

Abstract
Enzymes are long established as extremely efficient catalysts. Here, we show that enzymes can also be extremely efficient electrocatalysts (catalysts of redox reactions at electrodes). Despite being large and electronically insulating through most of their volume, some enzymes, when attached to an electrode, catalyze electrochemical reactions that are otherwise extremely sluggish (even with the best synthetic catalysts) and require a large overpotential to achieve a useful rate. These enzymes produce high electrocatalytic currents, displayed in single bidirectional voltammetric waves that switch direction (between oxidation and reduction) sharply at the equilibrium potential for the substrate redox couple. Notoriously irreversible processes such as CO2 reduction are thereby rendered electrochemically reversible—a consequence of molecular evolution responding to stringent biological drivers for thermodynamic efficiency. Enzymes thus set high standards for the catalysts of future energy technologies.
Keywords: electrocatalysis, catalysis, electrochemistry, electron transport, solar fuels
An electrocatalyst catalyzes a redox “half reaction” in which a chemical transformation is coupled to electron transfer at an electrode (1). The active sites of surface electrocatalysts such as platinum are integral to the electrode and contribute to the Fermi level, whereas molecular electrocatalysts are distinct entities with their own electronic and chemical properties. Molecular electrocatalysts can be attached to the electrode surface or diffuse freely in solution, but depend upon interfacial electron transfer (IET). Enzymes, a special category of molecular electrocatalysts, are distinguished by their extraordinary activities, yet limited by their size and fragility. Driven by industrial and technological needs for significant improvements in rates and efficiency, enzymes can provide crucial insights into the principles underpinning the design and performance of synthetic molecular electrocatalysts.
The efficiency of enzyme catalysis is widely accepted (2–4): Enzymes have highly selective substrate binding sites, avoid releasing reactive intermediates, and decrease activation energies (here, substrate refers to the species being transformed, not the supporting material). Traditional definitions of enzyme efficiency focus on how closely the rate approaches diffusion control (4). Recently, electrochemistry has revealed a hitherto unquantified dimension in enzyme catalysis; many redox enzymes minimize the energy needed to drive a reaction—a quantity that is easily visualized electrochemically and that we refer to as the overpotential requirement. The energy efficiency of enzyme catalysis is expected because biology must fully exploit available energy resources and minimize energy losses.
The performance of an electrocatalyst is readily visualized by cyclic voltammetry, a technique for driving reactions, measuring kinetics and thermodynamics, detecting activation/inactivation processes, and observing catalytic efficiency—all in a single experiment (5). To explain the term “overpotential requirement,” we refer to the two voltammograms in Fig. 1A. They depict the simple (uncatalyzed) interconversion of the oxidized (Ox) and reduced (Red) forms of a redox-active species. Importantly, in Fig. 1A and in all cases discussed here, both Ox and Red are present in solution: Thus, according to the Nernst equation, their activities (concentrations) define an important reference point, the equilibrium potential (Eeq). Electrochemically, Eeq is the open circuit potential established by the mixture of substrates, or the applied potential at which no net current flows. When the applied potential (E) is above Eeq (E > Eeq) Red is oxidized at the electrode, to adjust the concentrations of Ox and Red to the ratio set by the Nernst equation. When IET is too slow to maintain Nernstian equilibrium as E is changed, the voltammetry is termed irreversible (Fig. 1A, red): Very little current is observed until a sizeable overpotential (|E - Eeq|) is applied (for practical purposes, this is the overpotential requirement), and the exponentially increasing current is described by the Butler–Volmer equation (5). In the limiting case of an electrochemically reversible reaction (Fig. 1A, blue), IET is fast enough to maintain Nernstian equilibrium, and even a minuscule overpotential produces a significant net current that reflects the thermodynamics (the shifting Nernstian equilibrium), not the exponentially increasing rate of IET. The difference between the two cases is embodied in the exchange current (the magnitude of the equal oxidation and reduction currents that comprise the dynamic equilibrium at Eeq) (5). Irreversible systems have very low exchange currents and are unresponsive to potential changes close to Eeq; systems that approach the reversible limit have very high exchange currents and respond strongly.
Fig. 1.

Concepts applied in this article. (A) Steady-state electrochemical kinetics visualized by cyclic voltammetry. When both the oxidized and reduced forms of a redox-active species are present, a reversible electrochemical reaction (one with a large exchange current density) produces a single sigmoidal wave (blue) that cuts (without inflection) through the zero-current axis at the equilibrium potential (Eeq) and achieves a potential-independent limiting current in either direction at relatively low overpotential. Conversely, if the exchange current density is low, the current is negligible around Eeq and two sigmoidal waves (red), one for either direction, are separated in potential, emerging from the baseline with an exponential dependence on potential: A substantial overpotential is required to match the current produced by the reversible system. (B) Cartoon showing an adsorbed enzyme functioning as a molecular electrocatalyst.
To extend the concept of electrochemical reversibility to electrocatalysis by molecules attached to electrodes (see Fig. 1B), we adopt, pragmatically, the term “electrocatalytic exchange current” that embodies not only IET, but also the turnover of the catalytic center and (importantly for enzymes) intramolecular electron transfer, each of which may limit electrocatalysis. Note that the electrocatalytic exchange current remains defined at Eeq, not at the potential of any of the redox centers in the electrocatalyst. Thus, we refer to electrocatalysts with low electrocatalytic exchange currents (and large overpotential requirements) as irreversible, and electrocatalysts with high electrocatalytic exchange currents (and very little overpotential requirement in either direction) as reversible or efficient. In reality, electrocatalytic waveshapes rarely conform to the expectations for simple electrochemical systems, precluding simple quantitative definitions for terms such as the overpotential requirement. Finally, in electrocatalysis, as in simple electrochemistry, a potential-independent process eventually becomes rate limiting at high overpotential. It is highly desirable (returning to the traditional definition of enzyme efficiency; ref. 4) that the limiting current density (current per unit electrode area) should approach diffusion control. However, enzymes are large molecules, so they have large surface area requirements and low active-site densities; this feature must be taken into account when comparing the intrinsic abilities of enzyme and surface electrocatalysts.
Results from Enzyme Electrocatalysts
Fig. 2 shows voltammograms from several enzymes that contain the active sites shown in Scheme 1. Each enzyme, adsorbed on an electrode, catalyzes a half-cell reaction of technological importance, which does not occur without a catalyst and usually exhibits a large overpotential requirement.
Fig. 2.

Direct electrocatalysis by enzymes visualized using cyclic voltammetry. All enzymes were adsorbed on rotating-disc pyrolytic graphite edge electrodes and both the oxidized and reduced substrates are present (in each case, • indicates Eeq, referenced to Standard Hydrogen Electrode). (A) Reversible interconversion of H+ and H2 by hydrogenase-2 from Escherichia coli (pH 6, 10% H2 in Ar, 30 °C; ref. 48). (B) Reversible interconversion of CO2 and CO by CODH 1 from C. hydrogenoformans (pH 7, 50% CO in CO2, 25 °C; ref. 17). (C) Reversible interconversion of CO2 and formate by FDH1 from S. fumaroxidans (pH 6.4, 10 mM carbonate, 10 mM formate, 37 °C; ref. 16). (D) Reversible interconversion of NADH and NAD+ by the hydrophilic domain of mitochondrial complex I (pH 7.8, 1 mM NADH, 1 mM NAD+, 20 °C; ref. 22). Multiple scans are included in C and D to aid identification of the zero-current points. (E) Irreversible reduction of O2 to H2O by either Pt(111), or by bilirubin oxidase from Myrothecium verrucaria (pH 5.8, 100% O2, 20 °C; ref. 27; the Pt(111) voltammogram has been decreased in size to faciliate comparison).
Scheme 1.

Representations of the structures of the catalysts discussed in this article. Bound substrates or intermediates are in red and parts of the supramolecular structure that are implicated in the mechanism in gray. (1A) Active site of a [NiFe]-hydrogenase shown with a bridging hydride (8). (1B) Active site of a [FeFe]-hydrogenase, showing the likely position of bound H2 (8). (2) Active site of a CODH (structure from a crystallographically defined intermediate) (14). (3) Active site of a tungsten-containing FDH (15). (4) Active site of complex I with NADH stacked above the central ring of the flavin (20). (5) Active site of laccase shown with a bound O–O species assigned as peroxide (38). (6) Generic structure of the bisdiphosphine nickel complexes that are functional hydrogenase analogues (46).
Interconversion of H2 and H+.
Interconversion of H2 and H+ is central to H2 fuel cells and renewable H2 generation; it is also crucial in the metabolism of microbes that use metalloenzymes known as hydrogenases to catalyze H2 evolution (to relieve reductive stress) or H2 oxidation (H2 as an energy source).
The long-established electrocatalyst for H2 cycling, applied in proton-exchange membrane (PEM) fuel cells, is elemental platinum: H2 is reduced reversibly at Pt, with a high-exchange current density (at least 1 mA cm-2) (6, 7). However, if future transportation depended solely on such fuel cells, then demand for Pt would greatly exceed resources. In contrast, hydrogenases contain only common metals (iron and nickel) in their active sites (see Scheme 1, 1A and 1B) (8). Some hydrogenases oxidize H2 at ≫1,000 s-1 and, on a per active-site basis, their electrocatalytic activities may exceed that of Pt (7). Fig. 2A shows that electrocatalysis by a [NiFe]-hydrogenase is reversible—the single, bidirectional electrocatalytic wave cuts sharply through zero current at Eeq. Common features of all hydrogenase mechanisms proposed so far are η2 - H2 binding (Scheme 1, 1B), metal hydrido species (Scheme 1, 1A), and closely positioned Brønsted groups (linked to an H+-relay to solvent) for coupled H+ transfer (9).
Activation and Reduction of CO2 to CO or Formate.
At present, the only viable large-scale process for CO2 reduction is photosynthesis, though its overall efficiency is low. An efficient catalyst for converting CO2 directly to methanol or formate fuels, or to CO for Fischer–Tropsch hydrocarbon production, could revolutionize green energy technologies, hence there are extensive efforts to develop electrocatalysts that reduce CO2 (10–13). Normally, CO2 reduction proceeds via unstable intermediates such as 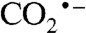 or HOCO• radicals; large overpotentials are required, mixtures of products are produced, and side reactions such as H2 formation compete. The most promising electrocatalysts so far are polymeric [Ru(bpy)(CO)2]n films that produce CO or formate, depending on the bipyridyl substituents, at modest overpotentials (10), and a pyridine system that can reduce CO2 to methanol, also at low overpotential (13).
or HOCO• radicals; large overpotentials are required, mixtures of products are produced, and side reactions such as H2 formation compete. The most promising electrocatalysts so far are polymeric [Ru(bpy)(CO)2]n films that produce CO or formate, depending on the bipyridyl substituents, at modest overpotentials (10), and a pyridine system that can reduce CO2 to methanol, also at low overpotential (13).
In contrast, biology has evolved specific enzymes for reducing CO2 to CO and formate, which behave as reversible electrocatalysts. Carbon monoxide dehydrogenases (CODHs) from anaerobic organisms contain a [Ni4Fe-4S] cluster (see Scheme 1, 2) (14) that catalyzes the interconversion of CO2 and CO (the C atom of the substrate is coordinated to the Ni, and the leaving O-atom coordinates as hydroxide to the “dangling” Fe). Formate dehydrogenases (FDHs) with tungsten-pterin active sites (see Scheme 1, 3) (15) catalyze the interconversion of CO2 and formate (16)—formally a hydride-transfer reaction, but the mechanism is unclear. Fig. 2 B and C show that CODH1 from Carboxydithermus hydrogenoformans and FDH1 from Syntrophobacter fumaroxidans are reversible electrocatalysts (16, 17); their single, bidirectional electrocatalytic waves cut sharply through zero current at Eeq.
Specific C–H Bond Transformations.
Electrochemically driven (C–H) hydride-transfer reactions, such as NADH oxidation, have many applications; but despite significant efforts, large overpotentials are required, and mixtures of products often result, for example, from the NAD• radical intermediate (18, 19). Two flavoenzymes in the mitochondrial electron-transfer chain, respiratory complexes I and II (the NADH: and succinate:ubiquinone oxidoreductases), catalyze reversible C–H bond formation. Reversible oxidation of NADH comprises removal of the C4 hydridic H atom from the pyridinium ring (stacked above the flavin isoalloxazine system; Scheme 1, 4) (20), whereas succinate oxidation to fumarate involves removal of H+ and H- to form a C = C bond (21). Both reactions exploit a flavin’s ability to mediate hydride and electron transfers. Fig. 2D shows that the membrane-extrinsic domain of complex I (subcomplex 1λ) is a reversible catalyst for NAD+/NADH interconversion (22). The membrane-extrinsic domain of complex II is also a reversible electrocatalyst (23).
Interconversion of O2 and H2O.
Interconversion of O2 and H2O is central to fuel cells and future energy technologies that use solar energy or surplus electricity to split water, but no reversible electrocatalyst (biological or artificial) is known. The best nonbiological electrocatalysts for the oxygen reduction reaction are Pt-based surfaces, but they have sizeable overpotential requirements [Pt3Ni(111) provides a 0.1-V improvement over Pt(111) alone; ref. 24]. The overpotential requirement for O2 reduction is a major reason why PEM fuel cells produce a voltage far short of the reversible cell potential (25). Enzymes perform better, and relatively efficient four-electron reduction of O2 to H2O is catalyzed by the “blue” Cu oxidases, laccase (26), and bilirubin oxidase (27). These enzymes contain a tri-Cu active site and a nearby blue Cu center that provides a fast electron relay (see Scheme 1, 5). Fig. 2E compares O2 reduction, at pH of approximately 6, by Pt(111) and by bilirubin oxidase adsorbed on graphite. Despite the high activity per enzyme molecule, the low number of active sites per unit electrode area gives a low current density. Both traces show irreversible electrocatalysis, but the enzyme’s overpotential requirement is significantly smaller than that of Pt.
Our enzyme electrocatalysts incorporate many common features. Their active sites are buried in the protein to exclude solvent, protect fragile inner coordination spheres, and provide highly organized outer-sphere (supramolecular) environments. Precisely positioned close-range functionalities deliver protons on demand and synchronize proton and electron transfers, while chains of H2O molecules and protein bases mediate long-range proton exchange with solvent. Multiple electron capacity is available in or close to the active site, and long-range electron transfer is handled by fast relays.
Principles for Minimizing Electrocatalytic Overpotential Requirements
For the simplest surface electrocatalysts, IET and substrate transformation occur via a single transition state, and the overpotential requirement corresponds to the activation energy. However, almost all redox enzymes catalyze the transfer of two or more electrons, coupled to transfer of protons and other species such as oxygen atoms. In these complex systems, an overpotential requirement can stem from various factors including poor interfacial or intramolecular electron-transfer kinetics, poorly synchronized coupled electron-transfer reactions with high-energy intermediates, and poor matching between the redox thermodynamics of substrate and catalyst.
Rate-Limiting Electron Transfer to/from the Active Site.
A molecular electrocatalyst facilitates two half cycles: the catalytic redox transformation and regeneration of the active site. Electrochemically reducing or oxidizing an enzyme’s active site requires both interfacial (potential-dependent) and intramolecular (potential-independent) electron transfer. Rates of long-range electron tunneling reactions are governed by electronic coupling between the donor and the acceptor, and by the activation energy, ΔGact, which in the semi-Classical Marcus model (28) is minimized when λ, the reorganization energy, is equal to -ΔG, the Gibbs free-energy change. Typically, an intramolecular electron-transfer pathway, such as the series of seven iron-sulfur clusters in respiratory complex I (20), connects the active site to a distal site closest to the surface (Fig. 1B). Intramolecular relays can be optimized by minimizing the tunnel barriers (distances between sites should be < 14 Å) (29), avoiding large free-energy changes (steps in potential) and minimizing reorganization energies (burying a site ensures λsolvent is small). If all the relay steps are fast, intramolecular electron transfer, even to a deeply buried active site, may have little impact on electrocatalysis. Alternatively, a particular relay site may constitute an electrocatalytic control center (30) that creates a potential-determining step: Electron exchange with the electrode up to, but not beyond, this point is fast and reversible. The rate of IET (between the distal relay site and the electrode) always depends on the electrode potential; the distal site must also be positioned close to the electrode surface to minimize the interfacial tunnel barrier. The fact that only a fraction of the possible orientations of an adsorbed enzyme fulfil this requirement modifies the dependence of IET rate on potential, accounting for an extended potential dependence in the electrocatalytic voltammetry (31, 32).
Proton and Electron-Transfer Reactions Within the Active Site.
Electron transfers within active sites drive chemical rearrangements that usually involve proton transfers. As protons can tunnel only very small distances (≪ 1 Å) the organization of the active site, and precise positioning of proton donors and acceptors, are critical (3): An excellent example is the bridgehead N atom positioned just above one of the Fe atoms in the active site of [FeFe]-hydrogenases (Scheme 1, 1B) (8). In electrocatalysis, both parts of a proton-coupled electron transfer (PCET) reaction must be synchronized (33, 34). Stepwise PCET reactions proceed via unstable (high energy) intermediates, as visualized through the Pourbaix (E-pH) diagram shown in Fig. 3A; on the other hand, they provide the basis for pumps and gates—devices that transduce or control energy flow in respiration and photosynthesis. The free intermediates provide limiting scenarios for explaining an overpotential requirement. Fig. 3A shows how the reduction and protonation of Ox to form RedH (at pH 7) occurs in a stepwise reaction, via the unstable Red intermediate. First, an overpotential is required to energize the system (1 → 3) and drive electron transfer to populate the Red state (3 → 4); the unstable Red intermediate is then protonated spontaneously (4 → 5) and energy is released as heat. When the two steps are entirely independent, the overpotential requirement is reflected in the difference between the pH-dependent Ox/RedH potential and the pH-independent Ox/Red potential. As the two steps become better kinetically coupled, the overpotential requirement decreases (note that kinetic coupling is a relative term; stepwise reactions appear better coupled on slower, less demanding catalytic timescales) (35). Only a truly concerted PCET reaction, which crosses the diagonal boundary line directly through a single transition state (1 → 2 in Fig. 3A), remains completely coupled under the most demanding conditions. Similar arguments apply when electron transfers are coupled to other chemical steps, such as transfer of a hydroxide or oxido group.
Fig. 3.

Overpotential requirements resulting from high-energy catalytic intermediates. (A) The Pourbaix diagram for a PCET reaction denotes the most stable species at each potential and pH. PCET reactions occur at intermediate pH, when only the reduced species (Red) is protonated. The inset shows a vertical slice through the main diagram at pH 7, showing how the energy of each state varies, relative to RedH. Concerted PCET reaction, 1 → 2 (no overpotential); stepwise reaction, 1 → 3 (overpotential), 3 → 4 (electron transfer), 4 → 5 (spontaneous H+ transfer). See text for details. (B) Frost (oxidation state) diagram showing the redox reactions of H+/H2, O2/H2O (49), and CO2/CO or formate (pH 7) (16, 17). The slope of the line connecting two species gives the reduction potential for their interconversion; intermediates above connecting lines are unstable with respect to disproportionation. The vertical displacement of an intermediate, above the line for the substrate reaction, indicates the overpotential requirement associated with its formation.
Multiple Electron and Proton Transfer Processes Within the Active Site.
Multiple electron–proton transfer reactions of small molecules are conveniently viewed using Frost (oxidation state) diagrams (36). Frost diagrams show how the free energy of the system varies with oxidation number (the slope of a line between two species gives the reduction potential for their interconversion) and refer to free species at equilibrium in aqueous solution (they are a limiting case of the diagrams used to describe bound species in heterogeneous catalysis; ref. 25). Fig. 3B shows Frost diagrams for some of the reactions discussed here. The energy-rich fuels and oxidants (H2, CO, HCOO-, and O2) appear toward the top, and the energy-poor products of fuel consumption (H2O, H+, and CO2) lower down. Between the fuels/oxidants and their products lie intermediates through which (formally) each species must pass; unstable intermediates lie above the line connecting reactant and final product, and their formation produces an overpotential requirement due to the difference between the potentials required to form the intermediate and the final product.
The cause of the large overpotential usually required to reduce CO2 to CO or formate is evident from Fig. 3B—CO2 reduction in two discrete one-electron steps requires formation of the 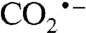 radical. Similarly, reduction of H2 in one-electron steps via an H-atom intermediate would require an overpotential as high as 1.7 V (see Fig. 3B), and uncatalyzed interconversion of NADH and NAD+ via the NAD• radical requires up to 0.7 V (18). Electrocatalysts avoid, or minimize, these overpotential requirements in two ways. First, they can kinetically couple the formation and onward reaction of the unstable intermediate: The limit of this approach, adopted for the interconversions of NAD+/NADH, H+/H2, and perhaps CO2/formate, is transfer of a hydride moiety (two electrons and one proton moving as a single entity). Second, the enzyme may bind and stabilize the intermediate, adjusting its Frost-diagram coordinate down toward the line joining reactant and final product.
radical. Similarly, reduction of H2 in one-electron steps via an H-atom intermediate would require an overpotential as high as 1.7 V (see Fig. 3B), and uncatalyzed interconversion of NADH and NAD+ via the NAD• radical requires up to 0.7 V (18). Electrocatalysts avoid, or minimize, these overpotential requirements in two ways. First, they can kinetically couple the formation and onward reaction of the unstable intermediate: The limit of this approach, adopted for the interconversions of NAD+/NADH, H+/H2, and perhaps CO2/formate, is transfer of a hydride moiety (two electrons and one proton moving as a single entity). Second, the enzyme may bind and stabilize the intermediate, adjusting its Frost-diagram coordinate down toward the line joining reactant and final product.
The interconversion of O2 and H2O is the most challenging reaction discussed here, as all free intermediates are unstable with respect to O2 and H2O (Fig. 3B). Although the direct reaction best avoids an overpotential requirement, it requires a single transition state in which the four electrons and four proton transfers are concerted with O–O cleavage—no known catalyst achieves this. More realistically, the reaction is split into two or more manageable stages, involving intermediates that are stabilized by binding them: This concept is important for developing synthetic O2 reduction electrocatalysts (25). For example, pausing at H2O2 relaxes the four-electron problem but introduces an overpotential requirement of up to 0.54 V in both directions (nevertheless, a lower cost than pausing at either radical state). Specific binding of the intermediate helps further: Indeed, spectroscopic studies of blue Cu oxidases have detected a peroxy- intermediate (37), and X-ray diffraction has revealed an O–O species coordinated between three Cu atoms (38) (see Scheme 1, 5).
Reduction Potential of the Catalyst Is Poorly Matched to That of the Substrate.
If the reduction potential of the active site (even when a substrate is bound) differs significantly from that of the reaction being catalyzed, then the catalyst will function only in one direction. For example, if the reduction potential of the active site is 0.5 V below that of the substrate, only a reductive electrocatalytic wave will be observed (close to the active-site potential) and no amount of overpotential will drive the oxidative reaction at any useful rate—an asymmetry that is not expected for surface electrocatalysts. The reduced active site resembles an unstable intermediate because it requires an overpotential (relative to Eeq) to drive its formation. If the potential mismatch is small, electrocatalysis will remain bidirectional but will be biased in one direction; in Fig. 2D, catalysis is biased toward NAD+ reduction. Viewed another way, the electrocatalytic exchange current is optimized when the reduction potential of the molecular electrocatalyst matches that of the reaction being catalyzed. Fig. 2C exemplifies the importance of a matched active-site potential, because CO2 is easily reduced by the tungsten center in Scheme 1 (4); in contrast, the corresponding higher-potential molybdenum center has only been observed to catalyze formate oxidation. In Fig. 2E, O2 reduction by bilirubin oxidase is irreversible, but the overpotential requirement results not from sluggish IET kinetics, but because the blue Cu (which relays electrons to the active site, Scheme 1, 5) is the electrochemical control center that determines the catalytic potential (27). Similarly, when a redox mediator is used to transfer electrons between enzyme and electrode, catalysis usually occurs at the mediator potential. For example, O2 reduction by laccase embedded in a hydrogel with covalently attached Os complexes occurs close to the reduction potential of the particular Os complex used (39).
Overpotentials in Biology
The electrocatalytic efficiency of an enzyme addresses only the oxidation or reduction of a single substrate, whereas biological redox reactions occur in pairs. A reversible enzyme-catalyzed redox reaction conserves the most energy, but many enzyme reactions are coupled to perform useful work. NADH oxidation by respiratory complex I is reversible (Fig. 2D), but when coupled to ubiquinone reduction (at a distant site) the approximate 0.5-V potential difference renders the redox reaction alone irreversible (and very inefficient). In the cell, most of the free energy is conserved by proton translocation across an energy transducing membrane, creating the proton-motive force that is used to power processes such as ATP synthesis. The complete proton-coupled redox reaction is reversible. Conversely, some enzymes have evolved to act irreversibly, to prevent back-reactions and maintain fluxes through pathways. Cytochrome c oxidase is the last enzyme in the mitochondrial respiratory chain; it reduces O2 to water in a series of stepwise electron–proton transfers that are tightly coupled to proton translocation (40). Only part of the energy from this reaction is conserved in proton pumping, and even the largest proton-motive forces available to biology are unable to drive the reverse reaction. Similarly, the photosynthetic Mn4CaO4 center uses a series of light-driven, mainly stepwise PCET reactions to oxidize water to O2, but it does not catalyze O2 reduction [electron transfer from O2 to P680+ (reduction potential ca. 1.25 V) through the Mn center is irreversible; ref. 41].
Translations into Emerging Energy Technologies
Maximal rates and efficiencies of energy conversion are highly desirable in emerging technologies, such as production of solar fuels. The energy-conserving efficiencies of electrocatalytic enzymes have been revealed in two demonstrations, each of which would not be possible if the catalysts exhibited significant overpotential requirements. First, a hydrogenase and a CODH coattached to conducting graphite platelets catalyze the water gas shift reaction (free-energy change of just 0.1 V) (42) (Fig. 4A): An aqueous suspension of platelets yields 2.5 mol H2 (mol hydrogenase)-1 s-1 at 30 °C, whereas the industrial reaction requires temperatures exceeding 200 °C for similar per-site activity. Second, enzymes attached to anatase nanoparticles provide excellent models for solar fuel conversion (Fig. 4B). As an alternative to UV (band gap) excitation, a coattached Ru-bipyridyl photosensitizer is used to populate the conduction band using visible light. Hydrogenases and CODH catalyze H2 production and CO production, respectively, despite the conduction band potential offering only small driving forces—a [NiFeSe]-hydrogenase uses approximately 0.16 V to drive H2 production at 50 mol H2 (mol hydrogenase)-1 s-1 (18,000 h-1) (43) and CODH uses approximately 0.05 V to catalyze CO production at 0.2 mol CO (mol CODH)-1 s-1 (720 h-1) (44).
Fig. 4.

Exploiting enzyme electrocatalysts and their design principles in technological applications. (A) The water–gas shift reaction is catalyzed, under ambient conditions, by two enzymes (a CODH and a hydrogenase), connected by coadsorbing them to conducting graphite platelets (potentials vs. Standard Hydrogen Electrode given for pH 6, 25 °C) (42). (B) Anatase (TiO2) nanoparticles comodified with an Ru-photosensitizer [which adsorbs light and donates electrons into the conduction band (CB)] and either a hydrogenase (for H2 production) or a CODH (for CO2 reduction) (43, 44). (C) A bioinspired electrocatalyst for interconverting H+ and H2: The design combines a catalytically competent active site that is protected from the aqueous phase but accessible to H+ and H2, and structures for relaying electrons rapidly between the electrode and the active site (47).
Can efficient electrocatalysts be designed using the principles of enzymes? Many d-block metal complexes have been synthesized to mimic the functions of redox enzymes, but they tend to lack critical features such as multielectron capacity and precisely positioned functional groups. They may not even work in water, which is certainly problematic for a water-splitting catalyst. We therefore highlight one particularly successful case, the step-by-step development of a reversible electrocatalyst for H2 production and oxidation, and show how its properties fit in with our discussions.
Among numerous functional analogs of hydrogenases, the most notable are a group of [Ni(diphosphine)2]2+ compounds (Scheme 1, 6) created by DuBois and coworkers (45, 46). The pendant N bases are well positioned for PCET, to allow smooth interconversion of H+ and η1 or η2-coordinated H2 species and mimic the assumed action of the bridgehead N atom in the [FeFe]-enzyme (Scheme 1, 1B). Compound 6 (Scheme 1, R = Ph or benzyl) catalyzes electrochemical H2 production with an overpotential requirement of approximately 0.2 V, but it must be dissolved in acetonitrile, with dimethylformamide as a proton donor (46). Subsequently, Artero and coworkers covalently linked compound 6 to conducting carbon nanotubes attached to an indium tin oxide electrode and embedded the assembly in Nafion, a proton-conducting polymer (47) (Fig. 4C). In contact with aqueous H2SO4, the integrated system functions as a reversible electrocatalyst for H2 production or oxidation, with bidirectional electrocatalytic voltammograms displaying a single wave that cuts sharply across the potential axis at Eeq. The Nafion performs the function of the protein, protecting the active site from H2O yet allowing facile H+ transfer, and the carbon nanotubes perform the function of the FeS relay (47).
Highly active and efficient redox catalysts are essential for future energy-capturing and energy-lean electrochemical/photoelectrochemical technologies. Much inspiration stems from these giant, unstable molecules that are so successful at redox catalysis yet unsuited for direct application: Enzymes reveal what is possible and provide test scenarios in which catalysis is so efficient that other factors become limiting. Bioinspired redox catalysts must do more than just mimic the inner sphere of an active site; they must provide a whole environment to minimize reorganization energies, synchronize electron and proton transfers, control the formation and stability of intermediates, and impose appropriate redox thermodynamics. In response to the need for low cost, renewable, robust, and efficient catalysts, these are tough but worthwhile challenges.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Savéant JM. Molecular catalysis of electrochemical reactions. Mechanistic aspects. Chem Rev. 2008;108:2348–2378. doi: 10.1021/cr068079z. [DOI] [PubMed] [Google Scholar]
- 2.Dewar MJS, Storch DM. Alternative view of enzyme reactions. Proc Natl Acad Sci USA. 1985;82:2225–2229. doi: 10.1073/pnas.82.8.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Warshel A, et al. Electrostatic basis for enzyme catalysis. Chem Rev. 2006;106:3210–3235. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- 4.Albery WJ, Knowles JR. Evolution of enzyme function and the development of catalytic efficiency. Biochemistry. 1976;15:5631–5640. doi: 10.1021/bi00670a032. [DOI] [PubMed] [Google Scholar]
- 5.Bard AJ, Faulkner LR. Electrochemical Methods. 2nd ed. New York: Wiley; 2001. [Google Scholar]
- 6.Sheng W, Gasteiger HA, Shao-Horn Y. Hydrogen oxidation and evolution reaction kinetics on platinum: Acid vs alkaline electrolytes. J Electrochem Soc. 2010;157:B1529–1536. [Google Scholar]
- 7.Vincent KA, Parkin A, Armstrong FA. Investigating and exploiting the electrocatalytic properties of hydrogenases. Chem Rev. 2007;107:4366–4413. doi: 10.1021/cr050191u. [DOI] [PubMed] [Google Scholar]
- 8.Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y. Structure/function relationships of [NiFe]- and [FeFe]-hydrogenases. Chem Rev. 2007;107:4273–4303. doi: 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]
- 9.Siegbahn PEM, Tye JW, Hall MB. Computational studies of [NiFe] and [FeFe] hydrogenases. Chem Rev. 2007;107:4414–4435. doi: 10.1021/cr050185y. [DOI] [PubMed] [Google Scholar]
- 10.Chardon-Noblat S, Deronzier A, Ziessel R, Zsoldos D. Selective synthesis and electrochemical behavior of trans(Cl)- and cis(Cl)-[Ru(bpy)(CO)2Cl2] complexes (bpy = 2,2′-Bipyridine). Comparative studies of their electrocatalytic activity toward the reduction of carbon dioxide. Inorg Chem. 1997;36:5384–5389. [Google Scholar]
- 11.Benson EE, Kubiak CP, Sathrum AJ, Smieja JM. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem Soc Rev. 2009;38:89–99. doi: 10.1039/b804323j. [DOI] [PubMed] [Google Scholar]
- 12.Peterson AA, Abild-Pedersen F, Studt F, Rossmeisl J, Nørskov JK. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ Sci. 2010;3:1311–1315. [Google Scholar]
- 13.Barton Cole E, et al. Using a one-electron shuttle for the multielectron reduction of CO2 to methanol: Kinetic, mechanistic, and structural insights. J Am Chem Soc. 2010;132:11539–11551. doi: 10.1021/ja1023496. [DOI] [PubMed] [Google Scholar]
- 14.Jeoung J-H, Dobbek H. Carbon dioxide activation at the Ni,Fe-cluster of anaerobic carbon monoxide dehydrogenase. Science. 2007;318:1461–1464. doi: 10.1126/science.1148481. [DOI] [PubMed] [Google Scholar]
- 15.Raaijmakers H, et al. Gene sequence and the 1.8 Å crystal structure of the tungsten-containing formate dehydrogenase from Desulfolvibrio gigas. Structure. 2002;10:1261–1272. doi: 10.1016/s0969-2126(02)00826-2. [DOI] [PubMed] [Google Scholar]
- 16.Reda T, Plugge CM, Abram NJ, Hirst J. Reversible interconversion of carbon dioxide and formate by an electroactive enzyme. Proc Natl Acad Sci USA. 2008;105:10654–10658. doi: 10.1073/pnas.0801290105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parkin A, Seravalli J, Vincent KA, Ragsdale SW, Armstrong FA. Rapid and efficient electrocatalytic CO2/CO interconversions by Carboxydothermus hydrogenoformans CO dehydrogenase I on an electrode. J Am Chem Soc. 2007;129:10328–10329. doi: 10.1021/ja073643o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moiroux J, Elving PJ. Mechanistic aspects of the electrochemical oxidation of dihydronicotinamide adenine dinucleotide (NADH) J Am Chem Soc. 1980;102:6533–6538. [Google Scholar]
- 19.Damian A, Omanovic S. Electrochemical reduction of NAD+ on a polycrystalline gold electrode. J Mol Catal A Chem. 2006;253:222–233. [Google Scholar]
- 20.Berrisford JM, Sazanov LA. Structural basis for the mechanism of respiratory complex I. J Biol Chem. 2009;284:29773–29783. doi: 10.1074/jbc.M109.032144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pankhurst KL, et al. A proton delivery pathway in the soluble fumarate reductase from Shewanella frigidimarina. J Biol Chem. 2006;281:20589–20597. doi: 10.1074/jbc.M603077200. [DOI] [PubMed] [Google Scholar]
- 22.Zu Y, Shannon RJ, Hirst J. Reversible, electrochemical interconversion of NADH and NAD+ by the catalytic (Iλ) subcomplex of mitochondrial NADH:ubiquinone oxidoreductase (complex I) J Am Chem Soc. 2003;125:6020–6021. doi: 10.1021/ja0343961. [DOI] [PubMed] [Google Scholar]
- 23.Hirst J, Sucheta A, Ackrell BAC, Armstrong FA. Electrocatalytic voltammetry of succinate dehydrogenase: Direct quantification of the catalytic properties of a complex electron-transport enzyme. J Am Chem Soc. 1996;118:5031–5038. [Google Scholar]
- 24.Stamenkovic VR, et al. Improved oxygen reduction activity on Pt3Ni(111) via increased surface site availability. Science. 2007;315:493–497. doi: 10.1126/science.1135941. [DOI] [PubMed] [Google Scholar]
- 25.Nørskov JK, et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J Phys Chem B. 2004;108:17886–17892. [Google Scholar]
- 26.Blanford CF, Heath RS, Armstrong FA. A stable electrode for high-potential, electrocatalytic O2 reduction based on rational attachment of a blue copper oxidase to a graphite surface. Chem Commun. 2007:1710–1712. doi: 10.1039/b703114a. [DOI] [PubMed] [Google Scholar]
- 27.dos Santos L, Climent V, Blanford CF, Armstrong FA. Mechanistic studies of the ‘blue’ Cu enzyme, bilirubin oxidase, as a highly efficient electrocatalyst for the oxygen reduction reaction. Phys Chem Chem Phys. 2010;12:13962–13974. doi: 10.1039/c0cp00018c. [DOI] [PubMed] [Google Scholar]
- 28.Marcus RA. Electron transfer reactions in chemistry: Theory and expt (Nobel lecture) Angew Chem Int Edit Eng. 1993;32:1111–1222. [Google Scholar]
- 29.Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 30.Léger C, et al. Enzyme electrokinetics: Using protein film voltammetry to investigate redox enzymes and their mechanisms. Biochemistry. 2003;42:8653–8662. doi: 10.1021/bi034789c. [DOI] [PubMed] [Google Scholar]
- 31.Léger C, Jones AK, Albracht SPJ, Armstrong FA. Effect of a dispersion of interfacial electron transfer rates on steady state catalytic electron transport in [NiFe]-hydrogenase and other enzymes. J Phys Chem B. 2002;106:13058–13063. [Google Scholar]
- 32.Reda T, Hirst J. Interpreting the catalytic voltammetry of an adsorbed enzyme by considering substrate mass transfer, enzyme turnover, and interfacial electron transport. J Phys Chem B. 2006;110:1394–1404. doi: 10.1021/jp054783s. [DOI] [PubMed] [Google Scholar]
- 33.Costentin C. Electrochemical approach to the mechanistic study of proton-coupled electron transfer. Chem Rev. 2008;108:2145–2179. doi: 10.1021/cr068065t. [DOI] [PubMed] [Google Scholar]
- 34.Warren JJ, Tronic TA, Mayer JM. Thermochemistry of proton-coupled electron transfer reagents and its implications. Chem Rev. 2010;110:6961–7001. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen K, et al. Atomically defined mechanism for proton transfer to a buried redox centre in a protein. Nature. 2000;405:814–817. doi: 10.1038/35015610. [DOI] [PubMed] [Google Scholar]
- 36.Frost AA. Oxidation potential-free energy diagrams. J Am Chem Soc. 1951;73:2680–2682. [Google Scholar]
- 37.Solomon EI, et al. O2 and N2O activation by binuclear, trinuclear, and tetranuclear Cu clusters in biology Acc. Chem Res. 2007;40:581–591. doi: 10.1021/ar600060t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferraroni M, et al. Crystal structure of a blue laccase from Lentinus tigrinus: Evidences for intermediates in the molecular oxygen reductive splitting by multicopper oxidases. BMC Struct Biol. 2007;7:60. doi: 10.1186/1472-6807-7-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gallaway JW, Calabrese Barton SA. Kinetics of redox polymer-mediated enzyme electrodes. J Am Chem Soc. 2008;130:8527–8536. doi: 10.1021/ja0781543. [DOI] [PubMed] [Google Scholar]
- 40.Kim YC, Wikström M, Hummer G. Kinetic gating of the proton pump in cytochrome c oxidase. Proc Natl Acad Sci USA. 2009;106:13707–13712. doi: 10.1073/pnas.0903938106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dau H, Zaharieva I. Principles, efficiency, and blueprint character of solar-energy conversion in photosynthetic water oxidation Acc. Chem Res. 2009;42:1861–1870. doi: 10.1021/ar900225y. [DOI] [PubMed] [Google Scholar]
- 42.Lazarus O, et al. Water-gas shift reaction catalyzed by redox enzymes on conducting graphite platelets. J Am Chem Soc. 2009;131:14154–14155. doi: 10.1021/ja905797w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reisner E, Powell DJ, Cavazza C, Fontecilla-Camps JC, Armstrong FA. Visible light-driven H2 production by hydrogenases attached to dye-sensitized TiO2 nanoparticles. J Am Chem Soc. 2009;131:18457–18466. doi: 10.1021/ja907923r. [DOI] [PubMed] [Google Scholar]
- 44.Woolerton TW, et al. Efficient and clean photoreduction of CO2 to CO by enzyme-modified TiO2 nanoparticles using visible light. J Am Chem Soc. 2010;132:2132–2133. doi: 10.1021/ja910091z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson AD, et al. Nature of hydrogen interactions with Ni(II) complexes containing cyclic phosphine ligands with pendant nitrogen bases. Proc Natl Acad Sci USA. 2007;104:6951–6956. doi: 10.1073/pnas.0608928104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DuBois MR, DuBois DL. The roles of the first and second coordination spheres in the design of molecular catalysts for H2 production and oxidation. Chem Soc Rev. 2009;38:62–72. doi: 10.1039/b801197b. [DOI] [PubMed] [Google Scholar]
- 47.Le Goff A, et al. From hydrogenases to noble metal free catalytic nanomaterials for H2 production and uptake. Science. 2009;326:1384–1387. doi: 10.1126/science.1179773. [DOI] [PubMed] [Google Scholar]
- 48.Lukey MJ, et al. How Escherichia coli is equipped to oxidize hydrogen under different redox conditions. J Biol Chem. 2010;285:3928–3938. doi: 10.1074/jbc.M109.067751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wood PM. The potential diagram for oxygen at pH 7. Biochem J. 1988;253:287–289. doi: 10.1042/bj2530287. [DOI] [PMC free article] [PubMed] [Google Scholar]