

Abstract
An efficient and selective four-electron plus four-proton (4e-/4H+) reduction of O2 to water by decamethylferrocene and trifluoroacetic acid can be catalyzed by a synthetic analog of the heme a3/CuB site in cytochrome c oxidase (6LFeCu) or its Cu-free version (6LFe) in acetone. A detailed mechanistic-kinetic study on the homogeneous catalytic system reveals spectroscopically detectable intermediates and that the rate-determining step changes from the O2-binding process at 25 °C room temperature (RT) to the O-O bond cleavage of a newly observed FeIII-OOH species at lower temperature (-60 °C). At RT, the rate of O2-binding to 6LFeCu is significantly faster than that for 6LFe, whereas the rates of the O-O bond cleavage of the FeIII-OOH species observed (-60 °C) with either the 6LFeCu or 6LFe catalyst are nearly the same. Thus, the role of the Cu ion is to assist the heme and lead to faster O2-binding at RT. However, the proximate Cu ion has no effect on the O-O bond cleavage of the FeIII-OOH species at low temperature.
Keywords: heme/copper, dioxygen reduction, ferric hydroperoxo, kinetic mechanism, enzyme model
The heme/copper (heme a3/CuB) heterodinuclear center in cytochrome c oxidases (CcO) (Fig. 1A) has attracted much interest, because this is the site where the four-electron and four-proton reduction of dioxygen to water takes place as the final stage of the respiration chain (Eq. 1).
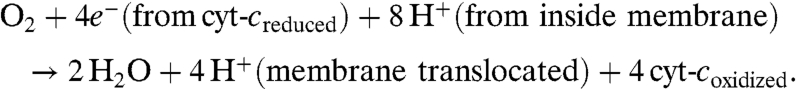 |
[1] |
Fig. 1.

(A) X-ray structures of the fully reduced bimetallic heme a3/CuB center in CcO from bovine heart (FeII⋯CuI = 5.19 Å) (3) [figure adapted from (8)]. (B) heme/Cu synthetic model for CcO (6LFeCu). (C) Cu-free version of synthetic model for CcO (6LFe).
This exergonic process, occurring without leakage of harmful partially reduced oxygen species, is coupled to the translocation of four additional protons across the membrane, generating a pH gradient and membrane potential which is harnessed through the subsequent synthesis of ATP (1–4).
In addition to protein crystallography, mechanistic enzymology, site-directed mutagenesis, and theoretical calculations, biomimetic inorganic modeling of the CcO active site has been employed. The goal is to provide insights and elucidate mechanisms of the four-electron reduction of O2 by coordination complexes including heme-Cu assemblies, as may be relevant to biological systems including CcO but also of technological significance such as in fuel cell chemistry (5–7).
A number of heme a3/CuB synthetic analogues have been developed to mimic the coordination environment of the heme a3/CuB bimetallic center in CcO (8–11). The functionality of these analogues has been investigated under two main complementary approaches. One focuses on the generation and characterization of stable O2-adducts and derived species in heme-copper synthetic assemblies (8, 9). This stoichiometric approach represents an efficient tool to probe the properties and plausibility of the intermediates relevant to the enzyme catalytic cycle and/or O-O reductive cleavage chemistry, the latter of critical importance in chemical and biochemical utilization of molecular oxygen. The second approach, based on electrochemical functional modeling, examines the capability of synthetic models to perform the catalytic four-electron reduction of O2 (10, 11). However, the solid supported state employed for such studies has precluded any spectroscopic monitoring or intermediates detection. This problem is a major obstacle for the development of kinetics and mechanistic studies in synthetic models or to answer important mechanistic questions such as the role of the CuB in the four-electron reduction of O2.
As an alternative to the limitations of the two approaches described above, we report herein an efficient 4e-/4H+ catalytic reduction of O2 to water, catalyzed by a heme/Cu functional model of CcO (6LFeCu, Fig. 1B) and its Cu-free version (6LFe, Fig. 1C). As described below, this homogeneous catalytic system has allowed us to develop a detailed kinetic description supported by spectroscopic detection of reactive intermediates overall providing unique mechanistic insights into the O-O reductive cleavage process (12).
We previously reported that the reduced form of  reacts with O2 to form a low-temperature stable
reacts with O2 to form a low-temperature stable 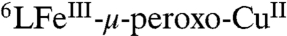
 complex (νO-O = 808 cm-1, Δ16/18O2 = -23 cm-1), deduced to be very similar to a crystallographically characterized FeIII-(μ-η2:η1-peroxo)-CuII complex reported by Naruta and coworkers (13). The
complex (νO-O = 808 cm-1, Δ16/18O2 = -23 cm-1), deduced to be very similar to a crystallographically characterized FeIII-(μ-η2:η1-peroxo)-CuII complex reported by Naruta and coworkers (13). The  complex thermally decomposes releasing
complex thermally decomposes releasing  equivalent (equiv) of O2 to generate an FeIII-μ-oxo-CuII complex
equivalent (equiv) of O2 to generate an FeIII-μ-oxo-CuII complex  (14–16). We find that the latter, as a convenient reagent, efficiently catalyzes the 4e-/4H+ reduction of O2 to water by decamethylferrocene (Fc*) as one-electron donor and trifluoroacetic acid (TFA) as a proton source (Eq. 2).
(14–16). We find that the latter, as a convenient reagent, efficiently catalyzes the 4e-/4H+ reduction of O2 to water by decamethylferrocene (Fc*) as one-electron donor and trifluoroacetic acid (TFA) as a proton source (Eq. 2).  2
2
Results and Discussion
Catalytic O2-Reduction at -60 °C.
When the time course for catalytic reaction Eq. 2 is followed by UV-visible spectroscopy at -60 °C (Fig. 2A), two main changes occur. There is a rise in the absorbance at 780 nm due to the formation of ferrocenium cation (Fc∗+) as product formed following electron-donation by Fc*. The strong absorbance at 415 nm corresponds to the Soret band of the heme in its steady-state form in the catalytic cycle. This reaction intermediate has been determined to be a unique 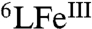 -hydroperoxo Cu (6LFeIII-OOH Cu) species (415, 538 nm);
-hydroperoxo Cu (6LFeIII-OOH Cu) species (415, 538 nm); 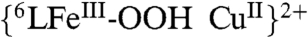 could be independently generated at low temperature (-80 °C) by the addition of an excess of TFA to the previously described peroxo complex
could be independently generated at low temperature (-80 °C) by the addition of an excess of TFA to the previously described peroxo complex  (418, 540, 558 nm) (Fig. S1). The hydroperoxo formulation is further supported by electrospray ionization mass spectrometry (ESI-MS) data and EPR spectroscopic monitoring of its formation (Figs. S2 and S3); the addition of an excess TFA to the EPR-silent
(418, 540, 558 nm) (Fig. S1). The hydroperoxo formulation is further supported by electrospray ionization mass spectrometry (ESI-MS) data and EPR spectroscopic monitoring of its formation (Figs. S2 and S3); the addition of an excess TFA to the EPR-silent 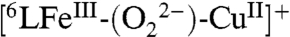 complex resulted in the display of a characteristic signal (g = 6.08) of a five coordinate high-spin heme and typical signal for the CuII moiety (A|| = 142 × 10-4 cm-1, A⊥ = 12 × 10-4 cm-1, g|| = 2.26 and g⊥ = 2.05)*. Hydroperoxo complex O-O reductive cleavage by 2Fc∗/3H+ (to two waters) leads to
complex resulted in the display of a characteristic signal (g = 6.08) of a five coordinate high-spin heme and typical signal for the CuII moiety (A|| = 142 × 10-4 cm-1, A⊥ = 12 × 10-4 cm-1, g|| = 2.26 and g⊥ = 2.05)*. Hydroperoxo complex O-O reductive cleavage by 2Fc∗/3H+ (to two waters) leads to 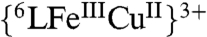 which undergoes further two-electron (2 Fc*) reduction to the fully reduced form
which undergoes further two-electron (2 Fc*) reduction to the fully reduced form 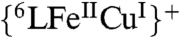 ; these observations also strongly support the (
; these observations also strongly support the ( ) steady-state species formulation (Figs. S4, S5).
) steady-state species formulation (Figs. S4, S5).
Fig. 2.

(A) UV-vis spectral changes during the four-electron reduction of O2 by Fc* (1.0 mM) catalyzed by 6LFeCu (1.0 μM) in the presence of TFA (1.0 mM) in air-saturated acetone ([O2] = 2.2 mM) at -60 °C. The insert shows the time profile of the absorbance at 780 nm relative to Fc∗+ formation. Note: a tiny amount of Fc∗+ is formed (see the nonzero absorbance) during the mixing time following combining catalyst and ferrocene solutions (in order to obtain a homogeneous solution) and before the first spectrum is recorded. Thus, for practical reasons, this first spectrum corresponds to time = 0. (B) Plots of kobs vs. [cat] for 6LFeCu and 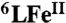 at -60 °C. (C) Plots of kobs vs. [Fc*] for 6LFeCu and 6LFe at -60 °C. (D) Plots of kobs vs. [TFA] for 6LFeCu and
at -60 °C. (C) Plots of kobs vs. [Fc*] for 6LFeCu and 6LFe at -60 °C. (D) Plots of kobs vs. [TFA] for 6LFeCu and 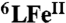 at -60 °C. (E) Plots of kobs vs. [O2] for 6LFeCu and 6LFe at -60 °C.
at -60 °C. (E) Plots of kobs vs. [O2] for 6LFeCu and 6LFe at -60 °C.
The stoichiometry of the overall reaction was confirmed by the observation that exactly four equiv of Fc∗+ form per mole of O2 reacted (i.e., under limiting [O2]). H2O2 as a product formed via the partial two-electron reduction of O2 was ruled out by iodometric titration experiments (Fig. S6); no hydrogen peroxide was detected. In addition, it was confirmed that O2 reduction by Fc∗ without catalysts (6LFeCu or 6LFe) is negligible under the same conditions (Fig. S6).
To help to examine the role of the Cu in this system, a Cu-free version (6LFe) (15) was also subjected to the same catalytic reaction conditions as 6LFeCu. Even in the absence of Cu the reaction proceeds through the 4e- process (Fig. S7). This result is in accordance with earlier electrochemical studies in which it was found that only the iron porphyrinate itself is essential for O2 four-electron reduction (17–20). In the case of  as well, the kinetics reveal that the FeIII-OOH complex represents the catalytic cycle steady-state species (Fig. S7A).
as well, the kinetics reveal that the FeIII-OOH complex represents the catalytic cycle steady-state species (Fig. S7A).
Further kinetic insights demonstrate the reactions being studied are zero-order with respect to Fc∗+ formation for both 6LFeCu (Fig. 2A, inset) and 6LFe (Fig. S7B) as catalysts. The derived rate constants increase linearly with an increase in catalyst concentrations (Fig. 2B) and also remain constant with a change in the concentration of Fc* (Fig. 2C). Further, and to our surprise, the observed zero-order rate constants (kobs) with 6LFeCu at -60 °C are exactly the same as those with the Cu-free complex 6LFe (Fig. 2B). Also, the rate of the reaction remains constant with an increase in [TFA] or [O2] (Fig. 2 D and E).
These data demonstrate that the rate of O2-reduction is not affected by the concentrations of O2, TFA, or Fc*. Thus, the rate-determining step of the catalytic reaction at low temperature is O-O bond cleavage in the steady-state FeIII-OOH species and this is followed by rapid electron transfer to complete the 4e- reduction of O2. The same kobs value observed for 6LFeCu and 6LFe suggests that the Cu is not bound to the FeIII-OOH moiety in  .
.
Catalytic O2-Reduction at 25 °C.
The spectral changes observed for the catalytic reaction (Eq. 2) at RT are quite different from those observed at -60 °C. At RT, the Soret band corresponding to the steady-state is observed at 422 nm (Fig. 3A), and this can be assigned to the reduced heme (FeII) (16). Further confirmation comes from spectral correlation with the species generated in the absence of O2 using Fc* in the presence of TFA (Figs. S5 and S8).
Fig. 3.

(A) Stopped-flow measurement of the absorbance changes during catalytic O2-reduction by Fc* (200 μM) with 6LFeCu (2.0 μM), TFA (4 mM) in O2-saturated acetone at 25 °C. The insert shows the time profile of the absorbance at 780 nm relative to Fc∗+ formation. See the Fig. 2A legend for the explanation of the nonzero ferrocenium concentration initially observed. (B) Plots of kobs vs. [cat] for 6LFeCu and 6LFe at 25 °C. (C) Plots of kobs vs. [Fc*] for 6LFeCu and 6LFe at 25 °C. (D) Plots of kobs vs. [TFA] for 6LFeCu and 6LFe at 25 °C. (E) Plots of kobs vs. [O2] for 6LFeCu and 6LFe at 25 °C.
In the presence of excess O2, the reaction kinetics are zero-order in Fc∗+ formation (Fig. 3A, inset), while the zero-order rate constant increases linearly with an increase in the catalyst concentration (Fig. 3B, Fig. S9), as expected. More interestingly, the rate constant also increases with an increase in the O2 concentration (Figs. 3E, Fig. S10), but remains constant with increases in the acid or Fc* concentrations (Fig. 3C). On the basis of these observations, and knowing that the reduced heme (6LFe) is the steady-state species observed for the catalytic cycle (vide supra), we can conclude that the rate-determining step at 25 °C is the O2-binding to the 6LFeCu catalyst complex.
As well, our kinetics investigations demonstrate that for the Cu-free catalyst, the reduced heme (6LFe) is the species found to be in steady-state. Thus, O2-binding is rate-determining. However, the rate constant determined (as measured by Fc∗+ formation) is approximately two times less than that observed with 6LFeCu (Fig. 3B). Consequently, the efficiency of the catalyst, as represented by the turnover frequency, is greater for 6LFeCu (TOF = 41 s-1) than that for 6LFe (TOF = 24 s-1). This result suggests that the role of the Cu, at ambient temperature, in this biomimetic catalytic system, is to assist the heme and lead to faster O2-binding during the catalytic cycle.
We can compare the results here with this heme-Cu CcO model compound to systems studied under similar conditions, in homogeneous solutions with ferrocene derivative reductant sources. 6LFeCu offers a significantly higher efficiency (2.2 × 106 s-1, 298 K; slope in Fig. 4C) than that observed for previously described cofacial dicobalt porphyrins (320 s-1) or with a mononuclear copper complex (17 s-1) (21–23).
Fig. 4.

Plots of kobs vs. temperature for the four-electron reduction of O2 by Fc* (1.0 mM) catalyzed by 6LFeCu or 6LFe (1.0 μM) in the presence of TFA (1.0 mM) in an air-saturated acetone. (B) Plots of the absorbance maximum vs. temperature corresponding to the Soret band of steady-state in the case of 6LFeCu and 6LFe. (C) Arrhenius plots obtained by conversion of plots (A).
Another role for Cu, as an electron storage and delivery site, was previously proposed by Collman and coworkers based on electrochemical studies (24). This difference may result from the fact that, in the electrochemical approach, the electron flow to the catalyst is controlled, in contrast with our solution homogeneous system. Also, an electrode material supported catalyst may possess a structure modified from that observed in solution (25).
Variable Temperature (VT) Studies.
As deduced from Fig. 4A, VT studies provide a clearer picture of the change in the rate-determining step for the catalytic 4e-/4H+ reduction of O2 by Fc* with TFA. At T < -5 °C, the O-O bond cleavage of FeIII-OOH is rate-determining.
Because the Cu is not involved here, the zero-order rates (kobs) for 6LFeCu are nearly the same as those of 6LFe (Fig. 4 A and C). Between -60 and -5 °C (Fig. 4C), the activation energy determined for  (9.4 ± 0.1 kcal mol-1) and 6LFe (9.9 ± 0.2 kcal mol-1) are essentially the same.
(9.4 ± 0.1 kcal mol-1) and 6LFe (9.9 ± 0.2 kcal mol-1) are essentially the same.
At higher temperature (T > -5 °C), however, the O2-binding to the reduced species (6LFeCu or 6LFe) becomes rate-determining. The difference in the kobs value between 6LFeCu and 6LFe becomes larger with an increase in temperature and at physiological temperature (37 °C) the kobs value for 6LFeCu becomes seven times larger than that of 6LFe.
Fig. 4B shows the change in Soret band as a function of reaction temperature. This phenomenon clearly represents a change in the identity of the steady-state complex for the low and higher temperature processes: The 415–417 nm absorption representing the FeIII-OOH complex in steady-state, shifts to 422–424 nm, identified as the reduced (FeII) heme complex. Notably, and showing the consistency of our findings and conclusions, the boundary temperature is about -5 °C for the different analyses represented by Fig. 4 A–C.
Kinetic analyses and Arrhenius plots (Fig. 4C) show that the catalytic O2-reduction using 6LFeCu gives an activation energy of 4.2 ± 0.1 kcal mol-1, between -5 and 35 °C. However, with the 6LFe catalyst, kobs values for O2-binding decrease with increasing temperature affording an apparent negative activation energy of -3.0 ± 0.1 kcal mol-1. Although the rate of Fc∗+ formation obeys approximately zero-order kinetics, a more careful examination of the data for 6LFe reveals that the apparent zero-order rate constant increases with increasing Fc* concentration (Fig. 3C, Fig. S11B). By contrast, for 6LFeCu, the zero-order rate constant remains the same under such conditions (Fig. 4C, Fig. S11A). These observations indicate that the binding of O2 to 6LFe may be in an equilibrium process and the subsequent electron-transfer reduction of a 6LFe-O2 intermediate competes with the back reaction (i.e., releasing O2). Because the O2-binding is likely an exothermic process, the back reaction (i.e., releasing O2) becomes faster than the electron transfer as the temperature increases. In such a case, for 6LFe, the overall reaction rate will decrease with an increase in temperature (Fig. 4A), thus the negative activation energy.
In light of all the results we suggest a mechanism for the 4e-/4H+ reduction of O2 catalyzed by our CcO active site model (6LFeCu) in the presence of Fc* and TFA in acetone, Fig. 5A. All species have been spectroscopically identified, and the interconversions characterized by the detailed kinetic studies. In the presence of acid, 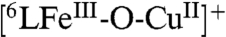 rapidly forms
rapidly forms 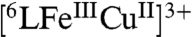 (409, 505 nm, Fig. S5) releasing water and the catalytic cycle starts via a fast reduction of the heme and then the Cu to generate the reduced complex
(409, 505 nm, Fig. S5) releasing water and the catalytic cycle starts via a fast reduction of the heme and then the Cu to generate the reduced complex 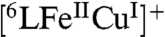 ; as discussed, this corresponds to the steady-state species at RT. The O2-binding in the next step is rate-determining at RT. The
; as discussed, this corresponds to the steady-state species at RT. The O2-binding in the next step is rate-determining at RT. The  complex thus generated undergoes a fast protonation to form
complex thus generated undergoes a fast protonation to form  corresponding to the low temperature steady-state species. An additional two equiv of Fc* and three protons are needed to complete the 4e-/4H+ reduction of O2 to water and regenerate the
corresponding to the low temperature steady-state species. An additional two equiv of Fc* and three protons are needed to complete the 4e-/4H+ reduction of O2 to water and regenerate the  catalyst.
catalyst.
Fig. 5.

The proposed mechanism of the four-electron reduction of O2 to water by Fc* in the presence of TFA in acetone catalyzed by: (A) the heme/Cu bimetallic center model of CcO (6LFeCu), (B) the Cu-free version (6LFe). In fact, it has not yet been established as to which side of the heme the O2-binding occurs.
In the case of 6LFe, the Cu-free catalyst version, the cycle (Fig. 5B) also starts via fast electron transfer from Fc* to the heme, forming  . In the absence of the Cu, however, as discussed, the subsequent O2-binding is slower than that observed for
. In the absence of the Cu, however, as discussed, the subsequent O2-binding is slower than that observed for  . Once an O2-adduct forms [formally an FeIII-superoxo species (418, 539 nm) (16)], subsequent electron transfer and protonation leads to FeIII-OOH, in steady-state at low temperature. Thus, the important role of Cu is to facilitate the O2- binding to
. Once an O2-adduct forms [formally an FeIII-superoxo species (418, 539 nm) (16)], subsequent electron transfer and protonation leads to FeIII-OOH, in steady-state at low temperature. Thus, the important role of Cu is to facilitate the O2- binding to  , directly increasing the rate of this reaction and stabilizing the resulting
, directly increasing the rate of this reaction and stabilizing the resulting  complex at ambient temperature. As seen for the 6LFeCu catalyst, the rate-determining step changes at low temperature, from O2-binding to O-O bond cleavage in the FeIII-OOH complex. Cu does not influence this latter step.
complex at ambient temperature. As seen for the 6LFeCu catalyst, the rate-determining step changes at low temperature, from O2-binding to O-O bond cleavage in the FeIII-OOH complex. Cu does not influence this latter step.
In fact, this conclusion about the role of Cu agrees with other notable findings: (i) O2-binding to copper complexes can occur at near diffusion-controlled rates (26, 27), (ii) CcO enzyme studies in fact implicate CuB as the entry point for O2 during catalysis (28–30).
Conclusion
In summary, we have here described a selective and efficient (turnovers > 1,000) four-electron reduction of O2 to water (without formation of H2O2) catalyzed by our CcO active site analogue (6LFeCu). Unprecedented direct observation of different reactive intermediates taking part in the catalytic cycle [i.e., 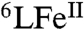 (CuI) and
(CuI) and  )], depending on the temperature of the system, combined with detailed kinetics studies of different steps of the catalytic reaction when comparing 6LFeCu and its Cu-free version allowed us to obtain unique mechanistic insights. Important conclusions that may relate to CcO chemistry are that in our model system the role for the Cu is to enhance the O2-binding and that it needs not contribute directly to the O-O cleavage process. As no model system can prove a mechanism for an enzyme, our chemistry provides that further systematic variations in the architecture or nature of the synthetic heme-copper catalyst [such as changes in the heme type, Cu-ligand denticity, N-donor type, neighboring groups which can H-bond or alter the local dielectric, or H• (H+ + e-) donors] can and will lead to further insights into O2 reductive activation relevant to CcO or fuel cell chemistry.
)], depending on the temperature of the system, combined with detailed kinetics studies of different steps of the catalytic reaction when comparing 6LFeCu and its Cu-free version allowed us to obtain unique mechanistic insights. Important conclusions that may relate to CcO chemistry are that in our model system the role for the Cu is to enhance the O2-binding and that it needs not contribute directly to the O-O cleavage process. As no model system can prove a mechanism for an enzyme, our chemistry provides that further systematic variations in the architecture or nature of the synthetic heme-copper catalyst [such as changes in the heme type, Cu-ligand denticity, N-donor type, neighboring groups which can H-bond or alter the local dielectric, or H• (H+ + e-) donors] can and will lead to further insights into O2 reductive activation relevant to CcO or fuel cell chemistry.
Materials and Methods
Materials.
Grade quality solvents and chemicals were obtained commercially and used without further purification unless otherwise noted. Decamethylferrocene (Fc*) (99%) was purchased from STREM, USA, TFA (99%) from Sigma Aldrich and NaI (99.5%) from Wako, Japan. Acetone was purchased from Wako, Japan and used whether without further purification for non-air-sensitive experiment or dried and distilled under argon then deoxygenated by bubbling with argon for 30–45 min for air-sensitive experiment. Preparation and handling of air-sensitive compounds were performed under MBraun UNilab inert atmosphere (< 1 ppm O2, < 1 ppm H2O) glove box filled with nitrogen. The complexes 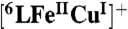 and
and 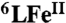 were prepared according to the literature procedure (14, 15).
were prepared according to the literature procedure (14, 15).
UV-Vis Spectroscopy Measurements.
Hewlett Packard 8453 diode array spectrophotometer with a quartz cuvette (path length = 10 mm) was used to examine the spectral change in the UV-visible. This instrument was coupled to Unisoku thermostated cell holder for low-temperature experiments. In a typical catalytic reaction, the quartz cuvette is loaded with 3 mL of 1∶1 Fc*/TFA (1 mM) in air-saturated solution of acetone. Then 30 μL of 0.1 mM solution of the catalyst in acetone is injected into the cuvette under vigorous stirring. The catalytic reaction is monitored by the increase in the absorbance at 780 nm corresponding to the formation of the ferrocenium cation (Fc∗+).
The limiting concentration of O2 in an acetone solution was prepared by a mixed gas flow of O2 and N2. The mixed gas was controlled by using a gas mixer (Kofloc GB-3C, KOJIMA Instrument Inc.), which can mix two or more gases at a certain pressure and flow rate.
Stopped-Flow Measurements.
Stopped-flow measurements were performed on a UNISOKU RSP-601 stopped-flow spectrophotometer with an MOS-type high selective photodiode array at various temperatures (248 K–298 K) using a Unisoku thermostated cell holder designed for low-temperature experiments. In a typical reaction, two reactant solutions for stopped-flow mixing were prepared. One is solution containing TFA and the catalyst in acetone, the other is solution Fc* in acetone. Rates of O2 reduction reactions were determined by monitoring the appearance of the absorption band at 780 nm due to the formation of Fc∗+.
ESI-MS Measurements.
The detailed information about ESI-MS is provided in SI Text.
EPR Measurements.
To a reaction solution of  (Fig. S3) or
(Fig. S3) or  (1.0 mM) (Fig. S14) in acetone solution, 10 equiv of TFA (10 mM) was added at RT. The resulting solution in the quartz ESR tube (3.0 mm i.d.) was frozen in liquid nitrogen. EPR spectrum recorded at 77 K was taken on a JEOL X-band spectrometer (JES-RE1XE) under nonsaturating microwave power conditions (1.0 mW) operating at 9.2025 GHz (Fig. S14) or a Bruker EMX spectrometer operating at X-band using microwave frequencies around 9.5 GHz (Fig. S3). The magnitude of the modulation was chosen to optimize the resolution and the signal to noise ratio (S/N) of the observed spectrum (modulation width, 20 G; modulation frequency, 100 kHz).
(1.0 mM) (Fig. S14) in acetone solution, 10 equiv of TFA (10 mM) was added at RT. The resulting solution in the quartz ESR tube (3.0 mm i.d.) was frozen in liquid nitrogen. EPR spectrum recorded at 77 K was taken on a JEOL X-band spectrometer (JES-RE1XE) under nonsaturating microwave power conditions (1.0 mW) operating at 9.2025 GHz (Fig. S14) or a Bruker EMX spectrometer operating at X-band using microwave frequencies around 9.5 GHz (Fig. S3). The magnitude of the modulation was chosen to optimize the resolution and the signal to noise ratio (S/N) of the observed spectrum (modulation width, 20 G; modulation frequency, 100 kHz).
Supplementary Material
Acknowledgments.
This research was supported by a Grant-In-Aid 20108010 (S.F.), the Global Centers of Excellence (COE) Program from the Ministry of Education, Culture, Sports, Science and Technology, Japan (to Z.H. and S.F.), by the National Institutes of Health (GM28962 to K.D.K.), and World Class University (WCU) Program R31-2008-000-10010-0 (to K.D.K. and S.F.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
*In 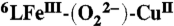 the high-spin FeIII (d5, S = 5/2) system is strongly antiferromagnetically coupled to the CuII ion (d9, S = 1/2) through the peroxo bridge, giving an overall S = 2 spin system and an EPR-silent spectrum. The addition of the excess of TFA causes the formation of the [FeIII-OOH CuII] in which the Fe and the Cu are no longer antiferromagnetically coupled and both the high-spin FeIII and the CuII ion centers independently display an EPR signal (Fig. S3).
the high-spin FeIII (d5, S = 5/2) system is strongly antiferromagnetically coupled to the CuII ion (d9, S = 1/2) through the peroxo bridge, giving an overall S = 2 spin system and an EPR-silent spectrum. The addition of the excess of TFA causes the formation of the [FeIII-OOH CuII] in which the Fe and the Cu are no longer antiferromagnetically coupled and both the high-spin FeIII and the CuII ion centers independently display an EPR signal (Fig. S3).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1104698108/-/DCSupplemental.
References
- 1.Ferguson-Miller S, Babcock GT. Heme/copper terminal oxidases. Chem Rev. 1996;96:2889–2908. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 2.Tsukihara T, et al. Structures of metal sites of oxidized bovine heart cytochrome c oxidase at 28 A. Science. 1995;269:1069–1074. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 3.Yoshikawa JS, et al. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science. 1998;280:1723–1723. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 4.Pereira MM, Santana M, Teixeira M. A novel scenario for the evolution of haem-copper oxygen reductases. Biochim Biophys Acta. 2001;1505:185–208. doi: 10.1016/s0005-2728(01)00169-4. [DOI] [PubMed] [Google Scholar]
- 5.Cracknell JA, Vincent KA, Armstrong A. Enzymes as working or inspirational electrocatalysts for fuel cells and electrolysis. Chem Rev. 2008;108:2439–2461. doi: 10.1021/cr0680639. [DOI] [PubMed] [Google Scholar]
- 6.Willner I, Yan YM, Willner B, Tel-Vered R. Integrated enzyme-based biofuel cells-A review. Fuel Cells. 2009;9:7–24. [Google Scholar]
- 7.Anson FC, Shi A, Steiger B. Novel multinuclear catalysts for the electroreduction of dioxygen directly to water. Acc Chem Res. 1997;30:437–444. [Google Scholar]
- 8.Kim E, Chufán EE, Kamaraj K, Karlin KD. Synthetic models for heme-copper oxidases. Chem Rev. 2004;104:1077–1134. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]
- 9.Chufán EE, Puiu SC, Karlin KD. Heme-copper/dioxygen adduct formation, properties, and reactivity. Acc Chem Res. 2007;40:563–572. doi: 10.1021/ar700031t. [DOI] [PubMed] [Google Scholar]
- 10.Collman JP, Boulatov R, Sunderland CJ, Fu L. Functional analogues of cytochrome c oxidase, myoglobin, and hemoglobin. Chem Rev. 2004;104:561–588. doi: 10.1021/cr0206059. [DOI] [PubMed] [Google Scholar]
- 11.Collman JP, Boulatov R, Sunderland CJ. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 11. San Diego, CA: Academic Press; 2003. pp. 1–49. [Google Scholar]
- 12.Collman JP, Ghosh S, Dey A, Decreau RA, Yang Y. Catalytic reduction of O2 by cytochrome c using a synthetic model of cytochrome c oxidase. J Am Chem Soc. 2009;131:5034–5035. doi: 10.1021/ja9001579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chishiro T, et al. Isolation and crystal structure of a peroxo-bridged heme-copper complex. Angew Chem Int Ed. 2003;42:2788–2791. doi: 10.1002/anie.200351415. [DOI] [PubMed] [Google Scholar]
- 14.Ju TD, et al. Dioxygen reactivity of fully reduced [LFeII⋯CuI]+ complexes utilizing tethered tetraarylporphyrinates: active site models for heme-copper oxidases. Inorg Chem. 1999;38:2244–2245. [Google Scholar]
- 15.Ghiladi RA, et al. Formation and characterization of a high-spin heme-copper dioxygen (peroxo) complex. J Am Chem Soc. 1999;121:9885–9886. [Google Scholar]
- 16.Ghiladi RA, Karlin KD. Low-temperature UV-visible and NMR spectroscopic investigations of O2 binding to (6L)FeII, a ferrous heme bearing covalently tethered axial pyridine ligands. Inorg Chem. 2002;41:2400–2407. doi: 10.1021/ic0103547. [DOI] [PubMed] [Google Scholar]
- 17.Shigehara K, Anson FC. Electrocatalytic activity of three iron porphyrins in the reduction of dioxygen and hydrogen peroxide at graphite cathodes. J Phys Chem. 1982;86:2776–2783. [Google Scholar]
- 18.Shi CN, Anson FC. Catalytic pathways for the electroreduction of oxygen by iron tetrakis(4-N-methylpyridyl)porphyrin or iron tetraphenylporphyrin adsorbed on edge plane pyrolytic graphite electrodes. Inorg Chem. 1990;29:4298–4305. [Google Scholar]
- 19.Ricard D, Andrioletti B, L’Her M, Boitrel B. Electrocatalytic reduction of dioxygen to water by tren-capped porphyrins, functional models of cytochrome c oxidase. Chem Commun. 1999:1523–1524. [Google Scholar]
- 20.Ricard D, L’Her M, Ricard P, Boitrel B. Iron porphyrins as models of cytochrome c oxidase. Chem Eur J. 2001;7:3291–3297. doi: 10.1002/1521-3765(20010803)7:15<3291::aid-chem3291>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 21.Fukuzumi S, Okamoto K, Gros CP, Guilard R. Mechanism of four-electron reduction of dioxygen to water by ferrocene derivatives in the presence of perchloric acid in benzonitrile, catalyzed by cofacial dicobalt porphyrins. J Am Chem Soc. 2004;126:10441–10449. doi: 10.1021/ja048403c. [DOI] [PubMed] [Google Scholar]
- 22.Rosenthal J, Nocera DG. Role of proton-coupled electron transfer in O-O bond activation. Acc Chem Res. 2007;40:543–553. doi: 10.1021/ar7000638. [DOI] [PubMed] [Google Scholar]
- 23.Fukuzumi S, et al. Mononuclear copper complex-catalyzed four-electron reduction of oxygen. J Am Chem Soc. 2010;132:6874–6875. doi: 10.1021/ja100538x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collman JP, et al. A cytochrome c oxidase model catalyzes oxygen to water reduction under rate-limiting electron flux. Science. 2007;315:1565–1568. doi: 10.1126/science.1135844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kadish KM, et al. Catalytic activity of biscobalt porphyrin-corrole dyads toward the reduction of dioxygen. Inorg Chem. 2009;48:2571–2582. doi: 10.1021/ic802092n. [DOI] [PubMed] [Google Scholar]
- 26.Fry HC, Scaltrito DV, Karlin KD, Meyer GJ. The rate of O2 and CO binding to a copper complex, determined by a “flash-and-trap” technique, exceeds that for hemes. J Am Chem Soc. 2003;125:11866–11871. doi: 10.1021/ja034911v. [DOI] [PubMed] [Google Scholar]
- 27.Lucas HR, Meyer GJ, Karlin KD. CO and O2 binding to pseudo-tetradentate ligand-copper(i) complexes with a variable N-donor moiety: kinetic/thermodynamic investigation reveals ligand-induced changes in reaction mechanism. J Am Chem Soc. 2010;132:12927–12940. doi: 10.1021/ja104107q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blackmore RS, Greenwood C, Gibson QH. Studies of the primary oxygen intermediate in the reaction of fully reduced cytochrome oxidase. J Biol Chem. 1991;266:19245–19257. [PubMed] [Google Scholar]
- 29.Oliveberg M, Malmström BG. Reaction of dioxygen with cytochrome c oxidase reduced to different degrees: indications of a transient dioxygen complex with copper-B. Biochemistry. 1992;31:3560–3563. doi: 10.1021/bi00129a002. [DOI] [PubMed] [Google Scholar]
- 30.Muramoto K, et al. Bovine cytochrome c oxidase structures enable O2 reduction with minimization of reactive oxygens and provide a proton-pumping gate. Proc Nat Acad Sci USA. 2010;107:7740–7745. doi: 10.1073/pnas.0910410107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.