

Abstract
Background
The mechanisms involved in hepatocellular carcinoma (HCC) establishing an immunologically tolerogenic tumor environment remain poorly characterized.
Aim
This study evaluates effector T cell responses and soluble IL-2 receptor alpha chains (sCD25) in relation to HCC stage/survival and characterizes the impact of sCD25 on effectors.
Methods
Effector cell responses with serum from HCC patients and in serum free conditions were assessed by IFN-γ ELISpot, proliferation and ATP production assays at baseline, after depletion of sCD25, and after supplementation with recombinant sCD25. Sera sCD25 were measured by ELISA and any relationship with stage/survival was determined.
Results
Hepatocellular carcinoma patients had marked global impairment in T cell responses at baseline which correlate with tumor burden and poor outcome. The impairment in immune responses is characterized by low IFN-γ production, cell proliferation, and ATP production. Effector responses are impaired by serum from HCC patients in a dose-dependent manner, implicating soluble factors in the observed immunosuppression. Significant elevations in serum levels of sCD25 are found in patients with HCC, which correlate with tumor burden and a worse survival. T cell reactivity is inversely proportional to serum level of sCD25. Impaired T cell responses improve with sCD25 depletion from HCC serum or IL-2 supplementation suggesting impairment in IL-2 signaling. In contrast, adding increasing doses of sCD25 suppresses effector T cells, which partly involves induction of apoptosis.
Conclusions
These findings show that HCC patients have blunted T cell immunity that is partly related to elevated levels of sCD25, supporting a novel immuno-inhibitory role for this soluble receptor.
Keywords: Soluble IL-2 receptor alpha chain (sIL-2RA), IL-2 signaling, T cell immunosuppression, Liver cancer, Serum, Soluble factors, Tumor tolerance
Introduction
Hepatocellular carcinoma (HCC) is a global health problem as it is the fifth most common cancer worldwide and the third leading cause of cancer-related death [1]. In the West, the incidence of HCC is increasing mainly due to the rising prevalence of hepatitis C (HCV) related cirrhosis and is the leading cause of death among patients with cirrhosis [2, 3]. The majority of patients with HCC are not candidates for curative treatments such as resection and liver transplantation by the time they present due to the extent of their tumor [4]. These patients have limited treatment options and even with the current treatments of transarterial chemoembolization and sorafenib, which have modest survival advantages, all eventually progress [5]. Therefore, improving our knowledge about the pathogenesis of this complex disease is critical in order to develop new treatment strategies.
A limitation of pre-clinical models in HCC is the lack of an intact immune system, and thus little has been defined regarding the immunopathogenesis of HCC [6]. Effector T lymphocytes are important in the control of tumor progression [7]. Lymphocytes that infiltrate tumor have been described in patients with HCC and their presence after resection indicates a better survival [8]. However, the progression of tumors despite the presence of infiltrating lymphocytes suggests the use of evasive mechanisms by HCC in order to avoid the host immune response. Our group and others have shown that one of the mechanisms used by HCC to escape immune surveillance is an increase in the suppressive CD4+CD25+ regulatory T cell (Treg) population in both the periphery and tumor micro-environment [9–12]. This increase in Tregs is linked to blunted anti-tumor immunity, tumor progression, and decreased patient survival. It is clear that many cancers, including HCC, establish a tolerogenic tumor environment that favors tumor development. Despite evidence showing that immunity impacts HCC disease progression and survival, the mechanisms involved remain poorly characterized.
In this prospective analysis of HCC patients, we have evaluated general effector T cell responses and soluble factors in relation to HCC staging and survival. We report a striking overall T cell suppression in HCC patients that correlates with tumor burden and poor survival. An analysis of the serum from HCC patients reveals that the soluble IL-2 receptor alpha chain (sIL-2Ralpha, sCD25) is partly involved in the observed T cell suppression. Elevated levels of soluble CD25 (sCD25) are found in the periphery of patients with HCC when compared with healthy subjects and patients with cirrhosis. The increase in sCD25 levels correlate with tumor burden and a worse survival. The impaired T cell responses improve with sCD25 depletion, while supplementation studies suggest sCD25 to have a suppressive effect on effector T cells. These findings support a need for targeted immunotherapy in HCC patients and suggest a novel immunosuppressive mechanism for sCD25.
Methods
Study Population
The study protocol was approved by the University of Florida IRB and included patients with HCC (n = 60), chronic HCV infection with (n = 20) and without cirrhosis (n = 40). Sex- and age-matched normal healthy controls (NHC; n = 30; 15 male, 15 female) were obtained from the local blood bank (Life South, Gainesville, FL). HCC was diagnosed according AASLD guidelines [13]. Demographic and clinical data were obtained from HCC patients. Tumor burden was defined using the Barcelona Clinic Liver Cancer (BCLC) staging system defined as: (stage A) single lesion <5 cm or 2–3 lesions <3 cm; (stage B) multiple lesions exceeding stage A criteria; (stage C) macroscopic vascular invasion or metastatic disease; and (stage D) metastatic disease with decompensated liver disease. The collected blood samples were immediately processed to obtain serum and peripheral blood mononuclear cells (PBMC). In addition, five patients with HCC undergoing liver transplantation had serum and explant tissue collected.
Cell Preparation: PBMC and CD4+CD25−
Peripheral blood mononuclear cells were isolated as described previously [14]. CD4+ T cells were isolated from PBMC by negative selection using immunomagnetic beads (Miltenyi Biotec, Auburn, CA). Negatively selected CD4+ cells were incubated with anti-CD25 microbeads (50 μg/108 cells) and the MS column (Miltenyi) was used to obtain the CD4+CD25− effector T cell responder population as previously detailed. The CD4+CD25− purity was >90%.
Cell Culture and Reagents
Peripheral blood mononuclear cells or CD4+CD25− cells were cultured in serum free medium (SFM; CTL media, Shaker Heights, Ohio) alone (negative control) or with positive controls (PHA 5 μg/ml, Candida albicans 10 μg/ml) in 5% CO2 at 37°C to assess T cell proliferation at 72 h by [3H]-thymidine incorporation and IFN-γ production at 48 h by ELISpot. NHC or HCC sera were added to the serum mixing studies to create a 10% culture concentration. HCC sera were depleted of sCD25 by sequential incubation at room temperature for 1 h with monoclonal murine coated plates (Bender Systems, Burlingame, CA). Supplementation studies were done using recombinant sCD25 (Bender Systems).
Cell Proliferation Assay
The [3H]-thymidine incorporation assay was performed as described previously [15]. Briefly, PBMC or CD4+CD25− cells (2 × 105/well) were seeded into 96-well plates in triplicate in 200 μl of SFM with various controls. After 72 h, [3H]-thymidine was added (1 μCi/well) for 18 h, and incorporation measured on a β-scintillation counter. Results were expressed as mean counts per minute with standard error (cpm ± SE).
Cylex® ImmuKnow™ Assay
The ImmuKnow™ assay was performed according to the manufacturer’s protocol and as described previously [15]. Briefly, whole blood samples were collected from HCC or NHC patients and tested within 10 h. Whole blood was diluted 1:4 with a sample diluent, added to a microtiter plate well, and incubated with or without PHA (2.5 μg/ml). After 16 h incubation, CD4 cells were positively selected with anti-human CD4 immunomagnetic beads (Dynabeads, Oslo, Norway) and a magnet tray (model 1050, Cylex®, Columbia, MD), washed to remove residual cells, and lysed to release intracellular ATP. Released ATP was measured (ng/ml) with a luciferin/luciferase system and a luminometer (Berthold, Knoxville, TN) and then calculated from a calibration curve.
ELISA for Soluble CD25
Serum samples were measured for sCD25 using ELISA according to manufacturer’s recommendations (Bender Systems). Microtiter plates were read at 450 nm using the SpectraMax 190 reader (Molecular Devices, Sunnyvale, CA). Measurements of sCD25 above the upper limit of the calibration range (20,000 pg/ml) were diluted by 1/2 using buffer from the manufacturer.
IFN-γ ELISpot
An established ELISpot was used to evaluate the IFN-γ T cell responses of PBMC cultured with HCC serum, HCC serum depleted of sCD25, and increasing doses of recombinant sCD25 [15]. Briefly, cells were incubated at 37°C and 5% CO2 for 48 h in SFM alone or with stimulatory agents (PHA, Candida) on a nitrocellulose plate (Millipore, Bedford, MA) coated with human anti-IFN-γ (15 μg/ml; Mabtech, Cincinnati, OH). After a series of washes, secondary antibody, enzyme and substrates were added according to manufacturer’s recommendations until spots developed. IFN-γ spots were counted using ELISpot Bio-reader® 4000 Pro-X (BioSys, Germany). Results are shown as mean IFN-γ spots per 2 × 105 PBMC/well with SE.
CD25 Quantitative Real Time-PCR
Total RNA was isolated from liver tissue with HCC and without using TRIzol (Invitrogen, CA, USA) and quantified using the Molecular Devices SpectraMax 190. cDNA was prepared with a high-capacity cDNA kit (Applied Biosystems, CA, USA). For each assay tested, 100 ng of cDNA was amplified with Applied Biosystems TaqMan Gene Expression Assay in 25-μl volumes in an Applied Biosystems 7500 Real-time PCR System with the following cycling conditions: 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. The relative expression of IL2-RA gene expression levels were measured by quantifying the cDNA relative to a calibrator sample, which in each case was the cDNA from the liver tissue without HCC. All quantitations were normalized by the expression of each target to GAPDH and then this normalized value was compared to the normalized expression in a calibrator to calculate the fold change value using the delta-delta-Ct method. Results are shown as relative IL2-RA mRNA levels.
Flow Cytometry
Apoptosis was measured with Annexin V staining. Briefly, NHC CD4+CD25− cells were cultured in SFM alone or with PHA and increasing concentrations of sCD25. After 48 h of incubation, the cells were stained using the Annexin V-FITC apoptosis detection kit according to the manufacturer’s instructions (BD Biosciences Pharmingen, San Jose, California). Apoptotic cells were detected using flow cytometry performed on a FACSCalibur. Thirty thousand-gated events were acquired for each condition, and data was analysed using CellQuest software.
Statistical Analysis
Data are expressed as box plots and mean ± SE. Statistical analyses were performed using the Mann–Whitney U-test and the Spearman’s rank correlation coefficient. Receiver operator characteristic (ROC) curves along with point of maximal accuracy and Kaplan–Meier survival curves were performed using MedCalc® for Windows, version 9.6.4.0 (MedCalc Software, Mariakerke, Belgium). All P values <0.05 were considered statistically significant.
Results
Clinical Characteristics of HCC Patients
The majority of patients (n = 60) were males (87%), with HCV as the underlying etiology of cirrhosis (62%). The majority of patients had compensated liver disease (Child Pugh: A 62%, B 27%) with a MELD <15 (78%). Using the BCLC staging system, 33% of patients presented with early HCC (stage A) and 40% of patients presented with advanced HCC (stage C; Table 1).
Table 1.
Baseline characteristics of the patients with hepatocellular carcinoma (HCC; n = 60) including biochemical parameters, underlying etiology of chronic liver disease, alpha-fetoprotein (AFP), Child-Pugh classification, model for end-stage liver disease (MELD) score, and the Barcelona clinic liver cancer (BCLC) stage
| Baseline parameters | Mean or n (% or range) |
|---|---|
| Age (years) | 60 (30–80) |
| Gender | |
| Male | 52 (87%) |
| Female | 8 (13%) |
| Etiology | |
| HCV | 37 (62%) |
| EtOH | 9 (15%) |
| HBV | 7 (12%) |
| NAFLD | 6 (10%) |
| AlATD | 1 (1%) |
| Liver biochemistries | |
| AST | 138 (26–510) |
| ALT | 93 (16–246) |
| ALK | 166 (52–482) |
| TB | 2.0 (0.3–2.1) |
| Albumin | 3.4 (2.4–4.5) |
| INR | 1.3 (0.9–2.0) |
| AFP | 6,474 (2.3–50,000) |
| <20 | 20 (33%) |
| 20–400 | 13 (22%) |
| >400 | 27 (45%) |
| Child-Pugh | |
| A | 39 (65%) |
| B | 19 (32%) |
| C | 2 (3%) |
| MELD | 11.2(6–9) |
| <15 | 47 (78%) |
| >15 | 13 (22%) |
| BCLC stage | |
| A | 20 (33%) |
| B | 13 (22%) |
| C | 24 (40%) |
| D | 3 (5%) |
Impaired CD4 T Cell Responses in HCC and Relationship with Stage and Survival
In this study, we evaluated the baseline (pretreatment) CD4 effector T cell responses in patients with HCC and determined their relationship with stage and survival. In order to characterize CD4 effector T cell responses in HCC patients an ex vivo analysis was performed prior to any treatment using an ATP activity-based assay (Cylex® ImmuKnow™). This in vitro assay is FDA-approved to monitor cellular immune function in patients on immunosuppressive medications and was chosen because of its ability to evaluate CD4 T cell reactivity using whole blood within 24 h. The CD4 T cell responses in HCC patients (n = 60) were compared to age/sex matched normal controls (NHC; n = 20) and patients with HCV related cirrhosis (n = 20; Fig. 1a). HCC patients had significantly lower general CD4 T cell responses (192 ± 20 ATP ng/ml; P < 0.01) when compared to HCV patients with cirrhosis (418 ± 46 ATP ng/ml) and normal subjects (515 ± 24 ATP ng/ml). An inverse correlation was found between CD4 T cell responses and tumor burden with those patients with advanced (stage C) and terminal HCC (stage D) having the lowest immune CD4 T cell reactivity (Fig. 1b: R = −0.64, P < 0.01). A correlation analysis between Child-Pugh status or MELD scores and CD4 T cell responses did not show a relationship suggesting that the observed correlation with tumor burden was not influenced by severity of cirrhosis in this cohort of patients. The correlation analysis between CD4 T cell responses and survival showed that higher CD4 T cell reactivity correlated with improved survival in HCC patients while lower CD4 T cell responses was associated with higher mortality (Fig. 1c: R = 0.51, P < 0.01).
Fig. 1.
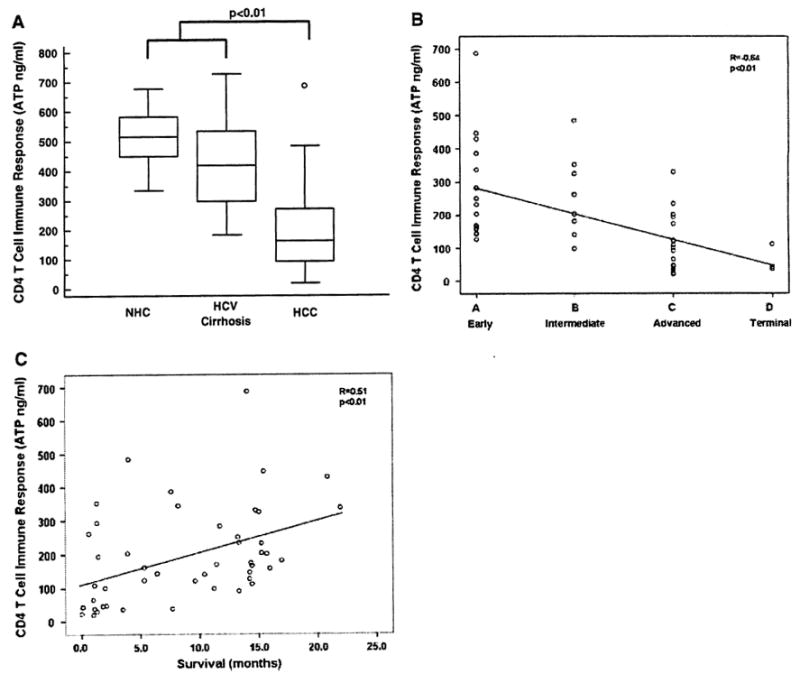
Impaired CD4 effector T cell responses in hepatocellular carcinoma (HCC). a Comparison of baseline (pretreatment) ex vivo general CD4 T cell responses for HCC patients (n = 60), age/sex matched normal controls (NHC, n = 20) and patients with HCV related cirrhosis (n = 20) using the ImmuKnow™ in vitro ATP based activity assay (Cylex®). CD4 T cell responses (ng/ml) for all three study groups are represented by the box-and-whisker plot. The horizontal lines illustrate the 25th, 50th, and 75th percentiles of the CD4 T cell reactivity. The vertical lines represent the 10th and 90th percentiles and the circles represent values outside these percentiles. In general, HCC patients showed statistically significantly lower general CD4 T cell responses when compared to HCV patients with cirrhosis and NHC (P < 0.01). b There was a significant negative correlation between general CD4 T cell responses and tumor stage as defined with the Barcelona clinic liver cancer (BCLQ staging classification (R = −0.64; P < 0.01). Patients with advanced HCC (stage C) and terminal HCC (stage D) had significantly lower CD4 T cell reactivity in contrast to patients with localized HCC (stage A and B) which showed higher levels of CD4 T cell responses, c A direct correlation was found between CD4 T cell responses and survival (R = 0.51, P< 0.01)
HCC Serum Suppresses Effector T Cell Proliferation, Cytokine Production, and ATP Production
Having shown that CD4 effector responses are impaired with increasing tumor burden, we then wanted to determine if serum from HCC patients can influence T cell activity. To evaluate the impact of serum derived from HCC patients on ex vivo effector T cell responses, a series of mixing studies using PBMCs and sera isolated from HCC patients were performed. PBMCs from patients with advanced stage C HCC (n = 5) were cultured with their own serum (autologous), different HCC patient’s serum (allogeneic HCC serum), healthy subject’s serum (allogeneic NHC serum) and serum free medium (SFM) in the presence of PHA (Fig. 2a). In general, HCC PBMCs showed impaired effector T cell proliferation as measured by [3H] thymidine incorporation in the presence of autologous and allogeneic HCC serum. The HCC PBMC cultures with autologous HCC serum had the lowest proliferative effector responses (1,524.0 ± 201.4 cpm, P < 0.01) followed by the cultures with allogeneic HCC serum (9,219 ± 200 cpm, P < 0.05) when compared to the cultures with allogeneic NHC serum (19,881.67 ± 1,642.2 cpm) and SFM (20,273.3 ± 1,059.7 cpm). A similar suppressive effect by HCC serum was seen on HCC PBMC using the IFN-γ ELISpot assay. Cultures with autologous HCC serum and HCC PBMC isolated from advanced HCC patients (n = 3) showed lower IFN-γ secretion when compared to cultures of HCC PBMC with allogeneic NHC serum (mean, 52 vs. 126 IFN-γ spots per 2 × 105 PBMCs, P < 0.01). Interestingly, HCC serum decreased IFN-γ production by NHC PBMCs (288 vs. 55, P < 0.01). To further confirm that HCC serum suppresses T cell responses in normal subjects, CD4 T cell ATP responses from NHCs (n = 5) were measured ex vivo after culture with a constant aliquot of HCC serum from three stages of HCC (Fig. 2b). The CD4 T cell reactivity of NHCs (564.6 ± 23.1 ATP ng/ml) decreased when cultured with serum from patients with early, intermediate, and advanced stage HCC in a graded fashion reflective of HCC stage (389.3 ± 20.4, 311 ± 6.5, 101.2 ± 7.0 ng/ml, P < 0.05). The decrease in CD4 T cell response is HCC specific and not related to the presence of cirrhosis since serum from HCV-related cirrhosis without HCC (559.5 ±9.51) resulted in equivalent T cell reactivity to cultures with serum from NHCs.
Fig. 2.
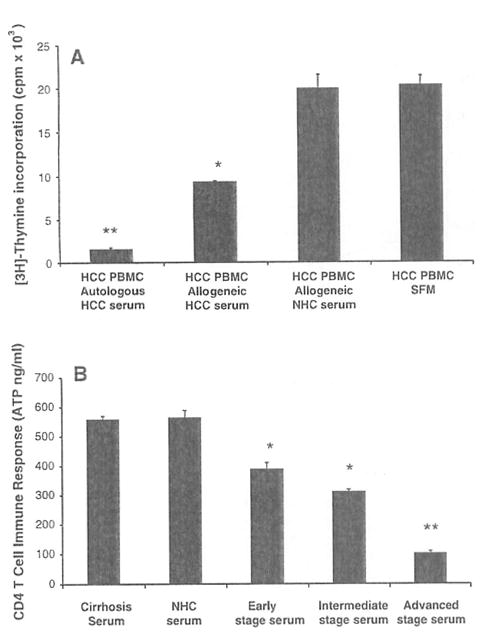
Hepatocellular carcinoma (HCC) serum suppresses effector T cell responses, a PBMCs from patients with advanced stage C HCC (n = 5) were cultured with autologous HCC serum, allogeneic HCC serum, allogeneic healthy subject (NHC) serum and in serum free medium (SFM) with PHA to measure effector proliferation by [3H] thymidine incorporation. Results are shown as mean ± standard error (SE) counts per minute (cpm × 103). The cultures with autologous and allogeneic HCC serum showed significantly lower proliferation when compared to the cultures with allogeneic NHC serum and SFM. b CD4 T cell immune responses from NHCs (n = 5) were measured ex vivo after culture with a constant aliquot of HCC serum from three stages of HCC (early, intermediate, and advanced) with the ImmuKnow™ ATP based assay. Results are shown as mean CD4 T cell immune response ± SE (ATP ng/ml). In general, the CD4 T cell reactivity by NHCs decreased in a graded fashion with worsening stage of HCC with maximal suppressive effect seen with the advanced HCC serum and the least suppressive effect seen with the early HCC serum. The suppressive effect of HCC serum appears to be HCC specific since it was not observed with serum from patients with HCV related cirrhosis (559.5 ± 9.51; n = 5). * P < 0.05, ** P < 0.01
Evaluation of Serum for Suppressive Molecules
After showing that serum from HCC patients is able to suppress effector T cells, the serum from normal subjects, patients with HCV-related cirrhosis, and HCC was evaluated to screen for significant differences in some well-known suppressive factors (TGF-β and IL-10) and sCD25. Only sCD25 showed a significant increase in serum levels among patients with HCC (data not shown), which led to more extensive evaluation of sCD25.
Serum Levels of sCD25 in HCC Patients
To better define the changes of sCD25 in HCC, we determined the serum levels in 60 clinically characterized patients with HCC and compared levels to age- and gender-matched healthy controls (NHC), patients with chronic HCV infection and patients with HCV cirrhosis (Fig. 3a). A comparison of the mean serum levels of sCD25 as measured by ELISA showed a five to sevenfold increase in HCC patients (20,701 ± 4,018 pg/ml; P < 0.01) when compared with healthy controls (2,953 ± 378 pg/ml, n = 30), patients with chronic HCV infection (4,604 ± 608 pg/ml, n = 40) and patients with cirrhosis (3,775.6 ± 534 pg/ml, n = 20).
Fig. 3.
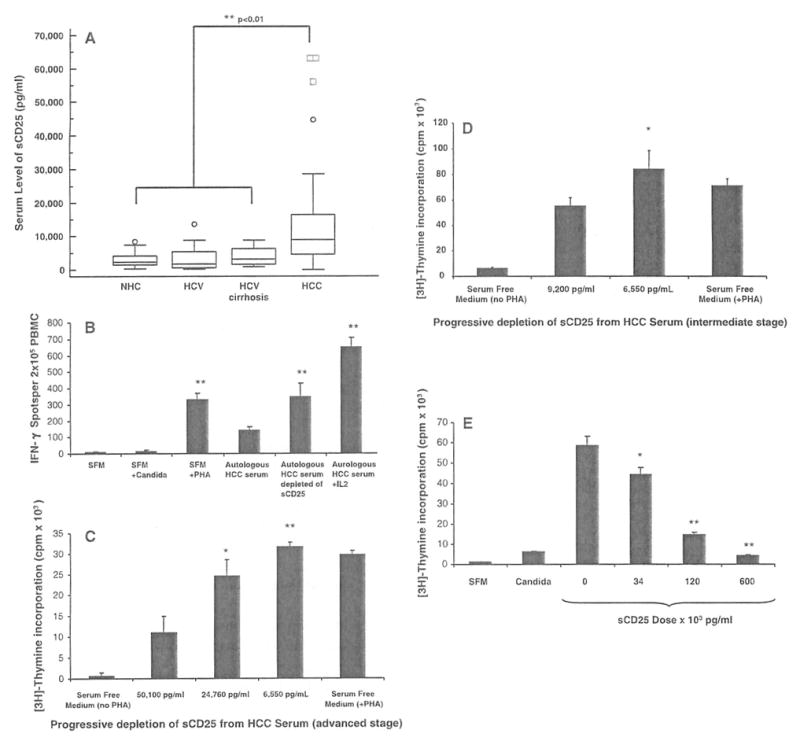
Serum levels of sCD25 and effect of sCD25 depletion and supplementation on T cell responses, a Levels of sCD25 in the serum of healthy subjects (NHC; n = 30), chronic HCV infection (HCV; n = 40), HCV-related cirrhosis (n = 20) and hepatocellular carcinoma (HCC) patients (n = 60) were determined using an ELISA assay. Data are expressed as box plots of the serum levels of sCD25 (pg/ml) for the four study groups. The average levels of sCD25 are significantly higher in HCC subjects (mean ± SE; 20,701 ± 4,018 pg/ml, P < 0.01) when compared to NHC (2,953 ± 378 pg/ml, P < 0.01) and disease controls (HCV 4,604 ± 608 pg/ml, P < 0.01; HCV cirrhosis 3,775.6 ± 534 pg/ml, P < 0.01). b Depletion of sCD25 enhances IFN-γ ELISpot responses. IFN-γ ELISpot responses were assessed using PBMC from patients with advanced HCC (n = 5) cultured with autologous serum with and without IL-2 (500 units/ml), autologous serum depleted of sCD25, and in serum free medium (SFM) with (PHA, Candida) and without stimulation. Results are expressed as means ± SE IFN-γ spots per 2 × 105 PBMCs. Depletion of sCD25 from the serum was performed with the use of the anti-sCD25 plated ELISA assay. The mean IFN-γ response obtained from the HCC PBMCs with autologous HCC serum prior to depletion (139 ± 24) was enhanced after depleting sCD25 from autologous HCC serum (348 ± 82, P < 0.01), when cultured with SFM plus PHA (329 ± 40, P < 0.01) and when high doses of IL 2 (500 units/ml) were added to the autologous HCC serum cultures (650 ± 57, P < 0.01). This analysis shows that the suppressive effects of HCC serum on IFN-γ T cell secretion seen with HCC PBMCs appears reversible when the content of sCD25 in the HCC serum was reduced and by exogenous IL-2. c Depletion of sCD25 enhances CD4 T cell proliferation. Mean proliferative responses (cpm × 103) of CD4+ CD25− T cells from normal healthy subjects (NHC, n = 5) when cultured with SFM, with HCC serum before sCD25 depletion with a sCD25 concentration of 50,100 pg/ml, and with HCC serum after two sequential depletions of sCD25 yielding two concentrations (24,760 and 6,550 pg/ml). Progressive sCD25 depletion from HCC serum enhanced the proliferative responses of NHC CD4+CD25− T cells (11,422 vs. 24,536 vs. 31,622 cpm). The degree of suppression showed a dose-dependent pattern that appeared related to the levels of sCD25 in the HCC serum, d Depletion of sCD25 from the serum in patients with intermediate stage HCC (n = 5) similarly enhances proliferation of CD4+CD25− T cells from NHC from 55,577 ± 6,295 cpm (before sCD25 depletion with a sCD25 concentration of 9,200 pg/ml) to 84,595 ± 13,922 (after sCD25 depletion with a sCD25 concentration of 113 pg/ml). e Supplementation of recombinant sCD25 suppresses CD4 T cell proliferation. Mean proliferation of CD4+CD25− T cells from NHCs (n = 5) when stimulated with PHA in serum free medium (SFM) with no sCD25 (0 pg/ml) and with increasing doses of sCD25 (34, 120 and 600 × 103 pg/ml) to characterize its effect on T cell proliferation by [3H] thymidine incorporation (mean ± SE cpm × 103). Recombinant sCD25 suppresses the mean proliferative response of CD4+CD25− T cells in a dose-dependent manner: 58,518 ± 4,709 cpm at baseline with no sCD25 (0 pg/ml) versus 44,109 ± 3,584 cpm (34 × 103 pg/ml) versus 14,667 ± 1,030 cpm (120 × 103 pg/ml) versus 4,331 ± 302 cpm (600 × 103 pg/ml). * P < 0.05, ** P < 0.01
Soluble CD25 and T Cell Function
An examination of the relationship between ex vivo CD4 T cell reactivity and serum level of sCD25 in the HCC patients (n = 60) revealed an inverse relationship (R = −0.31, P = 0.05). Low levels of serum sCD25 were associated with high CD4 T cell responses, while high levels of sCD25 were associated with low CD4 T cell reactivity.
Depletion of sCD25 from HCC Serum Enhances IFN-γ Production and Proliferation
Having shown that HCC patients have impaired effector T cell function and increased sCD25 levels, we investigated the effect of depleting sCD25 from HCC serum on HCC PBMC responses. When HCC PBMC from patients with advanced HCC (n = 5) were cultured with autologous HCC serum, low levels of IFN-γ ELISpot responses (139 ± 24 IFN-γ spots per 2 × 105 PBMCs) were observed in the presence of PHA (Fig. 3b). These low levels of IFN-γ ELISpot responses from the HCC PBMC increased when sCD25 were depleted from their autologous serum (348 ± 82, P < 0.01), when cultured with SFM (329 ± 40, P < 0.01) and when high doses of IL 2 (500 units/ml) were added to the autologous HCC serum cultures (650 ±57, P < 0.01). From these series of cultures we found that the impairment in IFN-γ T cell responses in patients with advanced HCC was reversible by reducing the amount of sCD25 in their respective HCC serum used in culture or by exogenous IL-2. In order to determine the susceptibility of NHC CD4+CD25− T cells to HCC serum and sCD25 mediated suppression, a series of cultures were performed before and after depletion of sCD25 using [3H] thymidine incorporation (Fig. 3c). This analysis showed that progressive depletion of sCD25 from HCC serum as measured by ELISA (50,100, 24,760, and 6,550 pg/ml) improved CD4+CD25− T cell proliferative responses (11,422 vs. 24,536 vs. 31,622 cpm). Similar results were observed with progressive depletion of sCD25 from serum with a baseline sCD25 concentration closer to the median of our HCC cohort, with enhancement of the baseline proliferation of 55,577 ± 6,295 cpm at a sCD25 concentration of 9,200 to 84,596 ± 13,922 pg/ml at a sCD25 concentration of 113 pg/ml (Fig. 3d). The decline in proliferation appeared related to the levels of sCD25 in the HCC serum used during the cultures.
Recombinant sCD25 Suppresses Proliferation and Cytokine Production
In order to confirm the suppressive effect of sCD25, a series of supplementation studies were performed using NHC CD4+CD25− T cells to assess the direct effect of increasing doses of recombinant sCD25 on T cell proliferation (Fig. 3e). The addition of increasing doses of recombinant sCD25 to CD4+CD25− T cells cultured with PHA resulted in dose-dependent suppression of effector proliferation (58,518 cpm at baseline with no sCD25 vs. 44,109 cpm with 34 × 103 pg/ml of sCD25 vs. 14,667 cpm with 120 × 103 pg/ml of sCD25 vs. 4,331 cpm with 60 0 × 103 pg/ml of sCD25). Similar sCD25 dose-related suppressive effects were seen with the addition of increasing doses of recombinant sCD25 on INF-γ production (480 SFUs at baseline with no sCD25 vs. 182 SFUs with 37.5 × 103 pg/ml vs. 52 SFUs with 50 × 103 pg/ml) by NHC PBMCs.
Soluble CD25 Induces Apoptosis
Having established that increasing doses of sCD25 impact effector T cell proliferation and EFN-γ production, we then evaluated the ability of sCD25 to induce apoptosis of CD4+CD25− T cells. We cultured isolated CD4+CD25− T cells from NHCs in SFM alone, in SFM with PHA but no sCD25 and in SFM with increasing doses of recombinant sCD25 (6,000 and 60,000 pg/ml) in the presence of PHA. Histograms with the mean fluorescence intensity for Annexin V staining are shown for the various culture conditions for a representative experiment in Fig. 4. A general induction of apoptosis was seen with increasing doses of sCD25 (27.7% with 0 pg/ml of sCD25 vs. 80.6% with 6,000 pg/ml of sCD25 vs. 90.2% with 60,000 pg/ml of sCD25). Trypan blue labeling showed that the overwhelming majority of the cultured cells in these studies were viable by their ability to exclude the trypan blue stain.
Fig. 4.
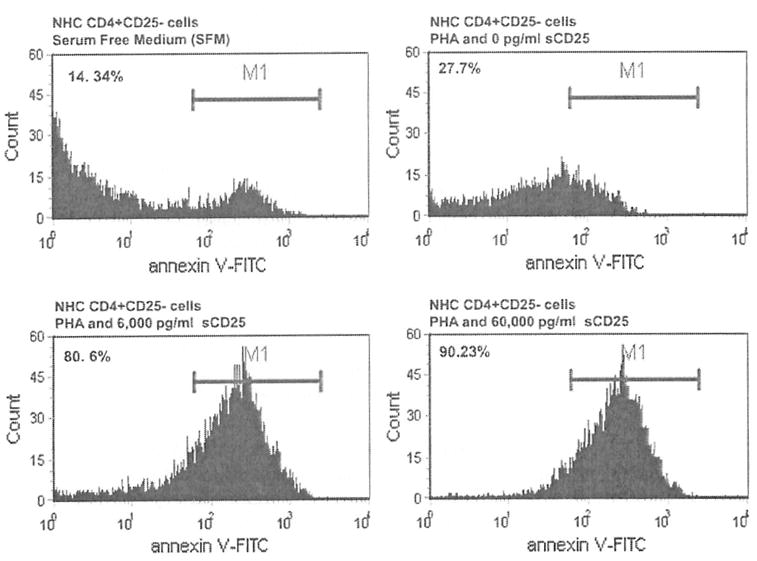
High doses of sCD25 induces T cell apoptosis. CD4+CD25− T cells from a normal healthy subject (NHC) were cultured in SFM alone, in SFM with PHA but no sCD25 and with increasing doses of recombinant sCD25 (6,000 and 60,000 pg/ml) in the presence of PHA in order to determine if apoptosis induction can explain the observed sCD25 mediated T cell suppression. Histograms with the mean fluorescence intensity for Annexin V staining are shown for a representative CD4+CD25− T cell experiment. Increasing doses of sCD25 induced apoptosis with the majority of apoptotic cells seen with the highest sCD25 concentration (27.7% with 0 pg/ml of sCD25 vs. 80.6% with 6,000 pg/ml of sCD25 vs. 90.2% with 60,000 pg/ml of sCD25)
Cellular Source(s) of Soluble CD25
In order to characterize the cellular source(s) of sCD25, a series of T cell cultures were performed using PBMC, CD4+CD25− T cells, and CD4+CD25+ T cells from NHC and HCC patients (Fig. 5a). Supernatants were collected and analyzed from each condition after polyclonal stimulation. From the analysis using the PBMC, CD4+CD25− T cell and CD4+CD25+ T cell cultures from NHC (n = 5), the PBMC cultures resulted in the highest levels of sCD25 detected in the supernatant (1,205 ±117 pg/ml) compared to modest levels by the CD4+CD25− T cell cultures (898 ± 44 pg/ml) and minimal levels in the CD4+CD25+ T cell cultures (131 pg/ml, lower limit of detection). In the cultures from the advanced HCC patients (n = 5), the highest levels of sCD25 were detected in the PBMC cultures (803 ± 84 pg/ml), while lower levels were observed in the CD4+CD25− T cell cultures (259 ± 19 pg/ml) and minimal levels in the CD4+CD25+ T cell cultures (131 pg/ml). Overall, this analysis revealed that the predominant cellular source of sCD25 in NHC was the CD4+CD25− T cell fraction. In contrast, in advanced HCC the major source of sCD25 was likely non-CD4 cell fractions, thus implicating antigen presenting cells as an important contributor to the sCD25 pool as has been previously described [24].
Fig. 5.
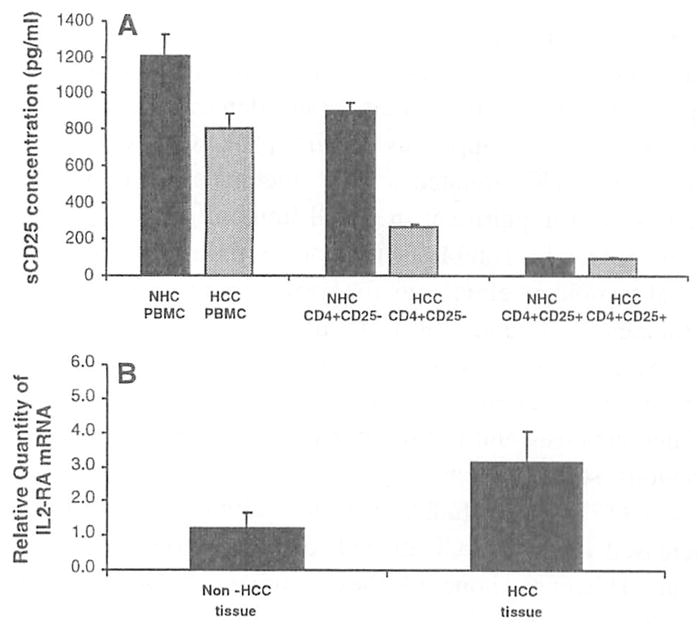
Sources of sCD25. a PBMC, CD4+CD25− T cells, and CD4+CD25+ T cells from NHC (n = 5) and patients with advanced HCC (n = 5) were stimulated with PHA and analyzed for their ability to secrete sCD25 into culture which was quantified by ELISA to characterize the cellular source of sCD25. PBMC (1.205 ± 117 pg/ml) and the CD4+CD25− T cell cultures (898 ± 44 pg/ml) from NHCs produced higher levels of sCD25 during the culture compared to CD4+CD25+ T cells (131 pg/ml, lower limit of detection of ELISA). In contrast, in the HCC cultures, the CD4+CD25− T cell cultures produced low levels of sCD25 (259 ± 19 pg/ml) while the PBMC cultures secreted higher levels of sCD25 (803 ± 84 pg/ml). This analysis suggested that the major source of sCD25 in the NHC cultures was the CD4+CD25− T cell fraction while in the HCC cultures non-CD4 T cells and/or antigen presenting cells may be the predominant source, b Real-time PCR was performed after isolating IL2-RA mRNA from liver tissue with HCC and without to determine if tumor is a source of sCD25. The mean level of IL2-RA mRNA from liver tissue with HCC was compared to liver tissue without HCC from the same patient (n = 6). There was a threefold increase in the IL2-RA mRNA found in the liver tissues with tumor when compared to sites without tumor (P < 0.05). Messenger RNA expression for IL2-RA was standardized to GAPDH mRNA expression and expressed as the fold-difference compared to normal for each tissue sample
HCC Tumor Microenvironment as a Source of Soluble CD25
To determine if the tumor microenvironment is also an important source of sCD25, liver tissue containing tumor as well as non-tumor sites were analyzed for CD25 mRNA. The mean level of IL2-RA mRNA from tumor liver tissue were compared to normal liver tissue from the same patient (n = 6). The relative expression of IL-2-RA mRNA was threefold higher in the liver tissue from HCC sites versus tissue sites without tumor (Fig. 5b).
Utility of Serum sCD25 as a Tumor Marker and Relationship with Stage and Survival
The demonstration of an overall elevation of sCD25 in the serum of HCC patients prompted an investigation to determine its relationship to HCC stage (using BCLC classification) and survival. A comparison between serum sCD25 levels and HCC tumor burden using the BCLC classification in the HCC patients showed a positive correlation (R = 0.35, P = 0.008). Serum levels of sCD25 were found to be the most elevated in patients with advanced HCC. To investigate the capability of sCD25 to distinguish HCC patients from controls (normal subjects and HCV with and without cirrhosis), a ROC curve analysis was generated to determine sensitivity and specificity (Fig. 6a). This analysis produced an AUC of 0.856 (95% CI 0.784–0.911; P = 0.0001). The maximal accuracy of the serum sCD25 to distinguish the presence of HCC from controls was seen at a cut-off of 5,800 pg/ml with a sensitivity of 69% and specificity of 86.5%. Using the cut-off serum value for sCD25 of 5,800 pg/ml divided the HCC patients into those with a serum level ≤5,800 pg/ml (group 1, n = 20) and those with a level >5,800 pg/ml (group 2,; n = 40). A comparison of the Kaplan–Meier survival curves of patients with a serum level of sCD25 of ≤5,800 pg/ml (group 1) versus those with a level of >5,800 pg/ml (group 2) by the log-rank test (P = 0.0135 with H.R. 0.391 ± 0.186–0.824) clearly showed a significant decline in survival in the HCC patients with higher sCD25 (Fig. 6b). An additional survival curve analysis performed without the four extreme data points did not reveal significant skewness related to these extreme values of sCD25.
Fig. 6.
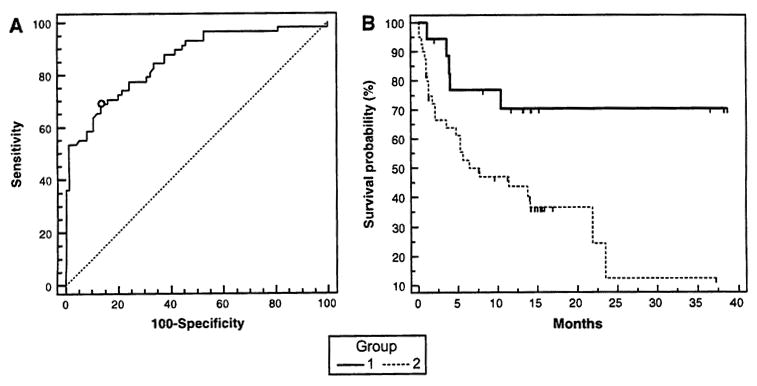
Serum sCD25 as a tumor marker and relationship with survival, a ROC curve analysis for sCD25 as a diagnostic test for HCC (n = 60) leads to an AUC of 0.856 (P = 0.0001) with a sensitivity of 69% and specificity of 86.5%, at a cut-off of 5,800 pg/ml. The negative control groups were composed of normal healthy controls (n = 30), patients with HCV (n = 40) and patients with HCV-related cirrhosis (n = 20). b Kaplan–Meier survival curves for patients with HCC in relation to higher and lower serum levels of sCD25. The survival curves of patients with a serum level of sCD25 of ≤5,800 pg/ml (group 1, n = 20) versus those with a level of >5,800 pg/ml (group 2, n = 40) was compared by the log-rank test (P = 0.0135 with H.R. 0.391 ± 0.186–0.824). The curves indicate the percentage of patients surviving at any given time. HCC patients with higher pretreatment serum levels of sCD25 have a worse survival compared to those with lower levels of sCD25 in their sera
Discussion
In this study, we show that HCC patients have a striking impairment in effector T cell response that correlates with advancing tumor stage and poor survival. Serum derived from HCC patients is able to suppress effector responses from healthy subjects in a dose dependent manner at multiple levels of T cell activation (ATP production, proliferation and IFN-γ secretion). These findings suggest that tumor related soluble factors are contributing to the blunted T cell immunity. An increasing body of evidence indicates that tumors create a suppressive immune network involving suppressive Tregs, tolerogenic dendritic cells, and myeloid-derived suppressive cells [16]. Our data also support that HCC-related soluble factors are involved in the observed impairment in T cell immunity.
In a search for soluble factors, our initial analysis did not reveal significant elevations for known immunomodulatory cytokines (IL-10 and TGF-β) in the serum of HCC patients. However, we found significant elevations of sCD25 in the sera of HCC patients compared with patients with HCV-related cirrhosis and normal controls. Our data agree with previous studies showing increased levels of sCD25 in cancer [17]. These studies in solid malignancies correlate increased levels of sCD25 with disease activity and outcome. However, none of these studies characterize the significance and biological role for the observed elevation in sCD25 in the pathogenesis of cancer. The inverse relationship between CD4 T cell activity and serum sCD25 level in HCC patients prompted us to investigate the role of sCD25 on effector T cell responses. The improvement in effector responses after depletion of sCD25 from HCC-derived serum suggests sCD25 is involved as a soluble mediator of T cell suppression. The lack of an improved T cell response using controls with HCV-related cirrhosis without cancer after depletion of sCD25 from their serum suggests that the major source and suppressive effect is related to the presence of HCC. The abrogation of the suppressive effects of the serum from HCC patients by depletion of sCD25 and the sCD25 dose-related suppression of T cells have important implications. These findings suggest a novel immune modulatory role for sCD25 in HCC which is supported by a recent study showing an inhibitory effect of sCD25 on IL-2 signaling by T cells in autoimmunity [18]. In this setting, sCD25 may have an antagonistic role where the high levels of sCD25 in the serum of HCC patients binds IL-2 and inhibits IL-2 dependent T cell activation and proliferation [19, 20]. The observation that maximal doses of IL-2 overcome the blunted T cell immunity related to HCC-derived serum supports the interpretation that effector T cells are suppressed in vivo in the presence of HCC and elevated sCD25. The poor T cell responses to Candida antigens in HCC patients compared to controls also supports that global T cell immunity is impaired in HCC. Furthermore, the data showing that the impairment of T cells in HCC patients is reversible, suggests that there may be a benefit to reducing sCD25 as part of a treatment strategy for HCC.
Soluble cytokine receptors are important regulators of inflammation and immune events through a number of different mechanisms [21]. While a functional role for sCD25 in regulating immune responses is not well described in the literature, there are examples of soluble receptors with functional properties that may serve as a model. For example, soluble IL-6 receptor (sIL-6R) exerts biologic activity by forming sIL-6R-IL-6 trans-signaling complexes leading to tumor progression in animal models [22]. Levels of IL-6 and sIL-6R are elevated in several solid malignancies including HCC where it may contribute to tumor progression in a paracrine or autocrine fashion [23]. In contrast to the agonistic sIL-6R-IL-6 complex, high concentrations of sCD25 appear to have antagonistic properties on T cell responses. In the context of our data, sCD25 may be exerting its inhibitory and apoptosis effect by competing with membrane bound CD25 for IL-2 binding thereby depriving effector T cells of IL-2 signaling [23]. This sCD25 mediated suppressive mechanism may potentially represent a mechanism of tumor escape from immune surveillance.
The correlation of sCD25 levels with tumor burden and overall survival suggests a direct contribution in the production of sCD25 by tumor cells. Our analysis using real time PCR showing higher levels of CD25 in HCC sites in comparison to non-HCC sites in the liver of the same patient serves as evidence that HCC expresses CD25 and may serve as a source of sCD25. Several studies have shown that non-lymphoid cancer cells such as malignant melanoma, kidney cancer, head and neck cancer, and esophageal carcinoma express CD25 on their surface and may contribute to the elevated sCD25 in malignancy [24]. Alternatively, the elevated levels of sCD25 may be the product of PBMC or T cell activation in response to HCC’s development and growth or the release from activated lymphoid cells infiltrating neoplastic tissues [24]. However, our findings show that HCC patients have poorly responding effector T cells with markedly impaired production of sCD25. For example, in our in vitro system when using PBMC from healthy subjects the source of sCD25 appears to be mainly from activation of CD4+CD25− T cells, while in subjects with advanced HCC the main source of sCD25 is the PBMC culture and not the CD4+CD25− effector T cells or CD4+CD25+ T cells. This change in the predominant cell population producing sCD25 implies that a non-T cell fraction plays a key role in sCD25 mediated T cell suppression in HCC that likely includes antigen presenting cells such as dendritic cells (DCs) [25]. Indeed, tumor associated DCs expressing membrane bound CD25 and secreting CD25 (sCD25) have been demonstrated to sequester IL-2 leading to inhibition of IL-2 dependent T cell proliferation. Further proof that HCC has a direct role in elevated levels of sCD25 is the finding that sCD25 decreases with resection or ablation of HCC (data not presented). Although the precise source of sCD25 remains unclear, it appears that high baseline or pretreatment levels of sCD25 in HCC patients reflects its biological aggressiveness by tumor stage and correlates with poor survival. Future studies will expand the analysis to include intrahepatic lymphocytes from tumor-bearing livers to evaluate their ability to secrete CD25.
Our ROC analysis show that a cut-off of >5,800 pg/m sCD25 has a diagnostic accuracy in detecting HCC with sensitivity and specificity rates of 69% and 87%, respectively. At present, serum AFP is the most widely used HCC marker, but guidelines have eliminated its recommendation due to its limited sensitivity and specificity. Further studies are needed to determine the diagnostic accuracy of sCD25 in discriminating among healthy controls, HCC and patients with cirrhosis. Our results are in agreement with literature indicating a potential role for sCD25 as a promising tumor marker in malignancies [24]. Our in vitro studies along with the results showing higher serum levels of sCD25 in HCC patients, brings forth an interesting hypothesis that levels of sCD25 above the range of 3,000–5,800 pg/ml may reflect physiologically a transitional state. For patients at risk for HCC within this range that are immunologically perturbed to produce higher levels of sCD25, the sCD25 mediated T cell suppression mechanism becomes relevant enough to contribute to tumor development and/or progression.
In summary, our data show that patients with HCC have an ineffective immune response characterized by impairment in effector T cell response and an elevation of sCD25, both of which correlate with tumor burden as well as poor outcome. The striking blunted effector responses appear to involve sCD25 as a novel soluble immuno-inhibitory molecule. The observation that sera from patients with HCC have higher levels of sCD25 when compared to patients with HCV-related cirrhosis opens a new venue to explore the possible use of this soluble receptor as a tool for diagnosis, monitoring disease and treatment. Taken collectively, our in vitro studies show that depletion of sCD25 enhances T cell function while supplementation decreases T cell activity. Ongoing studies are focused on the mechanisms involved in the impairment of effector T cells and sCD25 mediated suppression and its relationship to treatment response. The present classification models of HCC that use a strategy of sub-classifying HCC by molecular pathway profiling may benefit from the inclusion of an immunologic profile to further stratify this heterogeneous cancer. This may reveal a novel immunologic pathway that plays a role in tumor development/progression and may reveal novel immunotherapeutic targets in HCC.
Contributor Information
Roniel Cabrera, Email: cabrer@medicine.ufl.edu, Section of Hepatobiliary Diseases, Department of Medicine, University of Florida, 1600 SW Archer Rd., PO Box 100214, Gainesville, FL 32610-0214, USA.
Miguel Ararat, Section of Hepatobiliary Diseases, Department of Medicine, University of Florida, 1600 SW Archer Rd., PO Box 100214, Gainesville, FL 32610-0214, USA.
Mengde Cao, Section of Hepatobiliary Diseases, Department of Medicine, University of Florida, 1600 SW Archer Rd., PO Box 100214, Gainesville, FL 32610-0214, USA.
Yiling Xu, Section of Hepatobiliary Diseases, Department of Medicine, University of Florida, 1600 SW Archer Rd., PO Box 100214, Gainesville, FL 32610-0214, USA.
Clive Wasserfall, Department of Pathology, Immunology, and Laboratory Medicine, University of Florida, Gainesville, FL 32610-0214, USA.
Mark A. Atkinson, Department of Pathology, Immunology, and Laboratory Medicine, University of Florida, Gainesville, FL 32610-0214, USA
Chen Liu, Department of Pathology, Immunology, and Laboratory Medicine, University of Florida, Gainesville, FL 32610-0214, USA.
David R. Nelson, Section of Hepatobiliary Diseases, Department of Medicine, University of Florida, 1600 SW Archer Rd., PO Box 100214, Gainesville, FL 32610-0214, USA
References
- 1.Parkin DM, Bray F, Ferlay J, et al. Estimating the world cancer burden: Globocan 2000. Int J Cancer. 2001;94:153–156. doi: 10.1002/ijc.1440. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB, Davila JA, Petersen NJ, McGlynn KA. The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann Intern Med. 2003;139:817–823. doi: 10.7326/0003-4819-139-10-200311180-00009. [DOI] [PubMed] [Google Scholar]
- 3.Fattovich G, Giustina G, Degos F, et al. Morbidity and mortality in compensated cirrhosis type C: a retrospective follow-up study of 384 patients. Gastroenterology. 1997;112:463–472. doi: 10.1053/gast.1997.v112.pm9024300. [DOI] [PubMed] [Google Scholar]
- 4.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 5.Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 6.Newell P, Villanueva A, Llovet JM. Molecular targeted therapies in hepatocellular carcinoma: from pre-clinical models to clinical trials. J Hepatol. 2008;49:1–5. doi: 10.1016/j.jhep.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 7.Kalos M. Tumor antigen-specific T cells and cancer immunotherapy: current issues and future prospects. Vaccine. 2003;21:781–786. doi: 10.1016/s0264-410x(02)00598-4. [DOI] [PubMed] [Google Scholar]
- 8.Wada Y, Nakashima O, Kutami R, Yamamoto O, Kojiro M. Clinicopathological study on hepatocellular carcinoma with lymphocytic infiltration. Hepatology. 1998;27:407–414. doi: 10.1002/hep.510270214. [DOI] [PubMed] [Google Scholar]
- 9.Cao M, Cabrera R, Xu Y, et al. Hepatocellular carcinoma cell supernatants increase expansion and function of CD4+CD25+ regulatory T cells. Lab Invest. 2007;87:582–590. doi: 10.1038/labinvest.3700540. [DOI] [PubMed] [Google Scholar]
- 10.Ormandy LA, Hillemann T, Wedemeyer H, et al. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005;65 (6):2457–2464. doi: 10.1158/0008-5472.CAN-04-3232. [DOI] [PubMed] [Google Scholar]
- 11.Unitt E, Rushbrook SM, Marshall A, et al. Compromised lymphocytes infiltrate hepatocellular carcinoma: the role of T-regulatory cells. Hepatology. 2005;41:722–730. doi: 10.1002/hep.20644. [DOI] [PubMed] [Google Scholar]
- 12.Fu J, Xu D, Liu Z, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology. 2007;132:2328–2339. doi: 10.1053/j.gastro.2007.03.102. [DOI] [PubMed] [Google Scholar]
- 13.Bruix J, Sherman M. Practice guidelines committee, American association for the study of liver diseases. Management of hepatocellular carcinoma. Hepatology. 2005;42:1208–1236. doi: 10.1002/hep.20933. [DOI] [PubMed] [Google Scholar]
- 14.Nelson DR, Tu Z, Soldevila-Pico C, et al. Long-term interleukin 10 therapy in chronic hepatitis C patients has a proviral and antiinflammatory effect. Hepatology. 2003;38:859–868. doi: 10.1053/jhep.2003.50427. [DOI] [PubMed] [Google Scholar]
- 15.Cabrera R, Ararat M, Soldevila-Pico C, et al. Using an immune functional assay to differentiate acute cellular rejection from recurrent hepatitis C in liver transplant patients. Liver Transpl. 2009;15:216–222. doi: 10.1002/lt.21666. [DOI] [PubMed] [Google Scholar]
- 16.Hoechst B, Ormandy LA, Ballmaier M, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4+CD25+Foxp3+ T cells. Gastroenterology. 2008;135:234–243. doi: 10.1053/j.gastro.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 17.Murakami S. Soluble IL-2 receptor in cancer. Front Biosci. 2004;1(9):3085–3090. doi: 10.2741/1461. [DOI] [PubMed] [Google Scholar]
- 18.Maier LM, Anderson DE, Severson CA, et al. Soluble IL-2RA levels in multiple sclerosis subjects and the effect of soluble IL-2RA on immune responses. J Immunol. 2009;182:1541–1547. doi: 10.4049/jimmunol.182.3.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chopra RK, Powers DC, Kendig NE, Adler WH, Nagel JE. Soluble interleukin 2 receptors released from mitogen stimulated human peripheral blood lymphocytes bind interleukin 2 and inhibit IL2 dependent cell proliferation. Immunol Invest. 1989;18:961–973. doi: 10.3109/08820138909045783. [DOI] [PubMed] [Google Scholar]
- 20.Rubin LA, Jay G, Nelson DL. The released interleukin 2 receptor binds interleukin 2 efficiently. J Immunol. 1986;137(12):3841–3844. [PubMed] [Google Scholar]
- 21.Levine SJ. Mechanisms of soluble cytokine receptor generation. J Immunol. 2004;173:5343–5348. doi: 10.4049/jimmunol.173.9.5343. [DOI] [PubMed] [Google Scholar]
- 22.Becker C, Fantini MC, Schramm C, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 23.Giannitrapani L, Cervello M, Soresi M, et al. Circulating IL-6 and sIL-6R in patients with hepatocellular carcinoma. Ann N Y Acad Sci. 2002;963:46–52. doi: 10.1111/j.1749-6632.2002.tb04093.x. [DOI] [PubMed] [Google Scholar]
- 24.Bien E, Balcerska A. Serum soluble IL-2 receptor alpha in human cancer of adults and children: a review. Biomarkers. 2008;13:1–26. doi: 10.1080/13547500701674063. [DOI] [PubMed] [Google Scholar]
- 25.von Bergwelt-Baildon MS, Popov A, Saric T, et al. CD25 and indoleamine 2, 3-dioxygenase are up-regulated by prostaglandin E2 and expressed by tumor-associated dendritic cells in vivo: additional mechanisms of T-cell inhibition. Blood. 2006;108:228–237. doi: 10.1182/blood-2005-08-3507. [DOI] [PubMed] [Google Scholar]