Oxford, Clarendon Press; 1987. [Google Scholar]
- Jin ZZ, Ou XK. The diversity features of plant community types in the tropical rain forest vegetation of Xishuangbanna, Yunnan. Acta Bot Yunnan Suppl. 1997;1:1–30. [Google Scholar]
- Zhu H, Xu ZH, Wang H, Li BG. Tropical rain forest fragmentation and its ecological and species diversity changes in southern Yunnan. Biol Conserv. 2004;13:1355–1372. [Google Scholar]
- Dong XS, Zhou HN, Gong ZD, Dong LM, Wang XZ. Investigation of mosquito species in Yunnan Province with some new species. Chin J Vector Biol Control. 2004;5:186–188. [Google Scholar]
- Kealhofer L, Penny D. A combined pollen and phytolith record for fourteen thousand years of vegetation change in northeastern Thailand. Rew Palaeobot Palynor. 1998;103:83–93. [Google Scholar]
- Walton C, Handley JM, Collins FH, Baimai V, Harbach RE, Deesin V, Butlin RK. Genetic population structure and introgression in Anopheles dirus mosquitoes in Southeast Asia. Mol Ecol. 2001;10:569–580. doi: 10.1046/j.1365-294x.2001.01201.x. [DOI] [PubMed] [Google Scholar]