

Abstract
Carboxylic acids with known central nervous system and histone deacetylase (HDAC) inhibitory activities were converted to hydroxamic acids and tested using a suite of in vitro biochemical assays with recombinant HDAC isoforms, cell based assays in human cervical carcinoma HeLa cells and primary cultures from mouse forebrain, and a whole animal (Xenopus laevis) developmental assay. Relative to the parent carboxylic acids, two of these analogues exhibited enhanced potency, and one analogue showed altered HDAC isoform selectivity and in vivo activity in the Xenopus assay. We discuss potential uses of these novel hydroxamic acids in studies aimed at determining the utility of HDAC inhibitors as memory enhancers and mood stabilizers.
Keywords: HDAC inhibitors, hydroxamic acid, valproate, butyrate, phenylbutyrate, histone acetylation
At present there is great optimism for the development of inhibitors of histone deacetylases (HDACs) as novel therapeutics for memory and mood disorders. Much of this interest derives from studies reporting the enhancement of memory in rodent behavioral models by the short-chain carboxylic acid HDAC inhibitor sodium butyrate.1−5 The butyrate analogue phenylbutyrate, an FDA-approved drug known to inhibit HDACs,6 enhances cognition in an animal model of Alzheimer’s disease.7 Finally, another short-chain carboxylic acid, valproate (VPA), originally developed as an antiepileptic,8 has been shown to inhibit HDAC activity9,10 and is currently also in clinical use as a mood stabilizer for the treatment of bipolar disorder.11 However, these three compounds suffer from low potency and, in the case of VPA, a narrow therapeutic index.12
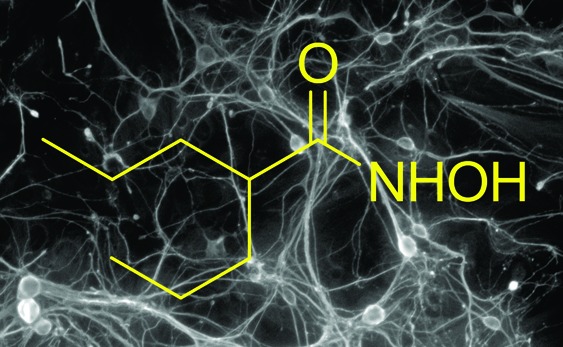
In an attempt to improve potency, these short-chain carboxylic acid HDAC inhibitors were converted to hydroxamic acid analogues. We reasoned that the carboxylic acid moiety itself is critical for inhibition of HDACs because the corresponding amide analogue of VPA is inactive.13 Hydroxamic acid is the zinc-chelating moiety from suberoyl hydroxamic acid, SAHA (Vorinostat; Merck), a highly potent HDAC inhibitor that has been approved by the FDA for the treatment of cutaneous T-cell lymphoma.14 Other hydroxamic acid HDAC inhibitors are currently in clinical development, including Panobinostat (LBH-589; Novartis AG), Belinostat (PDX-10; TopoTarget), and IF2357 (Italfarmaco SpA). Three hydroxamic acids (2,154,16617) were made from the corresponding carboxylic acids butyrate, phenylbutyrate, and valproate (1, 3, 5) by esterification with methanol followed by treatment of the resulting esters with hydroxylamine in methanol under basic conditions (Scheme 1).
Scheme 1.

Based on structural homology, zinc-dependent HDAC isoforms have been divided into three classes: I (HDACs 1, 2, 3, and 8); IIa (4, 5, 7, and 9); IIb (6 and 10); and IV (11). To determine the HDAC inhibitory activity of compounds 1−6, we used a panel of in vitro enzymatic trypsin-coupled assays with recombinant human HDACs 1−9. In these assays, HDAC isoforms were incubated with an acetyl-lysine tripeptide substrate linked to a coumarin fluorophore that is designed to mimic acetyl-lysine 12 of histone H4. Deacetylation of the substrate renders it sensitive to trypsin cleavage; this releases the aminomethylcoumarin (AMC) moiety that has greatly increased fluorescence in the free form. Critical to these studies was the use of a newly developed trifluoro-acetyl-lysine-AMC substrate for class IIa HDACs, which allows the measurement of the enzymatic activity of these HDACs free from contaminating activities of class I HDACs that have previously confounded many studies.18 With the concentrations of these substrates set to the Km for each enzyme, IC50 values were determined for each compound (Table 1).
Table 1. In Vitro Potency of Carboxylic and Hydroxamic Acid HDAC Inhibitorsa.
| class I |
class IIa |
class IIb | |||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC4 | HDAC5 | HDAC7 | HDAC9 | HDAC6 |
| 1 | 16 ± 11 | 12 ± 6 | 9 ± 6 | 15 ± 6 | >2000 | >2000 | >2000 | >2000 | >2000 |
| 2 | 7 ± 9 | 9 ± 10 | 10 ± 11 | 26 ± 20 | >2000 | 360 ± 240 | >1000 | >2000 | 2 ± 2 |
| 3 | 64 ± 18 | 65 ± 9 | 260 ± 160 | 93 ± 110 | >2000 | >2000 | >2000 | >2000 | 240 ± 95 |
| 4 | 0.6 ± 0.4 | 0.6 ± 0.8 | 2 ± 2 | 4 ± 1 | 140 ± 70 | 25 ± 13 | 150 ± 27 | 430 ± 304 | 0.5 ± 0.1 |
| 5 | 39 ± 5 | 62 ± 4 | 161 ± 81 | 103 ± 8 | >2000 | >2000 | >2000 | >2000 | >2000 |
| 6 | 560 ± 90 | 680 ± 280 | 340 ± 170 | 39 ± 9 | 170 ± 95 | 37 ± 4 | 99 ± 4 | 91 ± 30 | 16 ± 1 |
IC50 (μM) values for inhibition of enzymatic activity of HDACs 1−9. Data from two experiments (each performed in duplicate) are shown as mean ± SD.
The data in Table I indicate that carboxylic acids (1, 3, 5) are selective inhibitors of class I HDACs. Notably, in these new and improved assays, we found the potency of the inhibitors to be significantly greater than indicated in previous reports.13 In particular, butyrate (1) was the most potent of the carboxylic acids. While a previous report indicated that conversion of 1 to hydroxamic acid (2) increased its potency toward a mixture of HDACs isolated from rat liver,23 our data indicate increased potency toward class II enzymes, but no effect on inhibition of class I enzymes. The hydroxamic acid analogue of phenylbutyrate (4) had greatly enhanced potency toward all enzymes. In contrast, the hydroxamic acid of VPA (6; which we call VAHA) had reduced potency toward class I HDACs, and instead gained potency toward class II enzymes. This discovery is consistent with another recent report describing class IIa selective hydroxamates.19
We next tested the ability of compounds 1−6 to inhibit HDAC activity in cells (Figure 1). We used immunofluorescence assays to study the effects of these compounds on histone and non-histone substrates in human HeLa cells. Specifically, we measured acetylation of the N-terminal tails of histones H3 and H2A: in both cases, compound effects were similar (Figure 1; data for AcH3 not shown), and reflective of the in vitro potencies of the compounds toward class I HDACs. For a representative non-histone substrate, we used tubulin acetylation as a measure of inhibition of class IIb HDAC isoforms, as tubulin is deacetylated specifically by HDAC6.20 In these assays, the hydroxamic acids 2 and 4 were much more potent at inducing histone acetylation (Figure 1a) than their parent carboxylic acids. In contrast, VAHA (6) had no effect on histone acetylation, consistent with its low potency toward class I HDAC isoforms in vitro. However, 2, 4, and, to a lesser extent, 6 induced tubulin acetylation, consistent with their abilities to inhibit HDAC6 in vitro, whereas the carboxylic acids 1, 3, and 5 had no effect (Figure 1b). Thus, these data indicate that the hydroxamic acids 2, 4, and 6 are cell active HDAC inhibitors with altered potency or HDAC target selectivity that correlates with their in vitro activity.
Figure 1.

Induction of histone acetylation (a) or tubulin acetylation (b) in HeLa cells by carboxylic acid- and hydroxamic acid-based HDAC inhibitors. Histone H2A and tubulin acetylation were measured by immunofluorescence assays after 24 h compound treatments at the indicated doses.
Clinical use of VPA (5) is tempered by concern over its teratogenic effects,21 and the relationship of the isoform specificity of HDAC inhibitory effects of VPA to teratogenicity remains to be clarified. Thus, we tested the effects of VPA (5) and VAHA (6) in a frog embryo-based whole organism development assay (Figure 2). VPA (5), at 2 mM, induced dramatic developmental defects in frog embryos, including loss of anterior structures, shortening of the anterior−posterior axis, and heart looping and pigment defects, as reported previously.21,22 In contrast, at the same dose, VAHA (6) did not induce these developmental defects. VAHA (6) did induce tubulin acetylation, but not histone acetylation (Figure 2b), showing that it was active and selective in the embryos. These data further support the conclusion that conversion of VPA (5) to VAHA (6) results in a novel, class II selective HDAC inhibitor that is active in cells. Moreover, these data suggest that inhibition of class I HDACs by VPA (5) underlies its teratogenicity.
Figure 2.
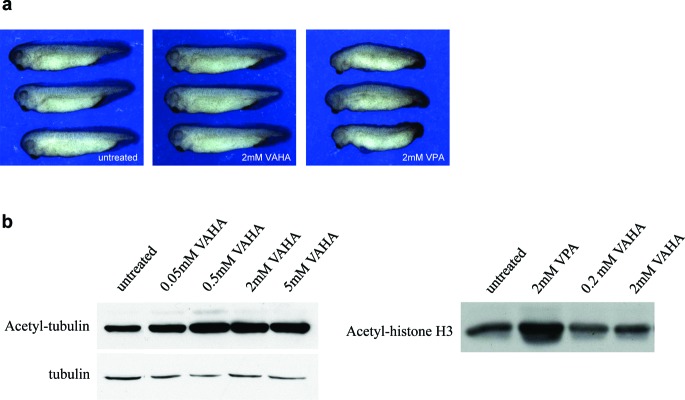
Induction of developmental defects in frog embryos by VPA and VAHA, and effect of VAHA on tubulin acetylation. (a) Xenopus developmental assay: left, untreated; center, VAHA (2 mM); right, 2 VPA (2 mM). (b) Induction of tubulin or histone acetylation in frog embryos by VAHA (6) or VPA (5). Western blot analysis of tubulin or histone acetylation in frog embryos treated at the indicated concentrations from the completion of neurulation (stage 18) through the tailbud stages (32) for 18 h (tubulin acetylation; as described in ref (21)) or 2 h (histone acetylation).
Finally, we tested the effects of compounds 1−6 on histone and tubulin acetylation in primary cultures of mouse forebrain neurons (Figure 3). The forebrain contains several regions (cortex, hippocampus, striatum) known to play key roles in mood and memory.24,25 At a concentration of 92.5 μM, the hydroxamic acid derivatives 2 and 4 induced robust histone acetylation, whereas the carboxylic acids (1, 3, 5), as well as VAHA (6), were inactive at this concentration. In contrast, VAHA (6) was able to induce tubulin acetylation in forebrain neurons, as did carboxylic acid 3.
Figure 3.

Induction of histone acetylation (a) and tubulin acetylation (b) in mouse primary forebrain cultures. Acetylation of histone H3, lysine 9 or tubulin was measured by immunofluorescence after 24 h compound treatments (compounds 1−6, 92.5 mM). * signifies statistical significance (p < 0.05).
In conclusion, here we report the synthesis and activity testing of novel hydroxamic analogues of short-chain carboxylic acid HDAC inhibitors. To date, the carboxylic acids butyrate (1) and phenylbutyrate (3) have been shown to enhance memory in rodent behavioral models.1−5 We propose that the hydroxamic acid analogues of butyrate and phenylbutyrate (2 and 4) might show even greater activity in these memory models, given their enhanced in vitro potency and cellular activity (Table 1 and Figure 1). The hydroxamic acid analogue of valproate, VAHA (6), had an in vitro and cellular activity profile consistent with it being a class II selective inhibitor. Interestingly, similar hydroxamic acid analogues of VPA have been shown to readily cross the blood−brain barrier,22 and VAHA has been shown to have anticonvulsant activity in vivo.17 Comparison of the activities of VPA (5) and VAHA (6) in rodent behavioral models could comprise an important test of the role of class I vs class II HDACs in regulation of mood and memory.
Supporting Information Available
Experimental procedures for the synthesis of hydroxamic acids 2, 4, and 6, and procedures for the in vitro HDAC inhibition assays, cellular histone and tubulin acetylation assays, and Xenopus embryonic development assays. This material is available free of charge via the Internet at http://pubs.acs.org.
This project has been funded in whole or in part with Federal funds from the National Cancer Institute’s Initiative for Chemical Genetics, NIH, under Contract No. N01-CO-12400. P.S.K. is supported by a grant from the NIH (1RO1MH58324). S.J.H is supported by a grant from the NIDA/NIH (5R01DA028301-02) and the Stanley Medical Research Institute.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Levenson J. M.; O’Riordan K. J.; Brown K. D.; Trinh M. A.; Molfese D. L.; Sweatt J. D. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 2004, 279, 40545–59. [DOI] [PubMed] [Google Scholar]
- Fischer A.; Sananbenesi F.; Wang X.; Dobbin M.; Tsai L. H. Recovery of learning and memory is associated with chromatin remodeling. Nature 2007, 447, 178–82. [DOI] [PubMed] [Google Scholar]
- Dash P. K.; Orsi S. A.; Moore A. N. Histone deactylase inhibition combined with behavioral therapy enhances learning and memory following traumatic brain injury. Neuroscience 2009, 163, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanko D. P.; Barrett R. M.; Ly A. R.; Reolon G. K.; Wood M. A. Modulation of long-term memory for object recognition via HDAC inhibition. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 9447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgore M.; Miller C. A.; Fass D. M.; Hennig K. M.; Haggarty S. J.; Sweatt J. D.; Rumbaugh G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2010, 35, 870–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea M. A.; Randolph V. M.; Hodge S. K. Induction of histone acetylation and growth regulation in eryrthroleukemia cells by 4-phenylbutyrate and structural analogs. Anticancer Res. 1999, 19, 1971–6. [PubMed] [Google Scholar]
- Ricobaraza A.; Cuadrado-Tejedor M.; Pérez-Mediavilla A.; Frechilla D.; Del Río J.; García- Osta A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease. Neuropsychopharmacology 2009, 34, 1721–32. [DOI] [PubMed] [Google Scholar]
- Loscher W. Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy. CNS Drugs 2002, 16, 669–94. [DOI] [PubMed] [Google Scholar]
- Phiel C. J.; Zhang F.; Huang E. Y.; Guenther M. G.; Lazar M. A.; Klein P. S. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 2001, 276, 36734–41. [DOI] [PubMed] [Google Scholar]
- Gottlicher M.; Minucci S.; Zhu P.; Kramer O. H.; Schimpf A.; Giavara S.; Sleeman J. P.; Lo Coco F.; Nervi C.; Pelicci P. G.; Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad P. M.; Das A.; Ashfaq M.; Wieck A. A review of valproate in psychiatric practice. Expert Opin. Drug Metab. Toxicol. 2009, 5, 539–51. [DOI] [PubMed] [Google Scholar]
- Walia K. S.; Khan E. A.; Ko D. H.; Raza S. S.; Khan Y. N. Side effects of antiepileptics-a review. Pain Pract. 2004, 4, 194–203. [DOI] [PubMed] [Google Scholar]
- Gurvich N.; Tsygankova O. M.; Meinkoth J. L.; Klein P. S. Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer Res. 2004, 64, 1079–86. [DOI] [PubMed] [Google Scholar]
- Kavanaugh S. A.; White L. A.; Kolesar J. M. Vorinostat: A novel therapy for the treatment of cutaneous T-cell lymphoma. Am. J. Health Syst. Pharm. 2010, 67, 793–7. [DOI] [PubMed] [Google Scholar]
- Fishbein W. N.; Daly J.; Streeter C. L. Preparation and some properties of stable and carbon-14 and tritium-labeled short-chain aliphatic hydroxamic acids. Anal. Biochem. 1969, 28, 13–24. [DOI] [PubMed] [Google Scholar]
- Huang F. C.; Shoupe T. S.; Lin C. J.; Lee T. D.Y.; Chan W. K.; Tan J.; Schnapper M.; Suh J. T.; Gordon R. J. Differential effects of a series of hydroxamic acid derivatives on 5-lipoxygenase and cyclooxygenase from neutrophils and 12-lipoxygenase from platelets and their in vivo effects on inflammation and anaphylaxis. J. Med. Chem. 1989, 32, 1836–42. [DOI] [PubMed] [Google Scholar]
- Levi M.; Yagen B.; Bialer M. Pharmacokinetics and antiepileptic activity of valproyl hydroxamic acid derivatives. Pharm. Res. 1997, 14, 213–7. [DOI] [PubMed] [Google Scholar]
- Bradner J. E.; West N.; Grachan M. L.; Greenberg E. F.; Haggarty S. J.; Warnow T.; Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6, 238–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier P.; Smil D. V.; Wahhab A.; Leit S.; Rahil J.; Zuomei L.; Deziel R.; Besterman J. M. Diphenylmethylene hydroxamic acids as selective class IIa histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 5684–8. [DOI] [PubMed] [Google Scholar]
- Hubbert C.; Guardiola A.; Shao R.; Kawaguchi Y.; Ito A.; Nixon A.; Yoshida M.; Wang X. F.; Yao T. P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–8. [DOI] [PubMed] [Google Scholar]
- Gurvich N.; Berman M. G.; Wittner B. S.; Gentleman R. C.; Klein P. S.; Green J. B. Association of valproate-induced teratogenesis with histone deacetylase inhibition in vivo. FASEB J. 2005, 19, 1166–8. [DOI] [PubMed] [Google Scholar]
- Eikel D.; Hoffmann K.; Zoll K.; Lampen A.; Nau H. S-2-pentyl-4-pentynoic hydroxamic acid and its metabolite S-2-pentyl-4-pentynoic acid in the NMRI-EXENCEPHALY-mouse model: pharmacokinetic profiles, teratogenic effects, and histone deacetylase inhibition abilities of further valproic acid hydroxamates and amides. Drug Metab. Dispos. 2006, 34, 612–20. [DOI] [PubMed] [Google Scholar]
- Skarpidi E.; Cao H.; Heltweg B.; White B. F.; Marhenke R. L.; Jung M.; Stamatoyannopoulos G. Hydroxamide derivatives of short-chain fatty acids are potent inducers of fetal globin gene expression. Exp. Hematol. 2003, 31, 197–203. [DOI] [PubMed] [Google Scholar]
- Clark L.; Chamberlain S. R.; Sahakian B. J. Neurocognitive mechanisms in depression: implications for treatment. Annu. Rev. Neurosci. 2009, 32, 57–74. [DOI] [PubMed] [Google Scholar]
- Dickerson B. C.; Eichenbaum H. The episodic memory system: neurocircuitry and disorders. Neuropsychopharmacology 2010, 35, 86–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.